
One-dimensional nanostructured design of Li1+x(Mn1/3Ni1/3Fe1/3)O2 as a dual cathode for lithium-ion and sodium-ion batteries†
Sujith
Kalluri
ab,
Wei Kong
Pang
ac,
Kuok Hau
Seng
a,
Zhixin
Chen
b,
Zaiping
Guo
*ab,
Hua Kun
Liu
a and
Shi Xue
Dou
a
aInstitute for Superconducting and Electronic Materials, University of Wollongong, NSW 2500, Australia. E-mail: zguo@uow.edu.au; Fax: +61-2-4221 5731; Tel: +61-2-4221 5225
bSchool of Mechanical, Materials and Mechatronics Engineering, University of Wollongong, NSW 2500, Australia
cAustralian Nuclear Science and Technology Organisation, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia
First published on 30th October 2014
Abstract
Potency of the cathode material is an important feature for upgrading lithium-ion/sodium-ion battery technology for next-generation applications such as in electrical grids and advanced electric vehicles. Various limitations related to electrochemical and socio-economic issues of these batteries are current research challenges. Amongst the various possible solutions to address such issues, developing nanostructured cathode materials, such as one-dimensional nanostructures, by versatile and easily scaled-up processes could be one of the options. Consequently, in the present study, Li1+x(Mn1/3Ni1/3Fe1/3)O2 one-dimensional nanofibers have been fabricated via a simple and low-cost electrospinning technique and used as a cathode material in lithium-ion batteries, which showed an improved initial reversible capacity (∼109 mA h g−1) and cyclic stability at the 0.1 C rate when compared to the performance of Li1+x(Mn1/3Ni1/3Fe1/3)O2 nanoparticles. On the other hand, the feasibility of this low-cost and eco-friendly material was also tested in sodium-ion batteries, and the same trend is observed. The enhanced electrochemical and structural features in both systems could be ascribed to the exceptional features of one-dimensional nanofibers such as efficient electron transport, facile strain relaxation, and short Li+/Na+ diffusion pathways.
1. Introduction
In the current regime of fossil fuel depletion, today's alternative, renewable energy, is being made from the warm sun and cool breezes by solar energy plants and wind mills, respectively.1–4 Energy storage systems, such as batteries with a desirable energy density, are necessary to store this renewable energy for future needs when and where it is required for a diverse range of applications from portable applications to electric vehicles (EVs) and electrical grids. There are various types of batteries, and amongst them, lithium-ion batteries (LIBs) discovered by the Sony Corporation in 1991 are commercially successful in portable applications such as mobile phones, lap-top computers, etc. They could also be possible candidates for applications such as EVs and electrical grids.5–7 The conventional and commercially available LIBs contain layered lithium cobalt oxide (LiCoO2) and graphite as cathode and anode materials, respectively.8,9 LiCoO2 is a viable cathode material with a theoretical capacity of 272 mA h g−1 and a limited reversible capacity of 140 mA h g−1 corresponding to 0.5 Li per LiCoO2 in the voltage range of 3–4.2 V. Nevertheless, LiCoO2 suffers from the high cost of Co, along with its toxic nature and lack of environmental friendliness.10 Hence, further optimization of LIBs, as per the large-scale commercialization needs of EVs and electrical grids, requires designing cathode materials to have not only better energy and power densities, combined with thermal safety, eco-friendliness and low-cost, but also excellent electrochemical properties such as calendar life and fast charging capability. Accordingly, global research has been focused on various types of cathode materials, from layered structures to spinels and phosphates, such as LiMO2 (M = Co, Mn, Ni, etc.), LiNi1−x−yCoxMnyO2, LiMn2O4, and LiFePO4, for their possible application as battery cathode materials in EVs.11–15 LiNiO2 and LiMnO2 are considered to be promising cathode candidates for LIBs due to their inexpensiveness and higher possible capacities of 160 mA h g−1 and 120–190 mA h g−1, respectively, but they suffer from various issues associated with the cost of their preparation techniques, the problems to do with the preparation of materials without cation mixing, structural degradation during the cycling process, and thermal instability.16–18 On the other hand, spinel LiMn2O4 and olivine LiFePO4 have enough potential or already commercialised cathode candidates, but they unfortunately suffer from poor long-term cycling stability due to Mn-ion Jahn–Teller distortion and poor conductivity, respectively.19,20 To overcome these issues, numerous approaches have been tried, such as cation substitution for Ni and Mn in layered structure compounds by foreign metallic ions such as Co, Al etc., along with alternative synthesis procedures under optimized conditions for better performance of these materials as cathode in LIBs. For instance, partial substitution for Ni by Co in LiNiO2 is reported to lead to a viable cathode candidate in terms of improved capacity values with better cycle life and low cost.21 Nevertheless, thermal safety issues still remain a major concern for their use in various applications.Extensive research in this direction has led to the simultaneous intermixing of Co, Ni, and Mn ions to form a layered structure with the molecular formula LiNixCoyMn1−x−yO2.22 For the optimized composition, synthesis conditions and voltage range during electrochemical measurements, various groups reported the most viable combination as a single phase LiNi1/3Co1/3Mn1/3O2 cathode with deliverable capacity values ranging from 160 to 200 mA h g−1,23–25 although this compound suffers from relatively poor cycling stability due to irreversible phase transitions during the charge–discharge process. For better cycling stability, a higher content of Co is usually required, thereby putting up the total cost and decreasing the rate capability for specific applications such as EVs and electrical grids. Owing to the significant synergic effects of Ni and Mn in the composition, costly and toxic ‘Co’ could be replaced by the low-cost and eco-friendly Fe element in the composition, although at the expense of relatively low capacity values. For example, Karthikeyan et al. successfully prepared LiMn1/3Ni1/3Fe1/3O2 material with a nanoparticle size of ∼200 nm by the co-precipitation method, but it unfortunately delivered a poor capacity value of ∼30 mA h g−1 at 0.5 C, which could be due not only to the inherent conducting nature of the material, but also possibly the electrolyte composition and its compatibility with the material, or the synthesis technique. Most importantly, the nanostructure of the material can never be ignored.26,27 There has been little further research on LiMn1/3Ni1/3Fe1/3O2 in terms of increasing its performance for commercial applications.
Nanostructured materials show enhanced electrochemical performance in batteries because of their short Li-ion diffusion pathways, reduced volume changes during the charge–discharge process, and better effective contact areas with the electrolyte.28,29 Nevertheless, the unfortunate self-agglomeration behaviour of nanoparticulates, either during the synthesis procedure or during the corresponding battery cycling process could cause increased inherent resistance, thereby resulting in poor electrochemical performance.30 Along the same lines, one-dimensional (1D) nanostructures have been proved to be viable candidates for battery applications with better performance due to their peculiar properties, such as resistance to self-agglomeration, improved well-guided charge transfer kinetics and high specific surface area.31,32 While scaling up such 1D nanostructures for specific applications such as EVs and electrical grids, the synthesis procedures cannot be ignored. Electrospinning is a cost-effective, easily scaled-up, and versatile synthesis procedure for continuous 1D nanofiber and other nanofibrous electrodes, which could provide a platform for better interfacial contact with the current collector, owing to their highly porous nature and directed charge transfer characteristics among the well-interconnected nanofibrous network.33–35 Inspired by these interesting features of 1D nanofibers and of LiMn1/3Ni1/3Fe1/3O2 as a possible cathode candidate because of its high theoretical capacity of ∼280 mA h g−1, herein we have made an attempt to fabricate LiMn1/3Ni1/3Fe1/3O2 nanofibers by the sol–gel assisted electrospinning technique and characterized their viability as a cathode candidate in LIBs. The sol–gel synthesis involves simultaneous chelation with cations, esterification, and polymerization of the polymer chelating agent (e.g. citric acid and polyvinylpyrrolidone), favouring cation mixing in most cases.36 This cation mixing could promote the structural stability of the material during the cycling process, however, with an expense of moderate electrochemical performance. In addition to LIBs, sodium-ion batteries (SIBs) also can be a great competitor for specific applications such as electrical grids, because of their similar electrochemistry with 0.3 V less redox potential than that of Li, eco-friendliness and most importantly the low-cost of sodium.37 Considering these interesting features of SIBs, the aforementioned cathode material has been used for both LIBs and SIBs to investigate the possible impact of its 1D nanostructured design on the dual functionality of the LiMn1/3Ni1/3Fe1/3O2 cathode for both battery technologies. With some compromise of the capacity values, which are relatively low, LiMn1/3Ni1/3Fe1/3O2 1D nanofibers showed a surprisingly improved performance in terms of their cycling stability when compared to that of the nanoparticulate system.
2. Experimental
2.1 Fabrication of LiMn1/3Ni1/3Fe1/3O2 nanofibers
The LiMn1/3Ni1/3Fe1/3O2 nanofibers were prepared by a sol–gel assisted electrospinning technique. The starting solution for the electrospinning was prepared from stoichiometric proportions of precursor salts, including lithium nitrate, manganese acetate, nickel acetate, and iron nitrate (all from Sigma Aldrich, USA, with the proportion of lithium nitrate made slightly higher (20%) than usual to compensate the high evaporation rate of Li at high calcination temperatures) in mixed solvents of absolute ethanol and N,N-dimethyl formamide (DMF, Sigma Aldrich) in the ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After thoroughly stirring for an hour, 9 wt% polyvinylpyrrolidone (PVP, 1300000 g mol−1, Sigma Aldrich) was used as the binder polymer for the electrospinning process and mixed with the resultant precursor solution. The resultant solution was stirred overnight and 10 ml of viscous precursor solution was thus obtained, which was fed into a plastic syringe with a 21G (guage) stainless steel needle. An electrospinning unit (NanoNC, South Korea) was used to fabricate the precursor nanofibers with optimized processing and solution parameters: applied potential: 20 kV, tip to target distance (TCD): 13 cm, flow rate: 0.9 ml h−1, and relative humidity (RH): 29–33%. The electrospinning process was carried out for 8 h continuously under ambient temperature conditions in a dry room, and the precursor nanofibrous sheets were thus obtained, which were vacuum dried at 120 °C for 3 h, followed by a stepwise calcination process: 1 °C min−1, 350 °C, 2 h; 2 °C min−1, 500 °C, 2 h; and 3 °C min−1, 900 °C, 4 h in an industrial oxygen atmosphere. The obtained LiMn1/3Ni1/3Fe1/3O2 nanofibers (LMNFO NFs) were free from binder polymers and organic residues.
:1. After thoroughly stirring for an hour, 9 wt% polyvinylpyrrolidone (PVP, 1300000 g mol−1, Sigma Aldrich) was used as the binder polymer for the electrospinning process and mixed with the resultant precursor solution. The resultant solution was stirred overnight and 10 ml of viscous precursor solution was thus obtained, which was fed into a plastic syringe with a 21G (guage) stainless steel needle. An electrospinning unit (NanoNC, South Korea) was used to fabricate the precursor nanofibers with optimized processing and solution parameters: applied potential: 20 kV, tip to target distance (TCD): 13 cm, flow rate: 0.9 ml h−1, and relative humidity (RH): 29–33%. The electrospinning process was carried out for 8 h continuously under ambient temperature conditions in a dry room, and the precursor nanofibrous sheets were thus obtained, which were vacuum dried at 120 °C for 3 h, followed by a stepwise calcination process: 1 °C min−1, 350 °C, 2 h; 2 °C min−1, 500 °C, 2 h; and 3 °C min−1, 900 °C, 4 h in an industrial oxygen atmosphere. The obtained LiMn1/3Ni1/3Fe1/3O2 nanofibers (LMNFO NFs) were free from binder polymers and organic residues.
2.2 Preparation of LiMn1/3Ni1/3Fe1/3O2 nanoparticles
The LiMn1/3Ni1/3Fe1/3O2 nanoparticles were prepared from the same precursor salts using a one-pot sol–gel process. The stoichiometric precursor solution was prepared and was first dried in a vacuum oven at 120 °C for 12 h, followed by a calcination process at 900 °C for 4 h in an industrial oxygen atmosphere. Thus LiMn1/3Ni1/3Fe1/3O2 nanoparticles (LMNFO NPs) were obtained.2.3 Sample characterizations
Both the LMNFO NF and the LMNFO NP samples were characterized for morphological properties and phase analysis using various characterization tools. X-ray powder diffraction (XRD, GBC MMA) equipped with Cu-Kα radiation was used to determine the structure and crystallography of LMNFO. Fullprof with visualization in WinplotR38–40 was employed to perform Rietveld analysis of the XRD data. The background coefficients, zero-shift, peak shape parameters, phase lattice, oxygen positional parameters, isotropic atomic displacement parameters, and occupancy factors of lithium and nickel at 3a and 3b sites were optimized in the refinements. The figures of merit for the refinements, including the weighted profile factor (Rwp) and goodness-of-fit (χ2), are provided in Fig. 1. Scanning electron microscopy (SEM, JEOL JSM-7500, Japan) and high-resolution transmission electron microscopy (HR-TEM, JEOL JEM-2010, Japan) were employed to reveal the morphology and related secondary structures. Digital micrograph software was used to obtain fast Fourier transform (FFT) analysis on selected areas of the HR-TEM images. Image J software was used to measure the fiber diameter and particle size distributions. The Brunauer–Emmett–Teller (BET, Quantachrome Nova 1000, USA) method was employed to calculate the specific surface areas of LMNFO NFs and LMNFO NPs. The thermal stability of these samples over the range of 50 °C to 1000 °C was analysed by thermogravimetric analysis (TGA, Mettler Toledo TGA/DSC1, Switzerland).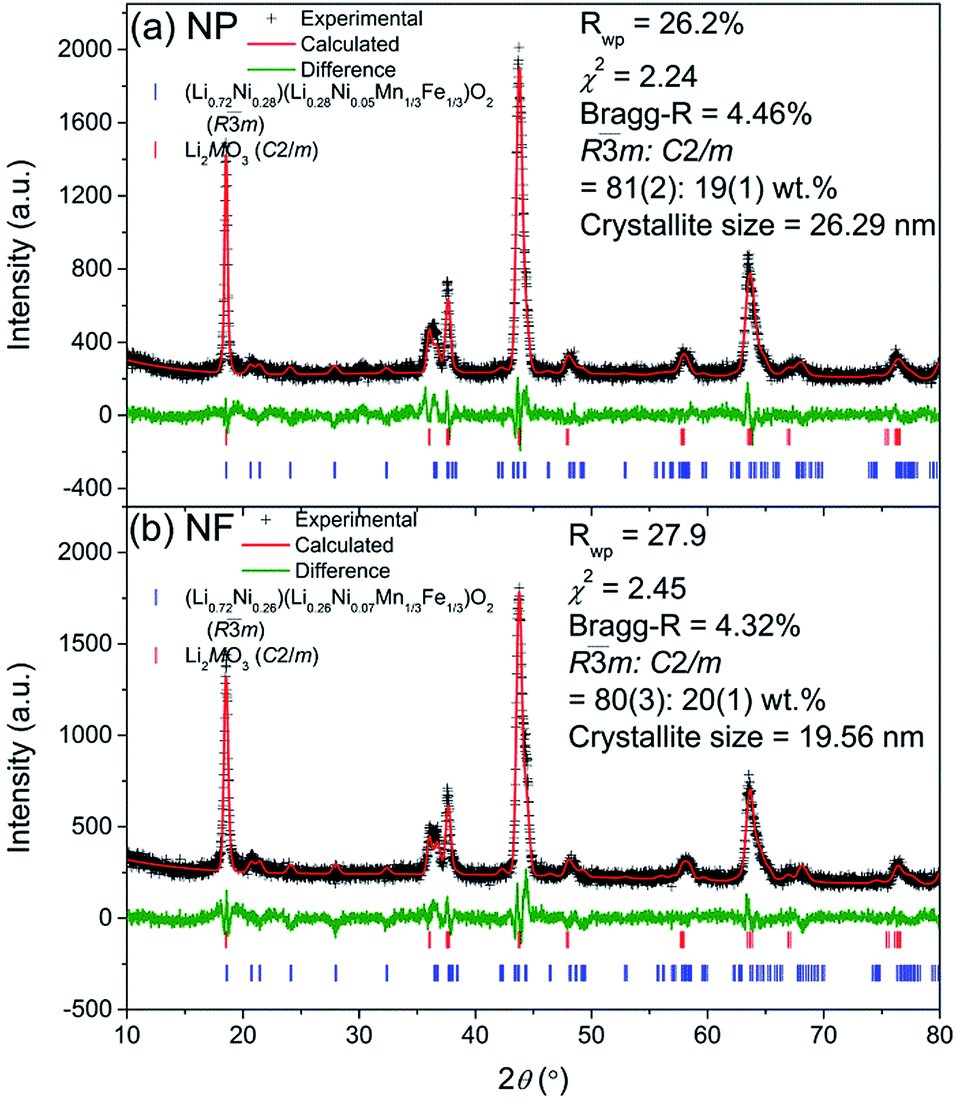 | ||
| Fig. 1 XRD Rietveld refinement profiles of (a) LMNFO NPs and (b) LMNFO NFs. | ||
2.4 Electrochemical characterizations
The electrochemical properties of all the prepared samples were characterized in CR2032 half-cell configured coin-cells assembled in an Ar-filled glove box (MBraun, Germany). The aforementioned cathodes, LMNFO NFs and LMNFO NPs, were prepared by thoroughly mixing the active material with carbon black (Super P, TIMCAL, Switzerland) as a conducting material and polyvinylidene fluoride (PVDF) as a binder material in the proportions of 80:10:10, respectively, in N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich) as a solvent. The resultant mixture was well ground in a planetary mixer (Kurabo Mazerustar, Japan), and the thus obtained slurries were coated on Al current collectors by using the doctor blade technique before drying overnight in a vacuum oven at 120 °C. Each cathode electrode was loaded with ∼1.5 mg cm−2 of active material, and half-cell type coin cells were assembled using disks of Li foil as negative electrodes, Celgard polypropylene film was used as the separator, and commercially available 1 M LiPF6 in ethylene carbonate (EC): diethyl carbonate (DEC) (1:1) was the electrolyte. The same procedure was repeated for the sodium-ion coin-cells with Na foil as the negative electrode and glass fiber film as the separator, with customized 1 M NaClO4 in EC:DEC as the electrolyte. Battery testing analysers (Land, China) were used in galvanostatic mode to measure the charge–discharge characteristics of both the samples. A Biologic VMP3 electrochemical workstation was used to obtain the cyclic voltammograms (CVs) and impedance spectroscopy Nyquist plots. The CV plots were obtained with a scan rate of 0.1 mV s−1 with Li-foil (or Na-foil) as the counter electrode. Impedance Nyquist plots were collected in the frequency range of 100 kHz to 100 mHz at open circuit potential.
3. Results and discussion
Rietveld refinement profiles using X-ray diffraction (XRD) of the LMNFO NFs and LMNFO NPs are shown in Fig. 1(a) and (b), respectively. The XRD data of the two samples appear very similar, indicating that the phase compositions are close. The main phase was modelled using a modified α-NaFeO2 structure with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group,41 and the second phase could be indexed using a C2/m phase,42 which we ascribe to a Li-rich component Li2MnO3. The phase fractions of the Rm and C2/m phases are also similar (80:20 wt%) in the two samples. The two layered compounds are structurally integrated at the atomic level, helping in structure stabilization especially during lithiation and delithiation, as suggested by the literature.43–46 The refined structure of the Rm phase is summarised in Table S1 (in the ESI†). Similarly, large differences in the Rm phase (003) (at ∼18.5°) and (104) (∼43.7°) reflection intensities are observed in both samples, suggesting that the main phase is a layered structure with a high level of cation mixing, 26–28% in Li/Ni as determined by the refinement analysis. Hence, the true composition of the cathode material can be represented by Li1+xMO2 (M = Li, Ni, Mn, Fe) or a composite containing a stabilizing component Li2MO3 (M = Ni, Mn, Fe) and an electrochemically active component (Li∼0.72Ni∼0.28) (Li∼0.28Ni0.05Mn1/3Fe1/3)O2 with a high level of Li/Ni cation mixing. The cation-mixing is planned and supposed to promote the cycling stability, by limiting the number of intercalatible and de-intercalatible lithium ions (charge carrier). The existence of Li2MO3 (M = Mn, Ni, Fe) and cation mixing between Li/Ni are supposed to be greatly useful in improving cycling stability. Overall, both NP and NF samples show similar structure and atomic arrangement of Rm, as shown in Table S1.† These similarities allow us to rule out the effects of phase compositions and crystallographic arrangement of the samples when doing the comparison of the NFs and NPs in terms of electrochemical performance. On the other hand, the lattice parameter a of the NP sample is larger, but the lattice parameter c is smaller than that of the NF sample. Moreover, the crystallite size of NFs (19.6 nm) is significantly smaller than that of the NP sample (26.3 nm). The smaller lattice parameter a and the smaller crystallite size suggest that the NF sample has better lithium diffusivity and hence, greater ionic conductivity, resulting in better capacity and rate performance.
m space group,41 and the second phase could be indexed using a C2/m phase,42 which we ascribe to a Li-rich component Li2MnO3. The phase fractions of the Rm and C2/m phases are also similar (80:20 wt%) in the two samples. The two layered compounds are structurally integrated at the atomic level, helping in structure stabilization especially during lithiation and delithiation, as suggested by the literature.43–46 The refined structure of the Rm phase is summarised in Table S1 (in the ESI†). Similarly, large differences in the Rm phase (003) (at ∼18.5°) and (104) (∼43.7°) reflection intensities are observed in both samples, suggesting that the main phase is a layered structure with a high level of cation mixing, 26–28% in Li/Ni as determined by the refinement analysis. Hence, the true composition of the cathode material can be represented by Li1+xMO2 (M = Li, Ni, Mn, Fe) or a composite containing a stabilizing component Li2MO3 (M = Ni, Mn, Fe) and an electrochemically active component (Li∼0.72Ni∼0.28) (Li∼0.28Ni0.05Mn1/3Fe1/3)O2 with a high level of Li/Ni cation mixing. The cation-mixing is planned and supposed to promote the cycling stability, by limiting the number of intercalatible and de-intercalatible lithium ions (charge carrier). The existence of Li2MO3 (M = Mn, Ni, Fe) and cation mixing between Li/Ni are supposed to be greatly useful in improving cycling stability. Overall, both NP and NF samples show similar structure and atomic arrangement of Rm, as shown in Table S1.† These similarities allow us to rule out the effects of phase compositions and crystallographic arrangement of the samples when doing the comparison of the NFs and NPs in terms of electrochemical performance. On the other hand, the lattice parameter a of the NP sample is larger, but the lattice parameter c is smaller than that of the NF sample. Moreover, the crystallite size of NFs (19.6 nm) is significantly smaller than that of the NP sample (26.3 nm). The smaller lattice parameter a and the smaller crystallite size suggest that the NF sample has better lithium diffusivity and hence, greater ionic conductivity, resulting in better capacity and rate performance.
Fig. 2(a) presents an SEM image of the well-interconnected, morphologically stable, as-spun precursor fibers with an average diameter of ∼475 nm, as shown in Fig. 2(c) and with the further step-wise calcination process, the binder polymer (PVP) and organic moieties from the precursor salts are decomposed, resulting in a decrease in the diameter of LMNFO NFs [Fig. 2(b)] to an average diameter of ∼200 nm [Fig. 2(d)]. Careful observation of LMNFO NFs reveals the ordered stacking as a chain of nanocrystallites along the unidirectional growth direction of the nanofibers, as shown in the inset of Fig. 2(b). For the baseline reference, LMNFO NPs were prepared by the conventional sol–gel method with an average particle size of ∼100 nm, as shown in Fig. S1 (in the ESI†).
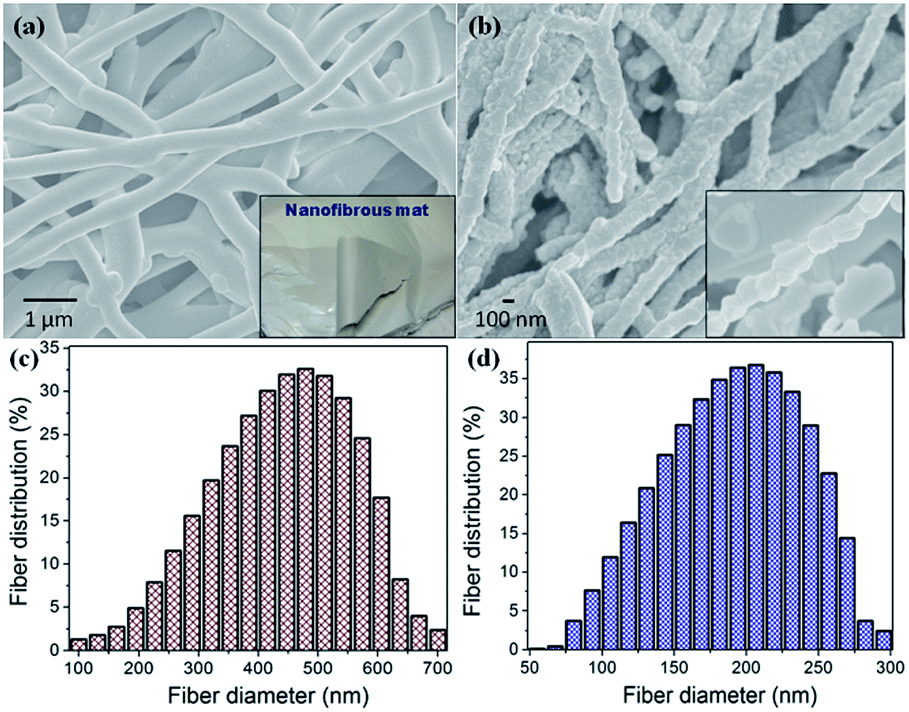 | ||
| Fig. 2 SEM images of (a) as-spun precursor fibers (inset: precursor nanofibrous mat), (b) LMNFO NFs after calcination (inset: high resolution image of NFs), (c) diameter size distribution of as-spun precursor nanofibers and (d) diameter size distribution of LMNFO NFs (after calcination). | ||
To reveal the secondary structure and crystallinity of the material, the as-prepared LMNFO NFs were characterized by HR-TEM. Fig. 3(a) shows an HR-TEM image of LMNFO NFs with nanocrystallites as secondary structures, which are joined together sequentially in an unidirectional fashion to form the hierarchical nanofibers, which is in good agreement with the high-resolution SEM images in terms of their morphology and secondary structure. Fig. 3(b) is an electron diffraction pattern that reveals the crystalline nature of the material, which features a hexagonal phase. To confirm this, selected area electron diffraction (SAED) patterns with fast Fourier transforms (FFTs) were collected from a selected region of the high-resolution image near the diffraction zone axis [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 100], and the results showed a well-defined hexagonal phase with lattice fringes that had a regular d-spacing value of 4.1 Å, as shown in Fig. 3(c) and its inset, which is in good agreement with the Rietveld analysis of the XRD data. Furthermore, the marked spots in the FFT pattern correspond to the (003) and (104) planes of the LMNFO crystal structure, which are consistent with the lattice fringe d-spacing values of 4.1 Å and 2.3 Å, respectively. Fig. 3(d) presents the energy dispersive X-ray (EDX) spectrum of LMNFO NFs, from which the individual elements Mn, Ni, Fe, and O can be identified. (Li cannot be detected, as Li Kα is too low; the spectrum also contains Cu from the usage of a Cu grid for the TEM analysis.) Brunauer–Emmett–Teller (BET) analysis was performed for LMNFO NFs and NPs to understand the influence of the nanostructures on the specific surface area and pore size distribution. Apparently, from the corresponding isotherms (Fig. S2 in the ESI†), it is evident that LMNFO NFs feature improved values in terms of specific surface area and pore size (78.6 m2 g−1 and 2 nm, respectively) when compared to those of LMNFO NPs (39.2 m2 g−1 and 5 nm, respectively). This high specific surface area of LMNFO NFs can promote good electrochemical performance in terms of facile ionic diffusion pathways and good penetration of the electrolyte into such a porous material. To evaluate the thermal stability of both samples, thermogravimetric (TGA) analysis was performed, and the corresponding curves (see Fig. S3 in the ESI†) confirm the excellent thermal stability of LMNFO NFs and NPs over the wide range of temperature from 50 to 1000 °C.
100], and the results showed a well-defined hexagonal phase with lattice fringes that had a regular d-spacing value of 4.1 Å, as shown in Fig. 3(c) and its inset, which is in good agreement with the Rietveld analysis of the XRD data. Furthermore, the marked spots in the FFT pattern correspond to the (003) and (104) planes of the LMNFO crystal structure, which are consistent with the lattice fringe d-spacing values of 4.1 Å and 2.3 Å, respectively. Fig. 3(d) presents the energy dispersive X-ray (EDX) spectrum of LMNFO NFs, from which the individual elements Mn, Ni, Fe, and O can be identified. (Li cannot be detected, as Li Kα is too low; the spectrum also contains Cu from the usage of a Cu grid for the TEM analysis.) Brunauer–Emmett–Teller (BET) analysis was performed for LMNFO NFs and NPs to understand the influence of the nanostructures on the specific surface area and pore size distribution. Apparently, from the corresponding isotherms (Fig. S2 in the ESI†), it is evident that LMNFO NFs feature improved values in terms of specific surface area and pore size (78.6 m2 g−1 and 2 nm, respectively) when compared to those of LMNFO NPs (39.2 m2 g−1 and 5 nm, respectively). This high specific surface area of LMNFO NFs can promote good electrochemical performance in terms of facile ionic diffusion pathways and good penetration of the electrolyte into such a porous material. To evaluate the thermal stability of both samples, thermogravimetric (TGA) analysis was performed, and the corresponding curves (see Fig. S3 in the ESI†) confirm the excellent thermal stability of LMNFO NFs and NPs over the wide range of temperature from 50 to 1000 °C.
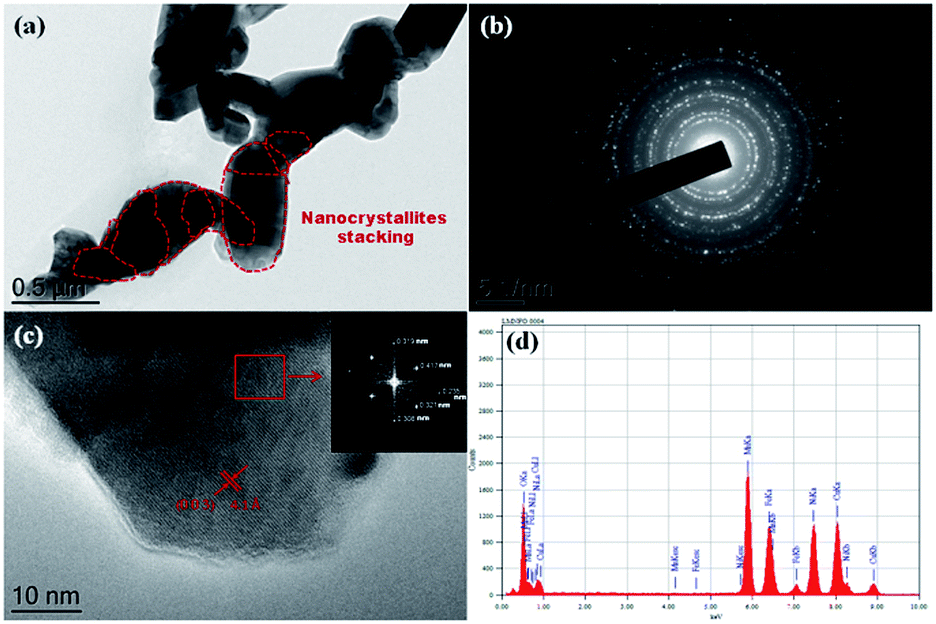 | ||
| Fig. 3 (a) TEM image of LMNFO NFs with well-ordered stacking of nanocrystallites (demarcated with red dotted lines); (b) electron diffraction pattern of LMNFO NFs; (c) high resolution TEM image of a single LMNFO crystallite in NFs with lattice fringes corresponding to the (003) plane and lattice d-spacing of 4.1 Å (inset: FFT pattern of a selected area in the image); and (d) EDX spectrum of LMNFO NFs. | ||
The LMNFO NF and LMNFO NP samples were electrochemically characterized by measuring their galvanostatic and potentiostatic curves using an automatic battery cycler and electrochemical workstation, respectively. The assembled coin-cells in the half-cell configuration were electrochemically tested in a voltage window of 2–4.5 V in order to avoid unnecessary degradation reactions associated with carbonate based electrolytes at high voltage. It is important to note that all electrochemical studies were performed with Li-foil (or Na-foil) as a counter electrode to test the compatibility of the LMNFO cathode in LIBs (or SIBs). Fig. 4(a) shows galvanostatic charge–discharge curves of LMNFO NFs and LMNFO NPs measured at 0.1 C vs. Li. They delivered an initial specific discharge capacity of ∼109 mA h g−1 and ∼96 mA h g−1, respectively, and retained the specific discharge capacity of ∼87 mA h g−1 and ∼62 mA h g−1, respectively, after 100 full charge–discharge cycles under the same experimental conditions. The uniform charge–discharge curves appear the same, as reported elsewhere,26,27 presenting the characteristic feature of layered structures with a single phase reaction. The constant plateau around 3.7 V during charging corresponds to oxidation of Ni with Ni2+/Ni3+/Ni4+ redox couples, which is also evident from our in situ neutron diffraction studies on an LMNFO cell.47 In charge–discharge behaviour, it is assumed that Mn oxidation couldn't take place or be involved in the electrochemistry, however, further observations on Mn redox couples are explained in the cyclic voltammetry section. During discharge, as there is no plateau observed around 4 V, it implies that the Fe3+/Fe4+ redox couple did not take place and instead, the stability of the crystal structure during Li-ion/Na-ion intercalation/de-intercalation in the charge–discharge process is facilitated at the expense of the capacity.27 The improved capacity values for LMNFO NFs could be attributed to the enhanced ionic conductivity due to the well-guided ionic transfer pathways during the cycling process and the high specific surface area of LMNFO NFs. Under the same experimental conditions at the current rate of 0.1 C and with Na-foil as a counter electrode, LMNFO NFs and LMNFO NPs showed an initial specific discharge capacity of ∼88 mA h g−1 and ∼74 mA h g−1, respectively, as shown in Fig. 4(b). The immense compromise in the capacity values (when compared to the Li ones) could be due to the difficulties in the ionic (Na and/or Li-ion) intercalation/de-intercalation process during electrochemical cycling because of the 3 times larger mass (23 g mol−1) and larger ionic radius (1.06 Å) of Na when compared to those of Li (mass: 6.9 g mol−1, ionic radius: 0.76 Å).37,48 Considering this experiment on LIBs and SIBs, there should be a significant impact of the type of nanostructure on the cycle life of LMNFO in both types of cells. To evaluate the cyclic stability of LMNFO NFs and LMNFO NPs in both LIBs and SIBs, assembled coin-cells were tested for 100 full charge–discharge cycles [see Fig. 4(c)], and the results were analysed. Irrespective of LIB or SIB, surprisingly, there is a significant influence of the 1D nanofiber morphology on the cycle life of LMNFO, although with the compromise in the capacity values (in SIBs) as mentioned earlier. In detail, at a current density of 0.1 C, LMNFO NF samples in LIBs were cycled for 100 full cycles and thereafter featured excellent cyclic performance, with a specific discharge capacity of ∼98 mA h g−1 and 10% capacity fading when compared to the performance of LMNFO NPs, with a specific discharge capacity of ∼69 mA h g−1 and 27% capacity fading. Seemingly, the LMNFO cathodes delivered less practical capacity values, whereas the cyclic performance is somehow acceptable. The improved cyclic performance is likely attributed to the stabilizing Li2MO3 (M = Mn, Ni, Fe) component and the limited lithium intercalation by cation mixing.47 As shown, there is also an obvious improvement in cycling performance of LMNFO NFs, compared to NPs, which could be ascribed to the impact of the high surface area, 1D nanofiber morphology in terms of their well-interconnected network with the electrolyte, due to their significant wettable properties and better charge transfer characteristics during the cycling process. It is likely that LMNFO NPs suffered from relatively poor cyclic stability because of their unfortunate self-agglomeration issue during the cycling process, which resulted in poor physical contact with the current collector, giving rise to internal resistance in the working cell. It is reported that 1D nanostructures such as nanofibers are free from such self-agglomeration due to their systematic nanocrystallite growth at localized sites along the direction of the nanofiber structure during the step-wise annealing process.49 The same trend in cycling performance is observed in the case of LMNFO NFs and LMNFO NPs in SIBs, although with loss of capacity. It is evidently worth noting that irrespective of the battery system (either LIB or SIB), the 1D nanostructure morphology could play a key role in the dual functionality of LMNFO in LIBs and SIBs.
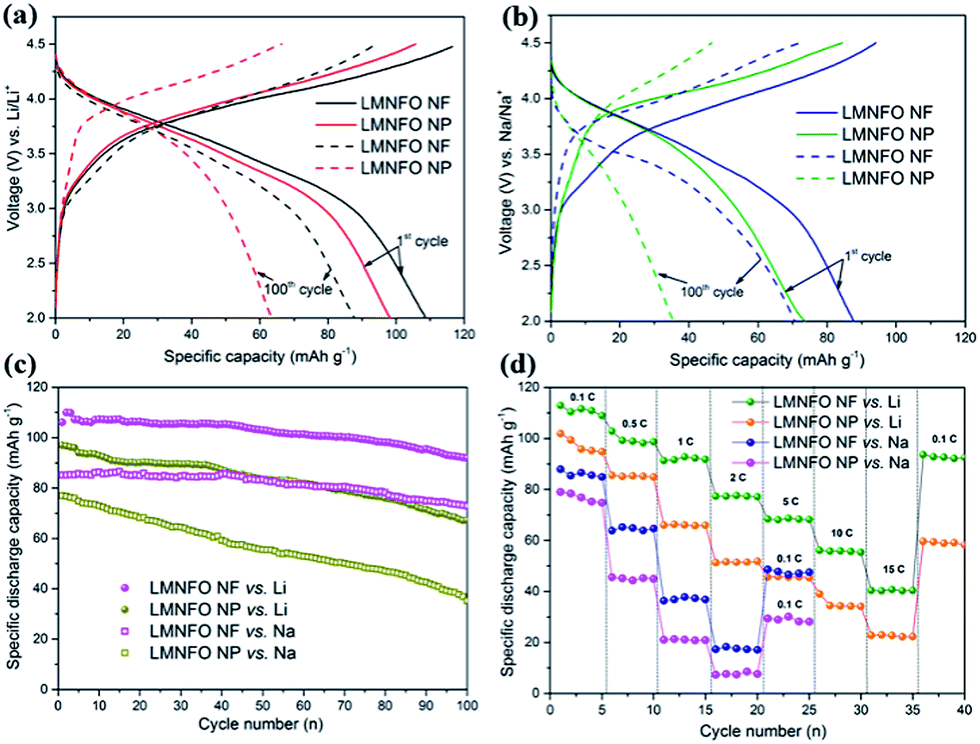 | ||
| Fig. 4 (a) & (b) Charge–discharge behaviour of LMNFO NFs and LMNFO NPs in LIBs and SIBs, respectively, in the initial cycle and 100th cycle at a 0.1 C current rate; (c) & (d) cycling performance and rate capability of LMNFO NFs and LMNFO NPs, respectively, in LIBs and SIBs in the voltage range of 2–4.5 V. | ||
To understand the high current operation of LMNFO, rate capability tests were conducted for both the LMNFO NF and NP samples in LIBs at various current rates in the range of 0.1 C to 15 C within a voltage window of 2–4.5 V, as presented in Fig. 4(d). As expected, LMNFO NFs showed improved rate performance when compared to that of LMNFO NPs, however, the capacity values are not promising at the very high rates of 5 C to 15 C i.e., 70–40 mA h g−1. With increasing current rate, the voltage plateau in the charge–discharge curves of both samples steadily diminishes, which could suggest that there is possible deterioration in the crystal structure of LMNFO during complicated ionic intercalation/de-intercalation processes, with corresponding phase transitions and gradual polarization of the electrodes during successive electrochemical cycles (see Fig. S4 in the ESI†). In the case of SIBs, LMNFO NFs showed a slight improvement in the rate performance when compared to that of LMNFO NPs; however, both electrodes suffered from severe capacity fading at current rates beyond 0.5 C [Fig. 4(d)]. This could be mainly attributable to the fundamental properties of the Na-ion with its larger mass and inability to shuttle quickly during intercalation/de-intercalation processes at relatively high rates (less shuttle time) during the cycling process.
To investigate the transitions of redox couples, cyclic voltammograms (CVs) were collected for both LMNFO NFs and LMNFO NPs in the voltage range of 2–4.5 V vs. Li/Li+ and Na/Na+ in LIBs and SIBs, respectively, at the scan rate of 0.1 mV s−1. Fig. 5(a) shows CVs of both samples in LIBs, where two pairs of redox couple peaks are observed in both anodic and cathodic sweeps at 4.2 V/3.6 V and 3.2 V/3.2 V, corresponding to Ni2+/Ni4+ and Mn3+/Mn4+ redox reactions.27,50 The oxidation/reduction of Mn ions would be partial, as is evident from the low intensity of the corresponding redox peak, when compared to that of Ni ions. This could be mainly attributed to the presence of the secondary Li2MO3 (M = Mn, Ni, Fe) phase (∼20%), which is electrochemically inactive in the current voltage range and instead promotes the structural stability of LMNFO during Li/Na-ion intercalation/de-intercalation processes by acting as a stabilization component. It is important to note that such an Li2MO3 (M = Mn, Ni, Fe) phase will be only electrochemically active at the voltage range >4.6 V by decomposing into Li2O and MnO2.45,46,51 On the other hand, Fig. 5(b) shows CVs of both samples in SIBs and repeats the trend found in LIBs with two pairs of redox couple peaks in both anodic and cathodic sweeps at 3.9 V/4.0 V and 3.2 V/2.9 V, corresponding to Ni2+/Ni4+ and Mn3+/Mn4+ redox reactions. The difference between the voltage values of the corresponding peaks in LIBs and SIBs is ∼0.3 V, which is obviously due to the difference in the redox potential values of LIBs (−3.04 vs. Li/Li+) and SIBs (−2.71 vs. Li/Li+).48 From both galvanostatic charge–discharge studies and CVs, it is evident that there is no oxidation/reduction process for the Fe ions in the current voltage range of tested cells; instead they could contribute to the structural stability of LMNFO during the cycling process at the expense of capacity.
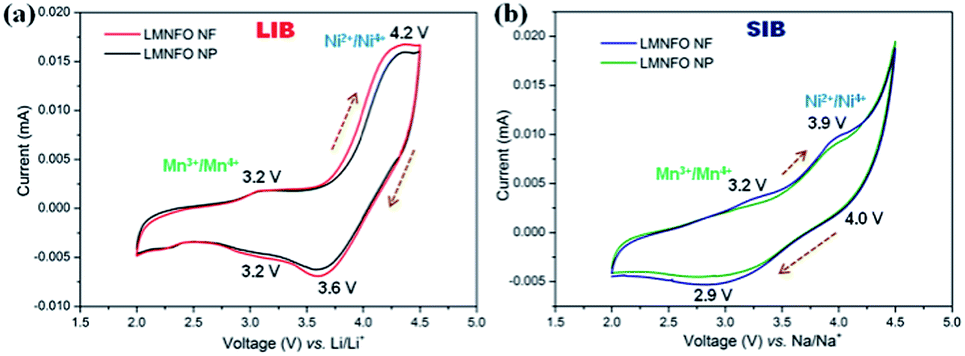 | ||
| Fig. 5 Cyclic voltammograms of LMNFO NFs and LMNFO NPs in the voltage range of 2–4.5 V vs. Li/Li+ (a) and Na/Na+ (b) at a 0.1 mV s−1 scan rate. | ||
As observed, the improved cyclic performance of LMNFO NFs when compared to that of LMNFO NPs in both LIBs and SIBs could be attributed to numerous aspects associated with the 1D nanostructure morphology and its structural stability. The structural stability of such hierarchical nanofibers corresponds to the formation of LMNFO nanocrystallites at localized sites along the one-dimensional growth direction of the nanofibers during the heat-treatment process to form LMNFO nanofibers from those of the precursor, which is clearly represented by the schematic illustration in Fig. 6. The secondary structure of nanofibers with hierarchical stacking of nanocrystallites is also revealed from SEM and high resolution TEM characterizations. Such a secondary structure in the nanofibers protects them from possible self-agglomeration during the cycling process.49,52 Such nanofibers with secondary structures can have better contact area with the electrolyte during electrochemical charge–discharge behaviour and could accelerate electrochemical behaviour. On the other hand, nanoparticles prepared by the sol–gel process tend to agglomerate during electrochemical cycling due to their high surface energy and non-uniform particle size distribution, which tends to impede electrochemical performance (see Fig. S5 in the ESI†). Conductivity profiles of such nanostructures vary greatly with their charge transfer mechanism during cycling processes. Irrespective of the LIB or SIB system, it is anticipated that LMNFO nanofibers facilitate facile ionic/electronic transmission in well-guided transfer pathways in a one-dimensional fashion (see Fig. 6) and thus enhance conductivity profiles. To further confirm this proposed mechanism, electrochemical impedance spectroscopy studies were performed for both LMNFO NFs and LMNFO NPs in LIBs and SIBs under open circuit voltage and in the frequency range from 100 kHz to 100 mHz. The corresponding impedance Nyquist plot of LMNFO NFs in an LIB shows a charge transfer resistance of 43 Ω, which is lower when compared to that of LMNFO NPs in an LIB (76 Ω) as shown in Fig. S6(a) (in the ESI†). In the case of SIBs [see Fig. S6(b)†], the same trend is observed, however, the charge transfer resistance of LMNFO NFs is relatively higher (480 Ω), which could be attributed to the larger ionic radius and mass of Na-ions, and thereby impede the facile charge transfer process. By considering the electrochemical performance of LMNFO NFs in both LIBs and SIBs, it is interesting to note that the one-dimensional nanomorphology of LMNFO and crystallographic aspects of the stabilization component Li2MO3 (M = Mn, Ni, Fe) play a key role in retaining the cyclic stability of LMNFO in either type of cell (LIB/SIB). Further research and development towards improving the dual functionality of such kinds of electrode materials in various energy storage devices are necessary.
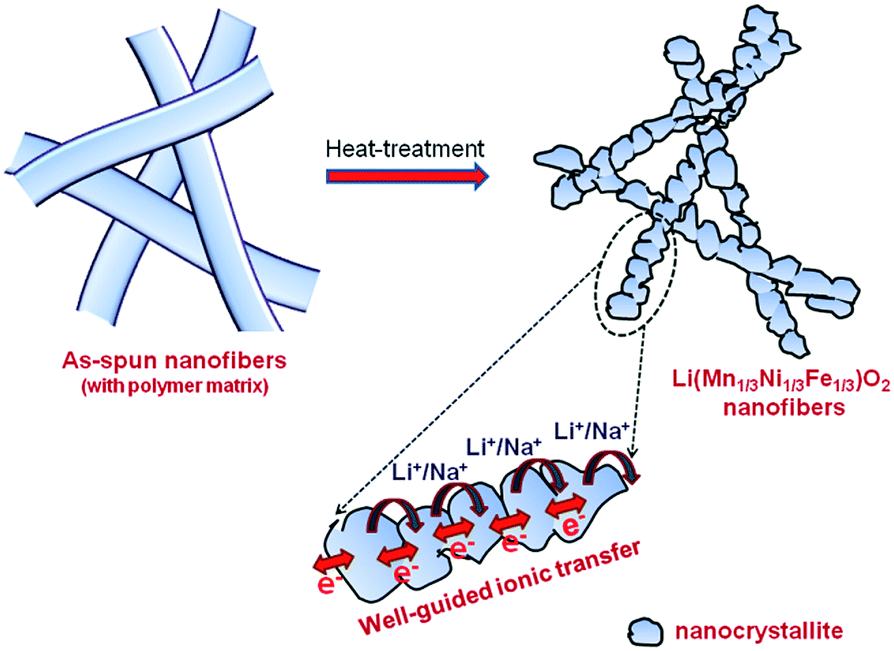 | ||
| Fig. 6 Schematic representation illustrating the fabrication of LMNFO NFs and proposed charge transfer kinetics in the secondary structure of the nanofibers. | ||
4. Conclusions
Low-cost, eco-friendly, and morphologically stable Li1+x(Mn1/3Ni1/3Fe1/3)O2 nanofibers were prepared by the electrospinning technique and were used as a cathode material in both LIBs and SIBs. In both systems, these fibers showed improved initial capacity values (∼109 mA h g−1vs. Li; ∼87 mA h g−1vs. Na) and good cycling stability when compared to the performance of nanoparticles, although at the expense of decreased capacity values in the SIB system. The significant improvement in electrochemical performance could be attributed to the high surface area, well-guided charge transfer kinetics with short ionic diffusion pathways, and the large effective contact area with the electrolyte during the cycling process. Such 1D nanostructured cathode materials could be the best alternatives to traditional ones in LIBs or SIBs.Acknowledgements
The authors would like to acknowledge the financial support provided by the Commonwealth of Australia and Automotive CRC 2020. The use of infrastructure and facilities at ISEM and the UOW Electron Microscopy Centre is gratefully acknowledged. We would also like to thank Dr Tania Silver for critical reading of the manuscript.Notes and references
- B. Scrosati, Nature, 1995, 373, 557 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271 CrossRef CAS.
- K. Kang, Y. S. Meng, J. Bréger, C. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- O. K. Park, Y. Cho, S. Lee, H.-C. Yoo, H.-K. Song and J. Cho, Energy Environ. Sci., 2011, 4, 1621 CAS.
- S.-Y. Chung, J. T. Bloking and Y.-M. Chiang, Nat. Mater., 2002, 1, 123 CrossRef CAS PubMed.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461 CrossRef CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364 CrossRef CAS PubMed.
- Y. Nishi, Chem. Rec., 2001, 1, 406 CrossRef CAS PubMed.
- L. M. Reddy, S. Nagarajan, P. Chumyim, S. R. Gowda, P. Pradhan, S. R. Jadhav, M. Dubey, G. John and P. M. Ajayan, Sci. Rep., 2012, 2, 960 Search PubMed.
- M.-J. Lee, S. Lee, P. Oh, Y. Kim and J. Cho, Nano Lett., 2014, 14, 993 CrossRef CAS PubMed.
- L.-H. Hu, F.-Y. Wu, C.-T. Lin, A. N. Khlobystov and L.-J. Li, Nat. Commun., 2013, 4, 1687 CrossRef PubMed.
- Y.-K. Sun, D.-J. Lee, Y. J. Lee, Z. Chen and S.-T. Myung, ACS Appl. Mater. Interfaces, 2013, 5, 11434 CAS.
- P. Kalyani and N. Kalaiselvi, Sci. Technol. Adv. Mater., 2005, 6, 689 CrossRef CAS PubMed.
- C. V. Rao, A. L. M. Reddy, Y. Ishikawa and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2011, 3, 2966 CAS.
- H. Arai, S. Okada, Y. Sakurai and J. Yamaki, Solid State Ionics, 1997, 95, 275 CrossRef CAS.
- R. Armstrong and P. G. Bruce, Nature, 1996, 381, 499 CrossRef.
- S. Chen, F. Cao, F. Liu, Q. Xiang, X. Feng, L. Liu and G. Qiu, RSC Adv., 2014, 4, 13693 RSC.
- W. Tang, Y. Hou, F. Wang, L. Liu, Y. Wu and K. Zhu, Nano Lett., 2013, 13, 2036 CrossRef CAS PubMed.
- Y. Wang, P. He and H. Zhou, Energy Environ. Sci., 2011, 4, 805 CAS.
- Y. Itou and Y. Ukyo, J. Power Sources, 2005, 146, 39 CrossRef CAS PubMed.
- L. Wang, J. Li, X. He and W. Pu, J. Solid State Electrochem., 2009, 13, 1157 CrossRef CAS.
- J. Xu, S. L. Chou, Q. F. Gu, H. K. Liu and S. X. Dou, J. Power Sources, 2013, 225, 172 CrossRef CAS PubMed.
- F. Wang, S. Xiao, Z. Chang, Y. Yang and Y. Wu, Chem. Commun., 2013, 49, 9209 RSC.
- K. C. Mahesh, H. Manjunatha, R. B. Shivashankaraiah, G. S. Suresh and T. V. Venkatesha, J. Electrochem. Soc., 2012, 159, A1040 CrossRef CAS PubMed.
- K. Karthikeyan, S. Amaresh, V. Aravindan, W. S. Kim, K. W. Nam, X. Q. Yang and Y. S. Lee, J. Power Sources, 2013, 232, 240 CrossRef CAS PubMed.
- K. Karthikeyan, S. Amaresh, G. W. Lee, V. Aravindan, H. Kim, K. S. Kang, W. S. Kim and Y. S. Lee, Electrochim. Acta, 2012, 68, 246 CrossRef CAS PubMed.
- H. K. Liu, Mater. Res. Bull., 2013, 48, 4968 CrossRef CAS PubMed.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414 CrossRef CAS PubMed.
- H. H. Liu, S. Surawanvijit, R. Rallo, G. Orkoulas and Y. Cohen, Environ. Sci. Technol., 2011, 45, 9284 CrossRef CAS PubMed.
- B. Su, Y. Wu and L. Jiang, Chem. Soc. Rev., 2012, 41, 7832 RSC.
- T. Song, L. Hu and U. Paik, J. Phys. Chem. Lett., 2014, 5, 720 CrossRef CAS.
- S. Kalluri, K. H. Seng, Z. Guo, H. K. Liu and S. X. Dou, RSC Adv., 2013, 3, 25576 RSC.
- S. Ramakrishna, K. Fujihara, W.-E. Teo, T. Yong, Z. Ma and R. Ramaseshan, Mater. Today, 2006, 9, 40 CrossRef CAS.
- S. Kalluri, K. H. Seng, W. K. Pang, Z. Guo, Z. Chen, H. K. Liu and S. X. Dou, ACS Appl. Mater. Interfaces, 2014, 6, 8953 CAS.
- M. P. Pechini, US Pat., 3.330697, 1967.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947 CrossRef CAS.
- D. Richard, M. Ferrand and G. J. Kearley, J. Neutron Res., 1996, 4, 33 CrossRef.
- J. R. Carvajal, Phys. B, 1993, 192, 55 CrossRef.
- T. Roisnel and J. R. Carvajal, WinPLOTR: a Windows Tool for Powder Diffraction Patterns Analysis, Presented at Materials Science Forum, Proceedings of the Seventh European Powder Diffraction Conference (EPDIC 7), 2000 Search PubMed.
- N. Terada, D. D. Khalyavin, J. M. Perez-Mato, P. Manuel, D. Prabhakaran, A. Daoud-Aladine, P. G. Radaelli, H. S. Suzuki and H. Kitazawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 184421 CrossRef.
- D. Wang, I. Belharouak, X. Zhang, Y. Ren, G. Meng and C. Wang, J. Electrochem. Soc., 2014, 161, A1 CrossRef CAS PubMed.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112 RSC.
- C. S. Johnson, J. S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, Electrochem. Commun., 2004, 6, 1085 CrossRef CAS PubMed.
- C. S. Johnson, N. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, Chem. Mater., 2008, 20, 6095 CrossRef CAS.
- A. Boulineau, L. Croguennec, C. Delmas and F. Weill, Chem. Mater., 2009, 21, 4216 CrossRef CAS.
- W. K. Pang, S. Kalluri, V. K. Peterson, S. X. Dou and Z. Guo, Phys. Chem. Chem. Phys., 2014, 16, 25377–25385 RSC.
- S. Engelke, Current and future sodium-ion battery research, http://storage4.eu/2013/03/current-and-future-sodium-ion-battery-research/.
- R. Hagen, A. Lepcha, X. Song, W. Tyrra and S. Mathur, Nano Energy, 2013, 2, 304 CrossRef PubMed.
- R. Kataoka, T. Mukai, A. Yoshizawa and T. Sakai, J. Electrochem. Soc., 2013, 160, A933 CrossRef CAS PubMed.
- M. Tabuchi, Y. Nabeshima, T. Takeuchi, H. Kageyama, K. Tatsumi, J. Akimoto, H. Shibuya and J. Imaizumi, J. Power Sources, 2011, 196, 3611 CrossRef CAS PubMed.
- L. Mai, L. Xu, C. Han, X. Xu, Y. Luo, S. Zhao and Y. Zhao, Nano Lett., 2010, 10, 4750 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table of Rietveld refinement parameters, BET N2 isotherms, TGA curves, electrochemical impedance plots, charge–discharge curves at various current rates of LMNFO NFs and LMNFO NPs; SEM image of LMNFO NPs. See DOI: 10.1039/c4ta04271a |
| This journal is © The Royal Society of Chemistry 2015 |