
Preparation of single-phase three-component alkaline earth oxide of (BaSrMg)O: a high capacity and thermally stable chemisorbent for oxygen separation†
Xuncai
Chen
a,
Taesung
Jung
b,
Jongho
Park
*b and
Woo-Sik
Kim
*a
aDepartment of Chemical Engineering, ILRI, Kyung Hee University, Yoing Kiheung-ku Seochun-dong, Kyungki-do 449-701, Republic of Korea. E-mail: wskim@khu.ac.kr
bKorea Institute of Energy Research, 71-2, Jangdong, Yuseong-gu, Daejeon, Republic of Korea. E-mail: jongho@kier.re.kr
First published on 28th October 2014
Abstract
This study presents a preparation method for a single-phase three-component alkaline earth oxide of (BaSrMg)O that is a high capacity and thermally stable chemisorbent for oxygen separation based on the redox reaction cycle of BaO + 1/2O2 ↔ BaO2. First, single-phase (BaSr)CO3 is co-precipitated based on the reaction of Ba2+ and Sr2+ with CO32− in a solution, and then transformed to single-phase (BaSrMg)CO3 with the addition of an Mg2+ solution. When varying the reaction conditions, such as the reactant concentrations of Ba2+, Sr2+, Mg2+, and CO32− and the reaction temperature, (Ba0.52Sr0.06Mg0.42)CO3 crystals are identified as the most stable phase. The single-phase (BaSrMg)CO3 is then converted into single-phase (BaSrMg)O by thermal decomposition under an H2 atmosphere at 750 °C. According to a TGA analysis, the chemisorption and desorption of oxygen in (BaSrMg)O are very fast at t80 = 3.9 min and t80 = 14 min, respectively. In addition, the chemisorption capacity of (BaSrMg)O is higher at 2.02 mmol g−1 at 700 °C when compared with the chemisorption capacity of BaO/MgO at 1.75 mmol g−1 (Jin et al., Ind. Eng. Chem. Res., 2005, 44, 2942). (BaSrMg)O is also thermally stable due to the inclusion of Mg. Thus, the chemisorption capacity of (BaSrMg)O is unchanged, even over 10 redox reaction cycles. Additionally, the transient oxygen pressure required for the redox reaction of BaO–BaO2 is shifted from 76 mmHg to 148 mmHg due to the inclusion of Sr in (BaSrMg)O. Consequently, the three component alkaline earth oxide (BaSrMg)O can be a highly effective sorbent for industrial applications to oxygen separation in terms of the process design and operation.
1. Introduction
Currently, almost 100 million tonnes of oxygen are consumed for industrial processes yearly.2 Additionally, this demand will only increase due to the involvement of oxygen in so many industrial processes, including glass melting, semiconductor manufacturing, food processing, metallurgical manufacturing (e.g., copper and steel production), cutting and welding, fuel combustion, GTL conversion, and wastewater treatment.3,4 Therefore, the development of a new approach for easy and cost-effective oxygen separation is an important challenge.Cryogenic distillation and physisorption are the two main methods presently applied to air separation.5 While cryogenic distillation is suitable for large-scale air separation, it requires high energy consumption and high capital and operating costs.6 Meantime, although physisorption is advantageous for processification, it has a poor selectivity for air separation as the physisorption enthalpies of oxygen and nitrogen are very similar.7–9 Thus, several recent studies have been focused on exploiting various oxygen-selective materials for air separation, such as perovskite, alkaline earth oxides, and metal–organic frameworks.7,10–13 Among these oxygen-selective materials, perovskite and alkaline earth oxides have been investigated for potential practical applications, as they can store oxygen in a lattice by sorption and then reversibly release the oxygen by desorption.14,15 As a result, a new method using alkaline earth oxides has been commercialized for air separation based on the BaO–BaO2 redox reaction cycle: BaO + 1/2O2 ↔ BaO2.1,10 In this method, BaO exhibits a high oxygen selectivity and sorption capacity. However, since BaO is very reactive, it reacts easily with its surroundings and is also sintered at an elevated temperature in the BaO–BaO2 redox reaction, resulting in a poor thermal stability and poor productivity for a multi-cyclic operation.1 Furthermore, the transition of the BaO–BaO2 redox reaction occurs at a low oxygen pressure, which requires a high operating cost. Thus, various attempts have already been made to overcome these drawbacks using additives as a promoter or stabilizer for the BaO–BaO2 redox reaction.16–18 For example, Jin et al.1 used MgO powders to improve the thermal stability of the BaO–BaO2 redox reaction, while Park et al.10 showed that the reaction rate and thermal stability of the BaO–BaO2 redox reaction were both enhanced by coating the BaO particles with MgO. Notwithstanding, the requirement of a low transition oxygen pressure for the BaO–BaO2 redox reaction remains an issue. Additionally, local sintering still occurs due to the non-uniform mixture of the MgO powders in the BaO particles.
There have been many recent attempts to develop single-phase multi-component alkaline earth oxides that are suitable for practical application to air separation with a high sorption capacity, high thermal stability, high selectivity, and high transition oxygen pressure. In general, the preparation of an alkaline earth oxide begins from the synthesis of an alkaline earth carbonate, as the alkaline earth carbonate is simply converted into an alkaline earth oxide, additionally, its composition and phase can be accurately controlled in an aqueous reaction.19 Therefore, various two-component alkaline earth carbonates have already been synthesized using a co-precipitation reaction,20–25 However, their methods have hardly produced single-phase alkaline earth carbonate (BaMg)CO3, but multi-phase ones, such as (BaMg)CO3 + MgCO3, (BaMg)CO3 + BaCO3, BaCO3 + LiCO3etc., frustrating their practical application. Furthermore, no studies have yet been reported on the synthesis of a three-component alkaline earth carbonate in a solution due to the complicated reaction, difficulty in controlling the reaction, and easy phase separation leading to multi-phase particles.
Accordingly, the present study presents a novel synthesis method for a single-phase three-component alkaline earth carbonate of (BaSrMg)CO3 as a precursor for the preparation of a single-phase three-component alkaline earth oxide of (BaSrMg)O. In addition, the formation mechanism of (BaSrMg)CO3 is also investigated. Here, it should be noted that the Mg and Sr in the alkaline earth components are considered as additives for thermal stabilization and a high transient oxygen pressure for the redox reaction of the sorbent. The effects of the reaction conditions, such as the Ba2+, Sr2+, Mg2+, and CO32− reactant concentrations and reaction temperature, on the synthesis of (BaSrMg)CO3 are also studied. Additionally, the redox reaction (chemisorption–desorption of oxygen) of (BaSrMg)O is characterized. Finally, the influence of Sr on the O2 chemisorption–desorption of (BaSrMg)O is investigated based on Raman characterization using 18O-labeling.
2. Experimental section
2.1 Preparation of (BaSrMg)O
The reagents of Ba(NO3)2, Sr(NO3)2, Mg(NO3)2·6H2O, and (NH4)2CO3 were all purchased from Sigma-Aldrich (ACS grade). 100 mL of a Ba(NO3)2 aqueous solution (84.8 g l−1) was mixed with an equal volume of a Sr(NO3)2 aqueous solution (17.1 g l−1) in a round-bottom flask equipped with a reflux condenser, and heated to 90 °C using a heating mantle. Next, 100 mL of a (NH4)2CO3 aqueous solution (40 g l−1) was added to the above mixture of Ba(NO3)2 and Sr(NO3)2 for the co-precipitation of (BaSr)CO3. After 2 hours, 100 mL of a Mg(NO3) aqueous solution (104.0 g l−1) was slowly fed into the above (BaSr)CO3 suspension at a flow rate of 1.67 mL min−1 for the re-coprecipitation of (BaSrMg)CO3. After finishing the feeding, the suspension was continuously stirred for 7 hours. Here, all the reactions were carried out at 90 °C. The (BaMgSr)CO3 crystals in the final suspension were filtered out using a filter paper and washed with distilled water. The filtered crystals were then dried in a convection oven at 110 °C overnight. Thereafter, the dried (BaMgSr)CO3 crystals were calcined in a tube furnace at 750 °C under a H2 environment (H2 flow rate = 100 mL min−1) for 4 hours to obtain (BaMgSr)O crystals.2.2 Analysis of (BaSrMg)CO3 and (BaSrMg)O
The size and shape of the (BaSrMg)CO3 and (BaSrMg)O crystals were examined using an FE-SEM (LEO SUPRA 55 microscope, Carl Zeiss, Germany). Their structure was analyzed using powder X-ray diffraction (M18XHF-SRA, Mac Science, Japan) with Cu Kα radiation (λ = 1.54056 Å). A (BaSrMg)CO3 particle was cut using a Cryo Ultra microtome (RMC PTPC ultramicrotome & photographic, Boeckeler, USA), and the chemical composition at the cross-section was analyzed using FE-SEM/EDS. During the synthesis, the cation concentrations in the solution (Ba2+, Mg2+, Sr2+) were measured using an ICP (Direct Reading Echelle ICP, Leeman, USA). The chemical compositions of (BaMgSr)CO3 and (BaMgSr)O were analyzed using EDS element mapping (Oxford INCA Resolution 30 mm2 136 eV at Mn Kα 5 B to 92 U) and ICP.2.3 O2 sorption/desorption of (BaSrMg)O
A thermogravimetric analyser (SDTQ600, TA, USA) was used to determine the sorption capacity of the (BaSrMg)O chemisorbent. About 50 mg of the (BaSrMg)O sample was placed in the TGA chamber and heated up to 700 °C at 5 K min−1. After stabilizing the temperature at 700 °C, a (1 atm) gas mixture (O2 and N2 gas) was injected into the TGA chamber. Here, the oxygen partial pressure was controlled from 80–760 mmHg using mass flow controllers (5850E, Brooks, Japan). The pressures of N2 and O2 were monitored using a Flow & pressure controller (GMC 1200, ISVT, Korea). Every 1 h, the oxygen and nitrogen partial pressure was changed for the sorption/desorption isotherm. Here, the total gas pressure was always fixed at 760 mmHg.2.4 Analysis using isotope of 18O
The chemisorption mechanism was studied using an oxygen isotope (18O2). The (BaSrMg)O sample was placed on a hot stage (700 °C) under vacuum conditions, and 18O2 gas slowly injected onto the hot stage (Linkam TS 1500, Linkam Scientific Instrument, UK) up to 1 atm. After 3 h, the sample was cooled to room temperature and analyzed as regards the chemisorption of 18O2 in the sample using a FT-Raman spectrometer and laser wavelength of 785 nm (Renishaw Micro Raman, Renishaw, UK).3. Results and discussion
Typical three-component (BaSrMg)CO3 crystals synthesized using the present method are shown in Fig. 1. According to the FE-SEM images, the crystals had an elongated hexagonal shape and were uniform in size at around 28 μm (Fig. 1a). The EDS spectrum revealed that a single crystal was uniformly composed of Ba, Sr, and Mg, at 51.49%, 6.58%, and 41.93%, respectively (Fig. 1b), which matched well with the average crystal composition analyzed using ICP (Ba = 51.42%; Sr = 6.57; Mg = 42.01%). Thus, the EDS and ICP analyses suggested that the (BaSrMg)CO3 crystals were uniformly single phase. The phase uniformity of the crystals was also confirmed by the EDS element mapping (Fig. 1), which showed that the three elements Ba, Mg, and Sr were uniformly distributed across the whole domain of a single crystal. In addition, the EDS spectrum demonstrated that the composition inside the crystal was equal to that at the crystal surface (ESI Fig. S1 and Table S1†). Thus, from the above results, it was concluded that the (BaSrMg)CO3 synthesized using the proposed method was uniformly single-phase crystals.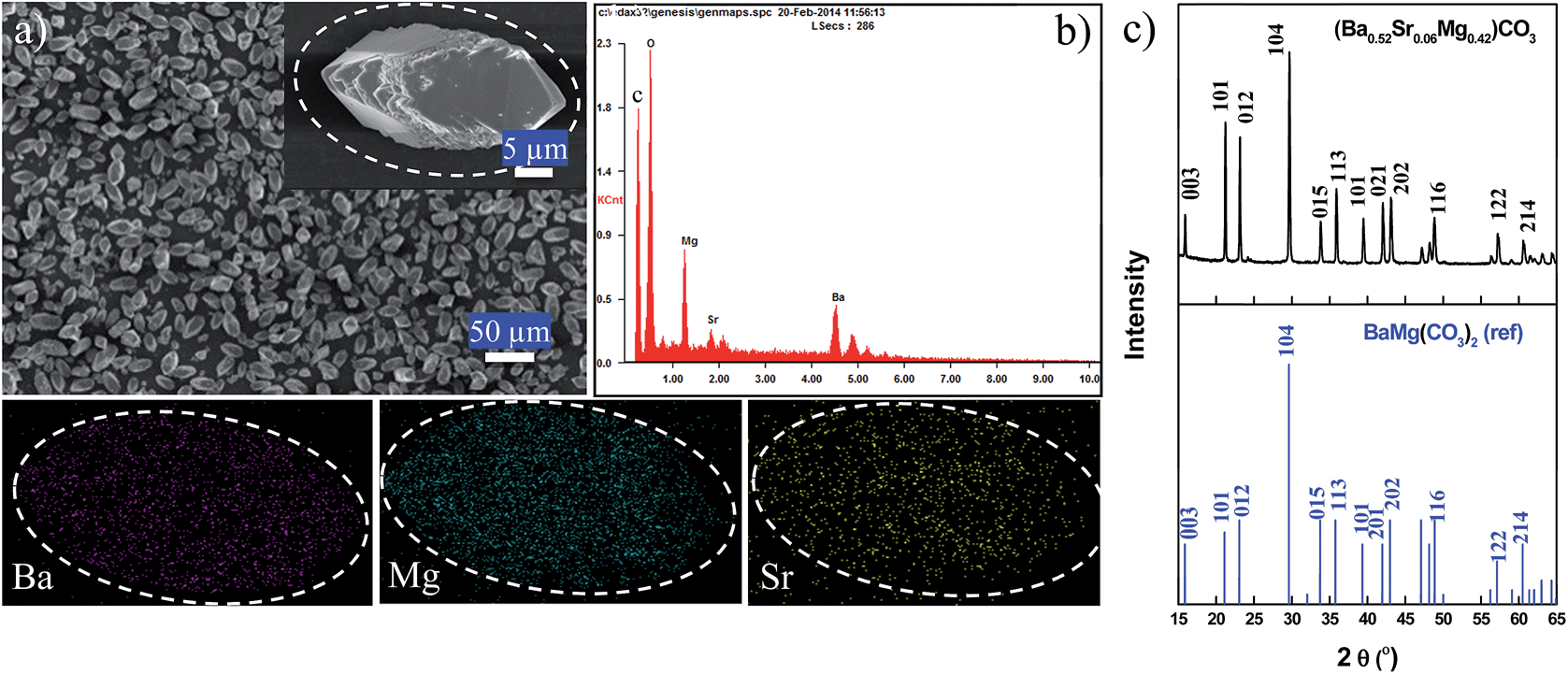 | ||
| Fig. 1 FE-SEM and XRD analysis of (BaSrMg)CO3 crystals (a) FE-SEM images of (BaSrMg)CO3 crystals, (b) element analysis on an isolated (BaSrMg)CO3 crystal using EDS-mapping (elements including Ba, Sr, and Mg), (c) XRD patterns of (BaSrMg)CO3 crystals. | ||
Fig. 1c shows the X-ray diffraction (XRD) patterns of (BaSrMg)CO3 with a main plane of (104). Although there are no reported references for (BaSrMg)CO3 crystals, the XRD patterns for the (BaSrMg)CO3 crystals were highly similar to those for the (BaMg)CO3 crystals, where both crystals were based on a main plane of (104) and all their characteristic peaks well matched with each other. This result is also reasonable when considering that only a small quantity of Sr (6 wt%) was incorporated in the crystals based on (BaMg)CO3. Therefore, the lattice parameters of the (BaSrMg)CO3 crystals, calculated based on a least-square analysis of the X-ray diffraction data as a = 5.0189 Å and c = 16.731 Å, were highly comparable to the parameters of the (BaMg)CO3 crystals (a = 5.02 Å and c = 16.75 Å). It is also interesting to note that the characteristic peak positions of the (BaSrMg)CO3 crystals were slightly shifted to a higher angle when compared to those of the (BaMg)CO3 crystals. This was due to the replacement of Ba2+ with Sr2+ in the crystal lattice. Since Ba2+ has a larger radius (1.6 Å) than Sr2+ (1.44 Å),26 when the Ba2+ in the crystal structure was replaced with Sr2+, the distance between the planes became smaller. Thus, according to Bragg's Law (2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ), this shifted the peak position to a higher angle. Additionally, it should be mentioned that the XRD patterns of the BaCO3, MgCO3, and SrCO3 crystals were significantly different from those of the (BaSrMg)CO3 crystals (ESI Fig. S2†).
sinθ = nλ), this shifted the peak position to a higher angle. Additionally, it should be mentioned that the XRD patterns of the BaCO3, MgCO3, and SrCO3 crystals were significantly different from those of the (BaSrMg)CO3 crystals (ESI Fig. S2†).
The synthetic mechanism of (BaSrMg)CO3 was investigated, as shown in Fig. 2 and 3. During the solid–liquid reaction between (BaSr)CO3-(s) and Mg(NO3)2-(l), the crystal shape and structure of the suspension solids were analysed using SEM and powder pattern XRD (Fig. 2 and 3a), while the solution composition of Ba, Sr and Mg in the suspension solution was analyzed using ICP (Fig. 3b). Here, the compositions of Ba and Sr in the reactant (BaSr)CO3 were around 80% and 20%, respectively (Table S2 and Fig. S3†). Initially, the typical rod-shape (BaSr)CO3 crystals were dominant until bulky hexagonal crystals of (BaSrMg)CO3 started to appear after about 1 h (Fig. 2a–c). After 2 h, there were more hexagonal crystals than rod-shape crystals (Fig. 2d), and after 4 h, there were no more rod-shape crystals and only bulky hexagonal crystals of about 30 μm (Fig. 2e and f). This change of (BaSr)CO3 crystals to (BaSrMg)CO3 crystals can be explained in terms of re-coprecipitation. That is, the (BaSr)CO3 crystals were dissolved out in the solution as their solubility was enhanced with the feeding of the Mg(NO3)2 solution. This in turn increased the cation concentration (Ba2+, Sr2+, and Mg2+) in the solution, which induced the three-component re-coprecipitation of (BaSrMg)CO3. As the (BaSrMg)CO3 crystals grew, the (BaSr)CO3 crystals were continuously dissolved out and eventually disappeared.
 | ||
| Fig. 2 Transient profile of (BaSr)CO3 + Mg(NO3)2 reaction at 90 °C monitored by FE-SEM. (a) FE-SEM image of (BaSr)CO3 crystals (b) product solids ((BaSr)CO3 and (BaSrMg)CO3) at 30 min, (c) product solids ((BaSr)CO3 and (BaSrMg)CO3) at 1 h, (d) product solids ((BaSr)CO3 and (BaSrMg)CO3) at 2 h, (e) product solids ((BaSr)CO3 and (BaSrMg)CO3) at 4 h, (f) product solids ((BaSr)CO3 and (BaSrMg)CO3) at 6 h. | ||
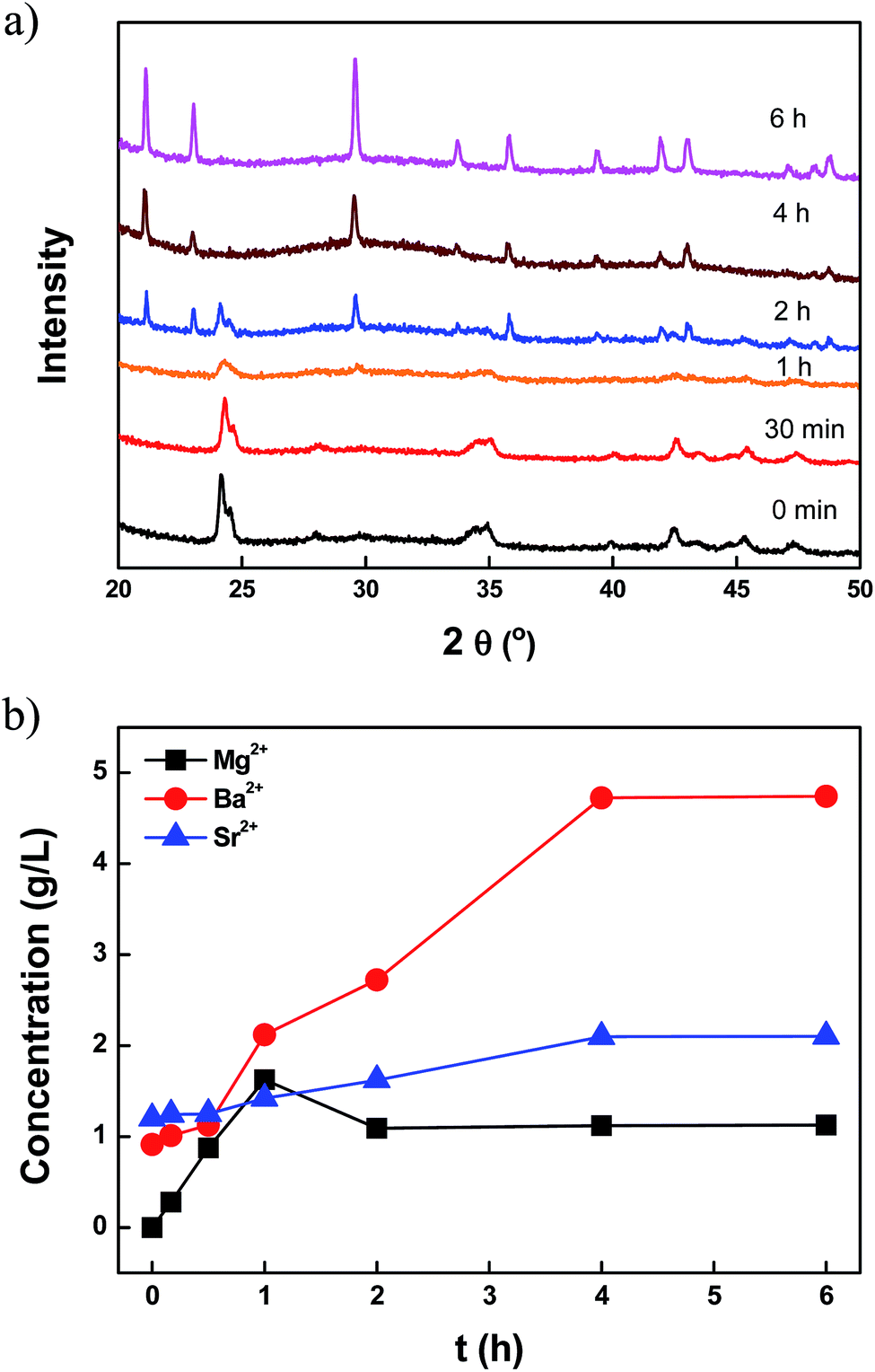 | ||
| Fig. 3 (a) Transient solid profile of (BaSr)CO3 + Mg(NO3)2 reaction at 90 °C monitored by XRD, (b) cation concentration profiles of Ba2+, Mg2+, and Sr2+ in solution during the (BaSr)CO3 + Mg(NO3)2 reaction at 90 °C. | ||
Using the XRD patterns, the transformation of the (BaSr)CO3 crystals into (BaSrMg)CO3 crystals was confirmed (Fig. 3a). For the first 30 min, the XRD patterns of the crystals matched the reference diffraction data for (Ba0.8Sr0.2)CO3 (JCPDS: 47-0223), even though the characteristic peak intensity gradually decreased due to the dissolution of the crystals. After 1 h, while the diffraction peaks of the (BaSr)CO3 crystals almost disappeared, new diffraction peaks for the (BaSrMg)CO3 crystals began to appear. After 4 h, all the characteristic peaks of the (BaSr)CO3 crystals completely disappeared and those of the new (BaSrMg)CO3 crystals were fully developed.
In addition, the cation composition in the solution was traced to confirm the mechanism of (BaSrMg)CO3 formation. As shown in Fig. 3b, when feeding the Mg(NO3)2 solution into the reactor, the cation concentrations of Ba2+ and Sr2+ in the solution monotonically increased and then levelled off after 4 h, as the Mg ions in the solution increased the solubility of the (BaSr)CO3, as previously reported by Davis et al.,27 Voronkov et al.,28 Berner et al.29 and Van Enckevort et al.30 Meanwhile, the Mg2+ concentration in the solution initially increased for 1 h, and then was followed by the sudden spontaneous nucleation of (BaSrMg)CO3, which resulted in a rapid reduction of the Mg concentration in the solution after 2 h. Thereafter, the Mg2+ concentration was slightly reduced until 4 h whereas there were significant changes in Ba2+ and Sr2+ concentrations. After 4 h, all ion concentrations levelled off, indicating the completion of the re-coprecipitation of (BaSrMg)CO3. It is interesting to note that the cation composition of the (BaSrMg)CO3 crystals was always about Ba2+ = 52%, Sr2+ = 6%, and Mg2+ = 42%, regardless of the initial reactant concentrations of (BaSr)CO3 and Mg(NO3)2, as shown in Table S3.† Thus, when changing the reactant molar ratio of Mg(NO3)2 to (BaSr)CO3, the solid–liquid reaction produced a significant variety of crystals. Here, (Ba0.8Sr0.2)CO3 crystals were used for the solid–liquid reaction. When the reactant ratio of [Mg(NO3)2]/[(BaSr)CO3] < 0.7, multi-phase crystals of (BaSr)CO3 and (BaSrMg)CO3 were obtained (Fig. 4a and b). This result was due to an incomplete conversion of (BaSr)CO3 crystals to (BaSrMg)CO3. When using EDS, it was confirmed that the (BaSr)CO3 in the multi-phase crystal product had a composition of Ba = 80% and Sr = 20%, the same as the reactant solid ((Ba0.8Sr0.2)CO3), however, the cation composition of (BaSrMg)CO3 in the product was almost the same as that of the above stable (BaSrMg)CO3 in Fig. 1 (ESI Fig. S4†). Yet, when [Mg(NO3)2]/[(BaSr)CO3] ≥ 0.7, the (BaSr)CO3 crystals were completely converted to stable (BaSrMg)CO3 crystals (Fig. 4c–e). The multi-phase crystals produced when varying the reactant molar ratio was also confirmed using XRD (Fig. 4f). At a reactant ratio of [Mg(NO3)2]/[(BaSr)CO3] < 0.7, the diffraction peaks for both (BaSr)CO3 and (BaSrMg)CO3 were present, however, the characteristic peaks for (BaSr)CO3 diminished when increasing the reactant ratio and completely disappeared with the reactant ratio above 0.7.
 | ||
| Fig. 4 Effect of reactant ratio [Mg(NO3)2]/[(BaxSry)CO3 ] on co-precipitation of (BaSrMg)CO3. Here, the x:y ratio of (BaxSry)CO3 was fixed at 0.8:0.2. (a) Reactant ratio of 0.5, (b) reactant ratio of 0.6, (c) reactant ratio of 0.7, (d) reactant ratio of 0.8, (e) reactant ratio of 0.9 and (f) XRD patterns of the product crystals. | ||
The influence of the cation ratio of Ba to Sr in the reactant (BaSr)CO3 on the synthesis of (BaSrMg)CO3 is investigated in Fig. 5. Here, the reactant ratio [Mg(NO3)2]/[(BaSr)CO3] was always fixed at 1.0. When the cation ratio of Ba to Sr (Ba/Sr) in the reactant (BaSr)CO3 was below 4.0, multi-phase crystals of (BaSrMg)CO3 and SrCO3 were coprecipitated (Fig. 5a and b). However, when increasing the Ba/Sr ratio above 4.0, single-phase crystals of (BaSrMg)CO3 were synthesized (Fig. 5c and d), as confirmed by an XRD analysis (Fig. 5e). The phase separation at a low Ba/Sr ratio was due to the excess Sr ions dissolved in the solution. That is, at a low Ba/Sr ratio, the relative amount of Sr to Ba and Mg was more than enough for the formation of (BaSrMg)CO3, resulting in the simultaneous precipitation of SrCO3. When compared with the carbonate compounds of BaCO3 and MgCO3, the low solubility of SrCO3 may also have contributed to its easy precipitation.31 It should also be mentioned that the crystal structure and shape of (BaSr)CO3 did not vary with the Ba/Sr ratio across the whole range of 1/9–9.0, as summarized in ESI Fig. S5 and S6.†
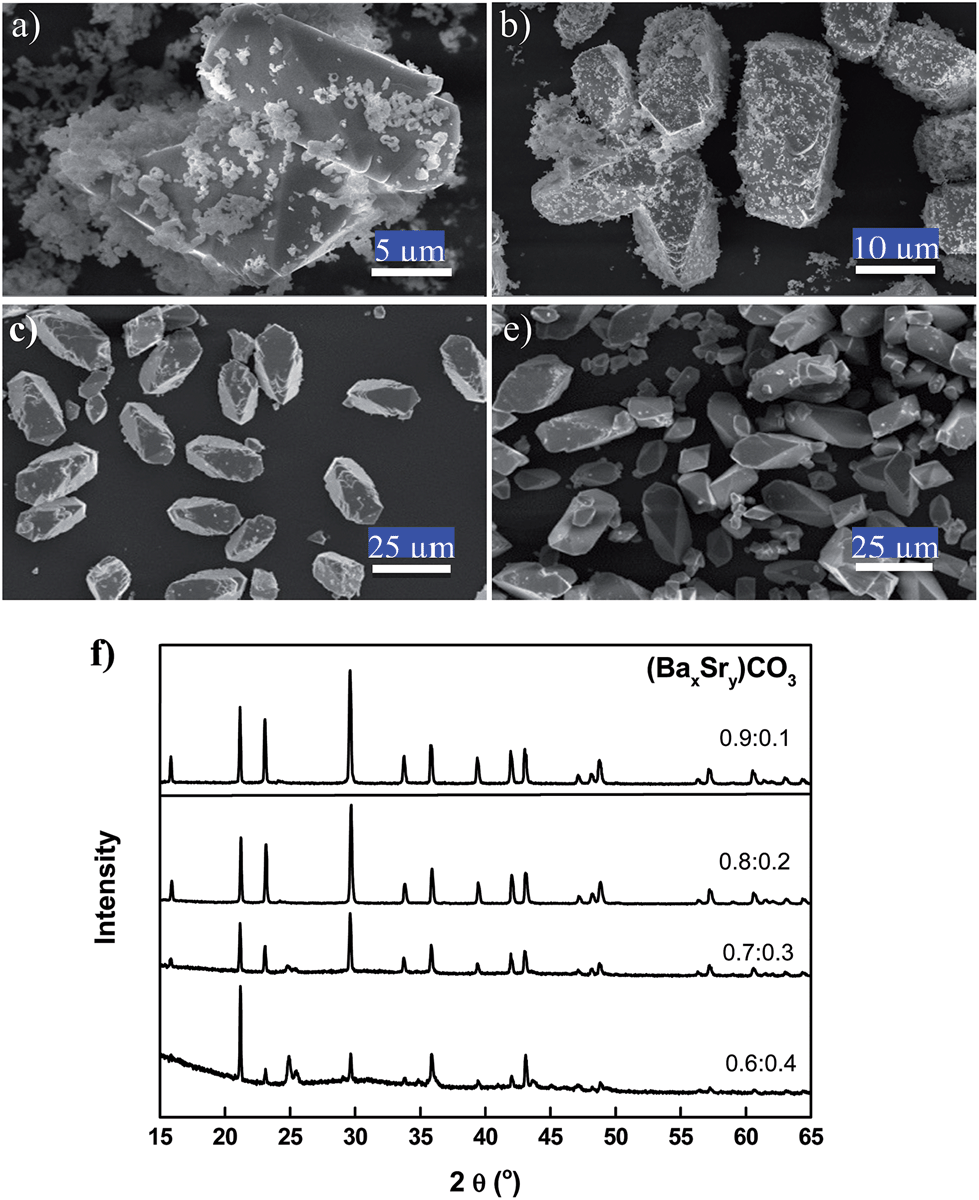 | ||
| Fig. 5 Effect of (BaxSry)CO3 with various x:y ratios on co-precipitation of (BaSrMg)CO3. Here, the reactant concentration ratio [Mg(NO3)2]/[(BaSr)CO3] was fixed at 1.0; (a) 0.6:0.4 (b) 0.7:0.3 (c) 0.8:0.2 (d) 0.9:0.1, and (d) XRD patterns of the product crystals. | ||
The influence of the reaction temperature on the preparation of the (BaSrMg)CO3 crystals was examined, as shown in Fig. S7.† According to the SEM images and XRD analysis, single-phase crystals of (BaSrMg)CO3 were produced at a reaction temperature above 70 °C, whereas below this temperature, multi-phase crystals of (BaSrMg)CO3 and (BaSr)CO3 were produced (Fig. S7a and S7b†) due to the dissolution of (BaSr)CO3 depending on the temperature. Thus, at a low temperature, since not enough (BaSr)CO3 was dissolved out in the solution due to its low solubility, the (BaSr)CO3 remained in the product suspension. In addition, the influence of the carbonate concentration [CO32−] on the synthesis of (BaSrMg)CO3 is summarized in Fig. S8 of the ESI.† Actually, the anion had no effect on the multi-component co-precipitation. Thus, the crystal shape, structure, and composition of (BaSrMg)CO3 remained unchanged across the whole range of carbonate concentrations from 0.04 g mL−1 to 0.12 g mL−1, yet the crystal size became smaller when increasing the carbonate concentration due to the promotion of (BaSrMg)CO3 crystal nucleation.
As shown in Fig. 6, the single-phase three-component alkaline earth carbonate (BaSrMg)CO3 was converted to single-phase three-component alkaline earth oxide (BaSrMg)O for use as a chemisorbent for oxygen separation based on the redox reaction cycle of BaO + 1/2O2 ↔ BaO2. While the (BaSrMg)O crystals were similar in shape and size to the (BaSrMg)CO3 crystals, they were covered with nanosize needles that were normally oriented to the crystal surface (Fig. 6a and b). These nano-needles on the crystal surface increased the specific reaction surface with O2 for chemisorption. Based on the EDS element mapping (Fig. 6b) and XRD pattern (Fig. 6c), the phase uniformity of the (BaSrMg)O crystals was confirmed.
 | ||
| Fig. 6 FE-SEM and XRD analysis of (BaSrMg)O crystals (a) FE-SEM images of (BaSrMg)O crystals, (b) SEM image of single (BaSrMg)O crystal, and element analysis of Ba, Mg, Sr and O by EDS-mapping, (c) XRD patterns of (BaSrMg)O crystals. (BaO reference JCPDS – 260177; MgO reference JCPDS – 450946; SrO reference JCPDS – 060520). | ||
The oxygen chemisorption of (BaSrMg)O was characterized using a thermogravimetric analyser (TGA), as shown in Fig. 7. According to the sorption isotherm of O2 at 700 °C (Fig. 7a), the oxygen chemisorption began to occur above an oxygen partial pressure of 148 mmHg, which is called the transient oxygen pressure and means the oxygen pressure required for the equilibrium of the redox reaction in (BaSrMg)O. Thus, the oxidation reaction (chemisorption) proceeds at an oxygen pressure above the equilibrium pressure, while reduction (desorption) prevails at an oxygen pressure below the equilibrium pressure. The transient oxygen pressure (148 mmHg) of (BaSrMg)O is much higher than transient oxygen pressure of BaO (76 mmHg),32 indicating that the three-component chemisorbent of (BaSrMg)O would be much more economical for industrial application than the single component chemisorbent of BaO. In addition, the sorption isotherm also revealed that the sorption capacity (2.02 mmol-O2 g−1) of (BaSrMg)O was much higher than that (1.75 mmol-O2 g−1) of (BaMg)O, as previously reported by Jin et al. and his co-workers.1,33 The chemisorption and desorption cycle is shown in Fig. 7b. Under the oxygen flow, the weight of the sorbent rapidly increased due to the fast oxidation reaction. Thus, the relaxation time, t80, was measured as 3.9 min for (BaSrMg)O, representing a much faster adsorption rate than that for (BaO2/MgO) prepared using hydrothermal (t80 = 4 min)and sol–gel (t80 = 6–9 min) methods, as reported by Park et al.10 The desorption was also much faster, where the desorption time, t80, was measured as 14 min under a N2 environment. Here, the relaxation time, t80, was defined as the time required for the fractional approach to reach 80% of the reaction from an equilibrium state to a new one, as reported by Lin et al.33Fig. 7c shows the multi-cycle chemisorption and desorption of the sorbent at 700 °C. No significant change in the chemisorption and desorption profiles was observed over 10 cycles, demonstrating the high thermal stability of the sorbent due to the incorporation of Mg in the crystals.
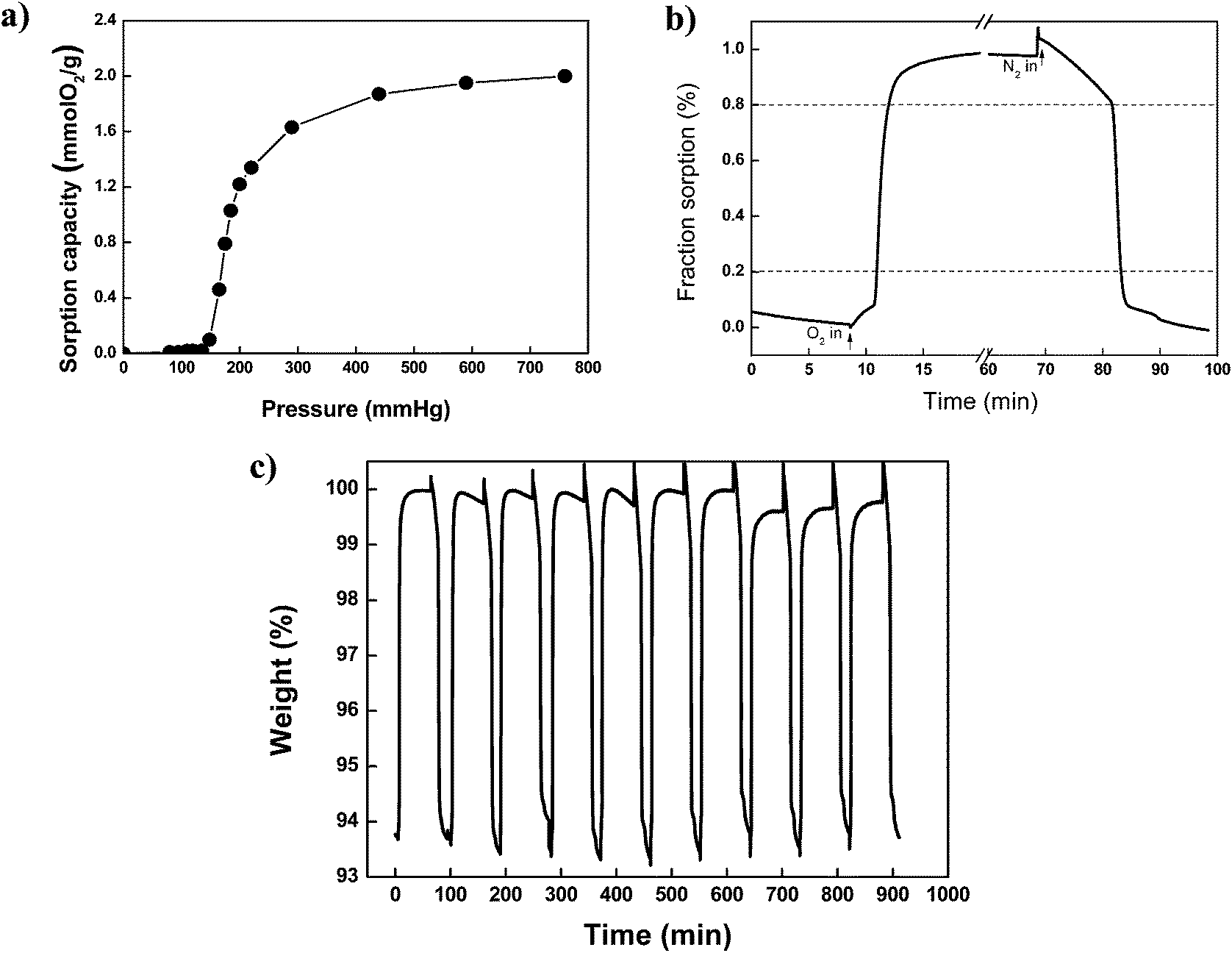 | ||
| Fig. 7 (a) Sorption capacity of O2 on (BaSrMg)O crystals, (b) one cycle of sorption and desorption, relaxation time, t80, was defined based on 80% weight change. (c) Multi sorption–desorption cycles of O2, all analyses were measured by TGA at 700 °C. | ||
As mentioned above, the high transient oxygen pressure for the redox reaction (BaO + 1/2O2 ↔ BaO2) of the sorbent was due to the incorporation of Sr in the crystal lattice ((BaSrMg)O). Since Sr belongs to the same family of alkaline earth metals as Ba, it easily replaced Ba in the crystal lattice (Ba–O–Sr). However, since the ionic radius of Sr2+ is smaller than that of Ba2+, Sr2+ formed a shorter and stronger bondage with oxygen (Sr–O) than Ba2+ (Ba–O). According to a report by de La Croix et al.,34 the lattice energy of Sr–O (−3240 kJ mol−1) is higher than that of Ba–O (−3021 kJ mol−1). In addition, due to the stronger electron attraction of Sr, the Ba–O bondage in Ba–O–Sr is slightly looser and longer when compared with that in Ba–O–Ba. Thus, it can be assumed that during the oxygen chemisorption, the Ba–O bondage in Ba–O–Sr was more easily broken for oxidation to Ba–O–O–Sr when compared with the oxidation of Ba–O–Ba to Ba–O–O–Ba. Inversely, during the oxygen desorption, Ba–O–O–Sr was more easily reduced to Ba–O–Sr when compared with the reduction of Ba–O–O–Ba to Ba–O–Ba. As a result, Ba–O–Sr and Ba–O–O–Sr could be in equilibrium at an even higher oxygen partial pressure than the Ba–O–Ba and Ba–O–O–Ba system. Consequently, the transient oxygen pressure for the redox reaction of (BaMgSr)O was much higher than that of BaO.
To confirm the above explanation, the (BaSrMg)O2 crystals were analysed using Raman spectroscopy. As shown in Fig. 8a, the O–O frequency of (BaSrMg)O2 occurring at 844 cm−1 was lower than the O–O frequency of BaO2 (847 cm−1),32 indicating that the O–O bonding in Ba–O–O–Sr was weaker than that in Ba–O–O–Ba. Using an isotope of oxygen (18O2), it was also found that all three absorption peaks of the three kinds of O–O bonding in (BaSrMg)O2, including 18O–18O, 18O–16O, and 16O–16O, appeared at 795, 820, and 844 cm−1, respectively, which were red-shifted from the corresponding absorption peaks at 799, 824, and 847 cm−1, respectively, in BaO2 (ref. 32) (Fig. 8b). This was due to the replacement of Ba with Sr that has a smaller radius, thereby allowing strong Sr–O bonding and reducing the O–O vibration energy in Ba–O–O–Sr. Thus, the small amount of Sr incorporated in the (BaSrMg)O crystals had a significant influence on the increment of the transient oxygen pressure required for the redox reaction cycle.
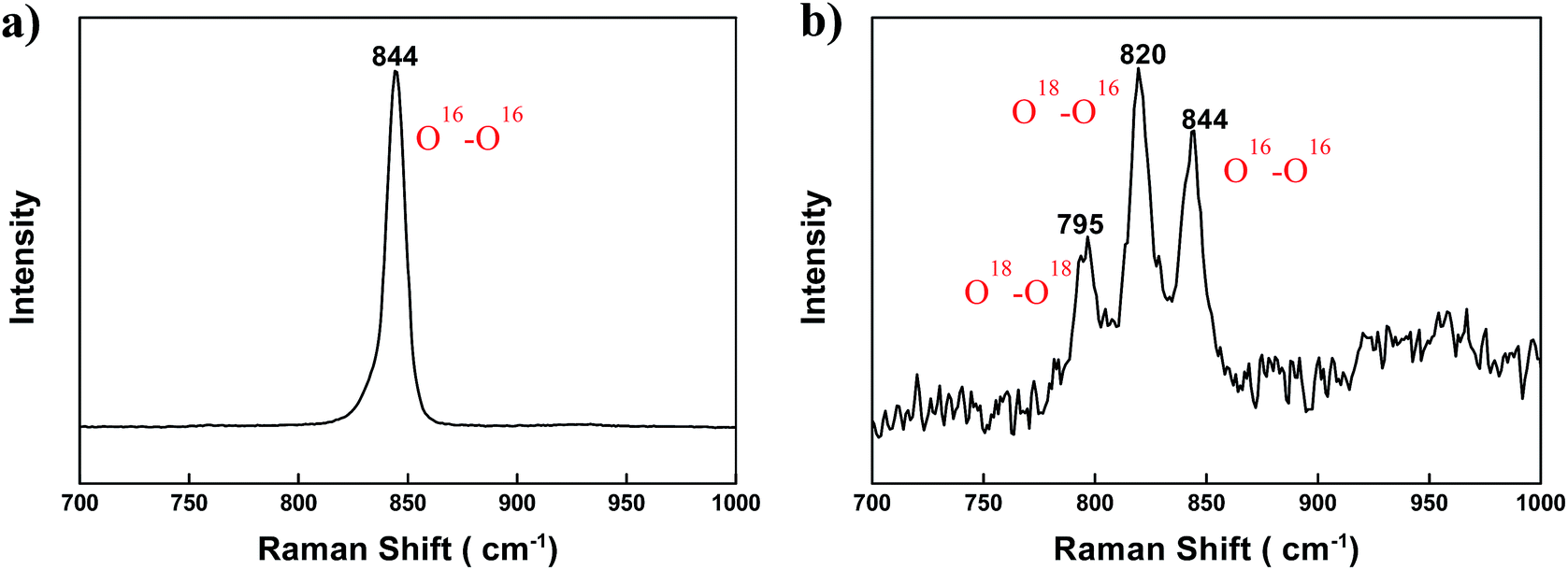 | ||
| Fig. 8 Raman spectra of (a) (BaSrMg)O2 crystals and (b) (BaSrMg)O2 crystals of the (BaSrMg)O crystals after chemisorption of 18O2 gas for 3 h. | ||
4. Conclusions
This study presented a method for preparing a highly capable and thermally stable chemisorbent of (BaSrMg)O. In addition, the formation mechanism was also discussed at length. First, the (BaSr)CO3 crystals are synthesized by co-precipitation and then transformed into (BaSrMg)CO3 crystals in the solution by the addition of an Mg2+ solution. Thus, when adding Mg2+, the Ba2+ and Sr2+ concentrations in the solution are increased due to the dissolution of (BaSr)CO3 in the solution, inducing three-component co-precipitation for the formation of single-phase (BaSrMg)CO3. The reactant concentrations of Ba2+, Sr2+, Mg2+, and CO32−, and the reaction temperature were found to have a significant influence on the formation of the (BaSrMg)CO3 crystals. However, the single-phase (BaSrMg)CO3 was always formed with a composition of 52% (Ba), 6% (Sr), and 42% (Mg), which was the most stable, regardless of the reaction conditions.The single-phase (BaSrMg)CO3 crystals were converted into single-phase (BaSrMg)O crystals by thermal decomposition under an H2 atmosphere at 750 °C. The (BaSrMg)O crystals showed a high chemisorption capacity of oxygen (2.02 mmol g−1) at 700 °C owing to the high fraction of Ba and a high thermal stability based on the inclusion of Mg in the crystals. Furthermore, the (BaSrMg)O crystals exhibited a high chemisorption rate (t80 = 3.9 min) and desorption rate (t80 = 14 min). In particular, the transient oxygen pressure for the redox reaction in (BaSrMg)O significantly increased from 76 mmHg to 148 mmHg at 700 °C, implying a high-strength chemisorbent for practical application to oxygen separation. This was because the replacement of Ba with Sr in the crystal lattice shifted the equilibrium state of the BaO–BaO2 redox reaction cycle to a high oxygen partial pressure. Therefore, the proposed method for the preparation of (BaSrMg)O is a promising platform for the elaboration of multi-component metal oxide materials. In addition, due to their high capacity, thermal stability, and high transient pressure, such materials represent a significant improvement in oxygen separation for industrial application.
Acknowledgements
This work was conducted under the framework of the Research and Development Program of the Korea Institute of Energy Research (KIER) (B4-2442-02).Notes and references
- C. Jin, T. Hirose and M. Koto, Ind. Eng. Chem. Res., 2005, 44, 2942 CrossRef CAS.
- T. F. Wall, Combustion processes for carbon capture, Proc. Combust. Inst., 2007, 31, 31 CrossRef PubMed.
- S. P. S. Badwal and F. T. Ciacchi, Adv. Mater., 2001, 13, 12 CrossRef.
- C. F. Watson, R. D. Whitley and M. L. Meyer, U.S. Patent 5529610, 1996.
- T. R. Gaffney, Curr. Opin. Solid State Mater. Sci., 1996, 1, 69 CrossRef CAS.
- Y. Cengel and M. Boles, Thermodynamics: an Engineering Approach, McGraw- Hill, New York, 5th edn, 2006 Search PubMed.
- P. D. Southon, D. J. Price, P. K. Nielsen, C. J. McKenzie and C. J. Kepert, J. Am. Chem. Soc., 2011, 133, 10885–10891 CrossRef CAS PubMed.
- S. Sircar, M. B. Rao and T. C. Golden, Adsorption and its Applications in Industry and Environmental Protection, Elsevier Science Publ B V, Amsterdam, 1999, vol. 120, p. 395 Search PubMed.
- A. F. Ismail and L. I. B. David, J. Membr. Sci., 2001, 193, 1 CrossRef CAS.
- J. H. Park, Y. S. Cho, K. B. Yi, S. S. Han and S. H. Cho, Appl. Surf. Sci., 2010, 256, 5528–5532 CrossRef CAS PubMed.
- V. V. Kharton and A. A. Yaremchenko, J. Membr. Sci., 1999, 163, 307–317 CrossRef CAS.
- E. A. Wang, Oxygen-Selective Adsorbents, USA Patent, Patent No.: US 6436171 B1, 2002.
- M. R. Tiné, Coord. Chem. Rev., 2012, 256, 316–327 CrossRef PubMed.
- Q. Yang, Y. S. Lin and M. Bülow, AIChE J., 2006, 52, 574–581 CrossRef CAS.
- Y. S. Lin, D. L. Maclean and Y. Zeng, USA patent, Patent No, 6059858, 2000.
- J. L. Moriaty, J. Iowa Acad. Sci., 1970, 77, 360 Search PubMed.
- L. G. Massey, P. B. Tarman and D. Ranwani, U. S. Patent, 3903010, 1975.
- M. A. Fahim and J. D. Ford, Chem. Eng. J., 1983, 27, 21 CrossRef CAS.
- J. H. Park, WO patent, Patent No: 2012102554A3, 2012.
- J. L. Moriaty, J. Iowa Acad. Sci., 1970, 77, 360 Search PubMed.
- W. C. Hood and P. F. Steidl, Am. Mineral., 1973, 58, 342–347 Search PubMed.
- J. Terada, J. Phys. Soc. Jpn., 1953, 8, 2 CrossRef.
- P. Pasierb, S. Komornicki, M. Rokita and M. Rekas, J. Mol. Struct., 2001, 596, 151–156 CrossRef CAS.
- S. Weinbruch, H. Buttner and M. Rosenhauer, Phys. Chem. Miner., 1992, 19, 289–297 CrossRef CAS.
- W. C. Hood, P. F. Steidl and D. G. Tschopp, American Mineralogist, 1974, 59, 471–474 CAS.
- Y. H. Chen, S. C. Yu, E. Huang and P. L. Lee, Phys. B, 2010, 405, 4386–4388 CrossRef CAS PubMed.
- K. J. Davis, P. M. Dove and J. J. De yoreo, Science, 2000, 290, 1134 CrossRef CAS.
- V. V. Voronkov and L. N. Rashkovich, Sov. Phys. Crystallogr., 1992, 37, 289 Search PubMed.
- R. A. Berner, Geochim. Cosmochim. Acta, 1975, 39, 489 CrossRef CAS.
- W. J. P. Van Enckevort and A. C. J. F. Van den Berg, J. Cryst. Growth, 1998, 183, 441 CrossRef CAS.
- D. Harris, L. K. Porter and E. A. Paul, Commun. Soil Sci. Plant Anal., 1997, 28, 747 CrossRef CAS.
- G. Mestl, M. P. Rosynek and J. H. Lunsford, J. Phys. Chem. B, 1998, 102, 154 CrossRef CAS.
- C. Jin, T. Suehiro, A. Kodama, M. Goto and T. Hirose, J. Chem. Eng. Jpn., 2001, 34, 279 CrossRef CAS.
- A. De La Croix, R. B. English and M. E. Brow, J. Solid State Chem., 1998, 137, 346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Table S1–S3, and Fig. S1–S8 (Section S 2) (PDF). CCDC 12-0530, 47-0223, 05-0378, 05-0418, 08-0479, 26-0177, 06-0502 and 45-0946. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ta05045b |
| This journal is © The Royal Society of Chemistry 2015 |