
One-pot synthesis of porous Pt–Au nanodendrites supported on reduced graphene oxide nanosheets toward catalytic reduction of 4-nitrophenol†
Jing-Jing
Lv
a,
Ai-Jun
Wang
*a,
Xiaohong
Ma
b,
Ru-Yi
Xiang
a,
Jian-Rong
Chen
a and
Jiu-Ju
Feng
*a
aCollege of Geography and Environmental Science, College of Chemistry and Life Science, Zhejiang Normal University, Jinhua 321004, China. E-mail: ajwang@zjnu.cn; jjfeng@zjnu.cn; Fax: +86 579 82282269; Tel: +86 579 82282269
bState Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China
First published on 27th October 2014
Abstract
In this work, a facile, convenient and effective one-pot wet-chemical method was developed for preparation of well-dispersed porous bimetallic Pt–Au alloyed nanodendrites uniformly supported on reduced graphene oxide nanosheets (Pt–Au pNDs/RGOs) at room temperature. The fabrication strategy was efficient and green owing to the use of cytosine as a structure-directing agent and weak stabilizing agent, without employing any organic solvent, template, seed, surfactant, or complicated apparatus. The as-synthesized Pt–Au pNDs/RGOs exhibited significantly enhanced catalytic performance toward the reduction of 4-nitrophenol, as compared to commercial Pt black and home-made Au nanocrystals.
1. Introduction
As well known, 4-aminophenol (4-AP) has broad applications as a photographic developer, hair-dying agent, anticorrosion-lubricant, and corrosion inhibitor.1,2 Furthermore, it is an important intermediate for preparation of a variety of analgesic and antipyretic drugs.3,4 Its synthesis usually involves the reduction of 4-nitrophenol (4-NP) by sodium borohydride (NaBH4) with the assistance of diverse catalysts such as Fe–FeO sludge,5 metal oxides,6 and especially metallic nanoparticles (NPs),7,8 because of their versatile and tunable size-, shape- and composition-dependent properties.9,10In particular, noble metals including Pt, Pd, and Au have always attracted significant attention owing to their broad applications,11–13 especially in catalysis.14,15 As a result, the size- and shape-controlled synthesis has recently stimulated tremendous research efforts, and thereby various morphologies have been fabricated, such as wires,16 rods,17 prisms,18 plates,19 polyhedrons,14 and branched nanostructures.20
Noble bimetallic nanocrystals (NCs) with highly branched structures have attracted increasing interest for their potential applications in organic reactions,21 fuel cells,22 and sensing devices.23 Accordingly, many synthetic approaches have been developed, such as hydrothermal method,24 electrochemical deposition,25 seed-mediated growth,26 and galvanic replacement.27 However, most of them are relatively time-consuming, high-cost, and involve complicated reaction steps.
It is desirable to deposit bimetallic NCs on an appropriate support such as a carbon-based material, with the purpose of reducing their usage and preventing their further aggregation, sintering, or leaching.28 Recently, graphene, as an oxygen-abundant carbon layered material, has been extensively employed as a promising support for bimetallic catalysts, owing to its excellent mechanical, electrical, and thermal properties, along with its high specific surface area.29 For example, Wang and co-workers fabricated three-dimensional Pt-on-Pd bimetallic nanodendrites supported on graphene as a catalyst for methanol oxidation.22 He and co-workers synthesized Au–Pd NPs/graphene oxide, which exhibited excellent catalytic activity for the reduction of 4-NP to 4-AP.30
Herein, we report a facile and convenient wet-chemical route to synthesize porous Pt–Au nanodendrites with highly branched nanostructures uniformly supported on reduced graphene oxide nanosheets (denoted as Pt–Au pNDs/RGOs). The catalytic activity of the as-prepared nanocomposites toward the reduction reaction of 4-NP in the presence of NaBH4 was examined.
2. Experimental section
2.1 Chemicals
Chloroplatinic acid (H2PtCl6·6H2O), chloroauric acid (HAuCl4), cytosine, hydrazine hydrate (16.5 M), graphite powder, 4-nitrophenol (4-NP), sodium borohydride (NaBH4), and commercial Pt black were purchased from Shanghai Aladdin Chemical Reagent Company (Shanghai, China). All other chemicals were analytical grade and used without further purification. All aqueous solutions were prepared with twice-distilled water.2.2 Synthesis of Pt–Au pNDs/RGOs
In a typical synthesis, GOs were firstly prepared by the modified Hummer's method.31 After this, 2.9 mL of H2PtCl6 (38.6 mM), 2 mL of HAuCl4 (24.3 mM), and 0.028 g cytosine (25 mM) were added to a 10 mL solution containing 1 mM GOs under stirring. Next, the pH value of the mixed solution was adjusted to 12 by using freshly prepared 0.1 M NaOH. Then, the mixture was placed into a water-bath (25 °C) and immediately turned black upon the addition of 100 μL hydrazine hydrate under stirring. Following this, the mixture was allowed to react continually for another 1 h. Finally, the black precipitate was collected by centrifugation, thoroughly washed with ethanol and water, and dried at 60 °C in a vacuum for further characterization.In the control experiments, Au NCs were fabricated in a similar way, as described previously.32 Moreover, the control experiments were carried out by changing the amount of cytosine or by preparation without GOs, while the other experimental conditions were kept the same.
2.3 Characterization
The morphology, composition, and crystal structures of the samples were characterized by transmission electron microscopy (TEM), high-resolution TEM (HRTEM) images, X-ray energy dispersive spectroscopy (EDS), and high angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) on a JEM-2100F HR transmission electron microscope coupled with an energy-dispersive X-ray spectrometer (Oxford-1NCA). TEM analysis was conducted at an accelerating voltage of 200 kV, and Cu grids were used as substrates. A small amount of the black sample was dispersed in ethanol under ultrasonication, and then a drop of the suspension was dropped onto the Cu grid for TEM observation.The crystal structures were obtained by X-ray diffraction (XRD) on a Bruker-D8-AXS diffractometer system equipped with Cu Kα radiation (Bruker Co., Germany). X-ray photoelectron spectroscopy (XPS) measurements were conducted with a ThermoFisher-ESCALab 250 (ThermoFisher, E. Grinstead, UK), using Al Kα X-ray radiation (1486.6 eV) for excitation. Raman spectra were recorded on a Renishaw Raman system model 1000 spectrometer equipped with a charge-coupled device (CCD) detector. Thermogravimetric analysis (TGA) was carried out under a flow of air on a NETZSCH STA 449C analyzer. The samples were heated from 25 to 900 °C with a heat rate of 10 °C min−1.
2.4 Catalytic measurements
The measurements for the catalytic reduction of 4-NP by Pt–Au pNDs/RGOs, Pt black, and Au NCs were conducted by UV-Vis spectroscopy in a quartz cuvette on a Lambda 950 UV/Vis/NIR spectrophotometer (Perkin Elmer Corporation). A total amount of 300 μL of 4-NP (0.7 mM) was diluted with 1 mL of water, followed by the addition of 1 mL of freshly prepared ice-cold NaBH4 solution (0.5 M). Subsequently, a certain amount of the as-prepared catalyst suspension was injected into the cuvette to trigger the reaction, and the UV-Vis spectral changes were monitored at different time intervals. All of the experiments were conducted at room temperature, if not stated otherwise.3. Results and discussion
As illustrated in Fig. 1A, the typical product contains many nanodendrites uniformly anchored on RGOs (labeled with red arrows). The as-prepared nanodendrites possess interconnected porous structures (Fig. 1B and C), and their sizes range from 51 to 91 nm in diameter, with an average diameter of 70 nm (inset of Fig. 1B).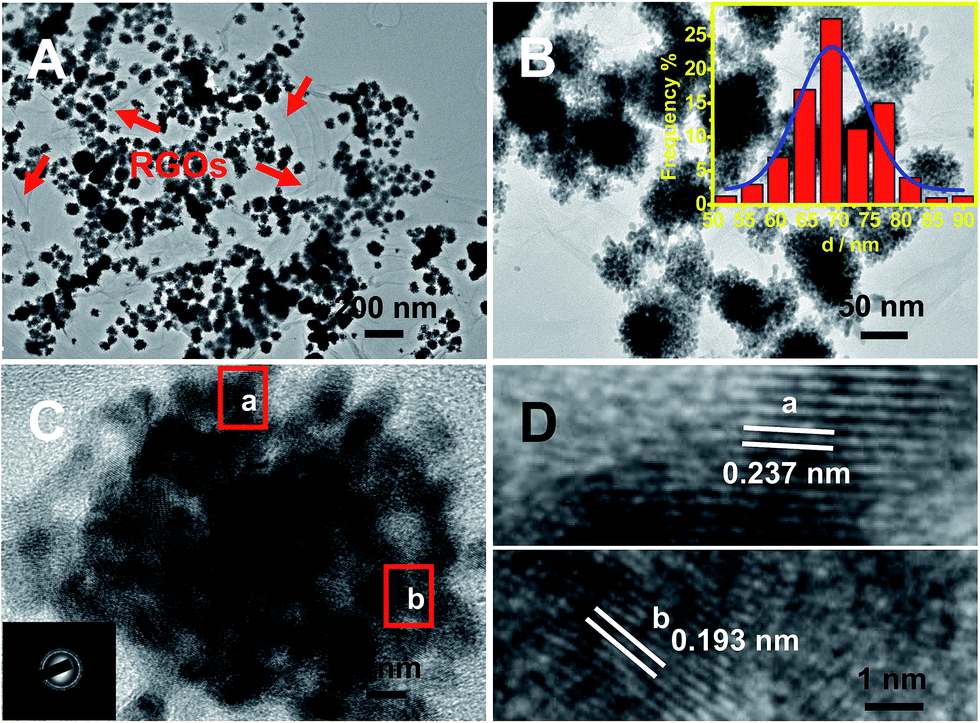 | ||
| Fig. 1 Low (A), medium (B), and high (C and D) magnification TEM images of Pt–Au pNDs/RGOs. The insets of (B) and (C) show the corresponding size-distribution histogram and SAED pattern, respectively. The products were obtained with 25 mM cytosine. | ||
The selected-area electron diffraction (SAED) pattern displays the characteristics of the face-centered cubic (fcc) feature of Pt/Au (inset of Fig. 1C), revealing the polycrystalline nature of the as-synthesized Pt–Au nanodendrites.33 Moreover, their high crystallinity is strongly demonstrated by the HRTEM images taken from the marked regions (Fig. 1D), in which well-defined lattice fringes are observed, with interplanar spacing distances of 0.237 and 0.193 nm, which are well-matched with the (111) and (200) crystal planes, respectively.34,35
The chemical composition and elemental distribution of Pt–Au pNDs/RGOs were characterized by EDS-mapping (Fig. 2A–E) and their EDS spectrum (Fig. 2F). The porous interconnected dendritic structures of Pt–Au pNDs can be clearly observed in the HAADF-STEM image (Fig. 2A), and the homogeneous distribution of Pt and Au elements throughout Pt–Au pNDs is confirmed by the respective elemental mapping images (Fig. 2B–D). These observations confirm the alloyed nature of Pt–Au pNDs,36 as further demonstrated by the line scanning profiles (Fig. 2E),37 where Pt and Au are homogeneously distributed in an individual nanodendrite. The Pt/Au atomic ratio is 50.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49.4 based on the data from the line scanning profiles and EDS spectrum (Fig. 2F). This value is in good agreement with the initial ratio of the precursors (H2PtCl6:HAuCl4 = 50:50), showing the effectiveness of the reduction strategy in the preparation of Pt–Au pNDs.
:49.4 based on the data from the line scanning profiles and EDS spectrum (Fig. 2F). This value is in good agreement with the initial ratio of the precursors (H2PtCl6:HAuCl4 = 50:50), showing the effectiveness of the reduction strategy in the preparation of Pt–Au pNDs.
 | ||
| Fig. 2 HAADF-STEM (A) and HAADF-STEM-EDS mapping (B–D) images, line scanning profiles (E), and EDS spectrum (F) of Pt–Au pNDs/RGOs. | ||
The XRD pattern of Pt–Au pNDs/RGOs (Fig. 3A, curve a) was obtained to provide more structural information, using GOs as a reference (Fig. 3A, curve b). As illustrated, four representative diffraction peaks emerge at 38.8°, 44.9°, 65.9°, and 79.4° for Pt–Au pNDs/RGOs, which are indexed to the (111), (200), (220), and (311) planes of the fcc structure of Pt/Au metal.38 It is worth noting that these peaks are observed coincidently for bulk Pt (JCPDS 04-0802) and Au (JCPDS 04-0784), further confirming the formation of a uniform Pt–Au alloy.39 Additionally, unlike Pt–Au pNDs/RGOs, there is a sharp peak located at 10.8° for GOs, which is assigned to the (001) planes of graphite oxide.40 The difference is ascribed to the significant reduction of GOs and the exfoliation of the layered graphene.41
 | ||
| Fig. 3 XRD (A) and Raman (B) spectra of Pt–Au pNDs/RGOs. | ||
This conclusion is strongly supported by the Raman spectrum of Pt–Au pNDs/RGOs (Fig. 3B, curve a), using GOs as a standard (Fig. 3B, curve b). Clearly, there are two predominant peaks at ca. 1610 and 1350 cm−1, which correspond to the well-documented G and D bands.42 The G band is associated with the vibrations of the graphite sp2 carbon domains, while the D band comes from the vibrations of the sp3 carbon atoms in the disordered graphene nanosheets.43 The D/G intensity ratio (ID/IG) is usually used as an indicator for quantifying the level of chemical modification of the graphitic carbon sample.44 The ID/IG value is 1.1 for Pt–Au pNDs/RGOs, which is higher than that of GOs (0.9), demonstrating the efficient removal of oxygenated functional groups from GOs during the present synthesis.45
XPS analysis further verified the efficient removal of oxygenated functional groups from GOs and the formation of Pt–Au pNDs/RGOs (Fig. 4). Specifically, there are four representative peaks located at 284.7, 285.6, 286.5, and 288.3 eV in the high-resolution C 1s spectrum (Fig. 4A), which are attributed to the C–C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO groups, respectively.46 Impressively, the intensity of the C–C band is much higher than that of GOs (Fig. S1†), revealing that the oxygenated functional groups are effectively reduced in the reaction, causing the formation of many defects on the RGOs. These defects would provide binding sites for the subsequent deposition of Pt–Au pNDs.47
O and O–CO groups, respectively.46 Impressively, the intensity of the C–C band is much higher than that of GOs (Fig. S1†), revealing that the oxygenated functional groups are effectively reduced in the reaction, causing the formation of many defects on the RGOs. These defects would provide binding sites for the subsequent deposition of Pt–Au pNDs.47
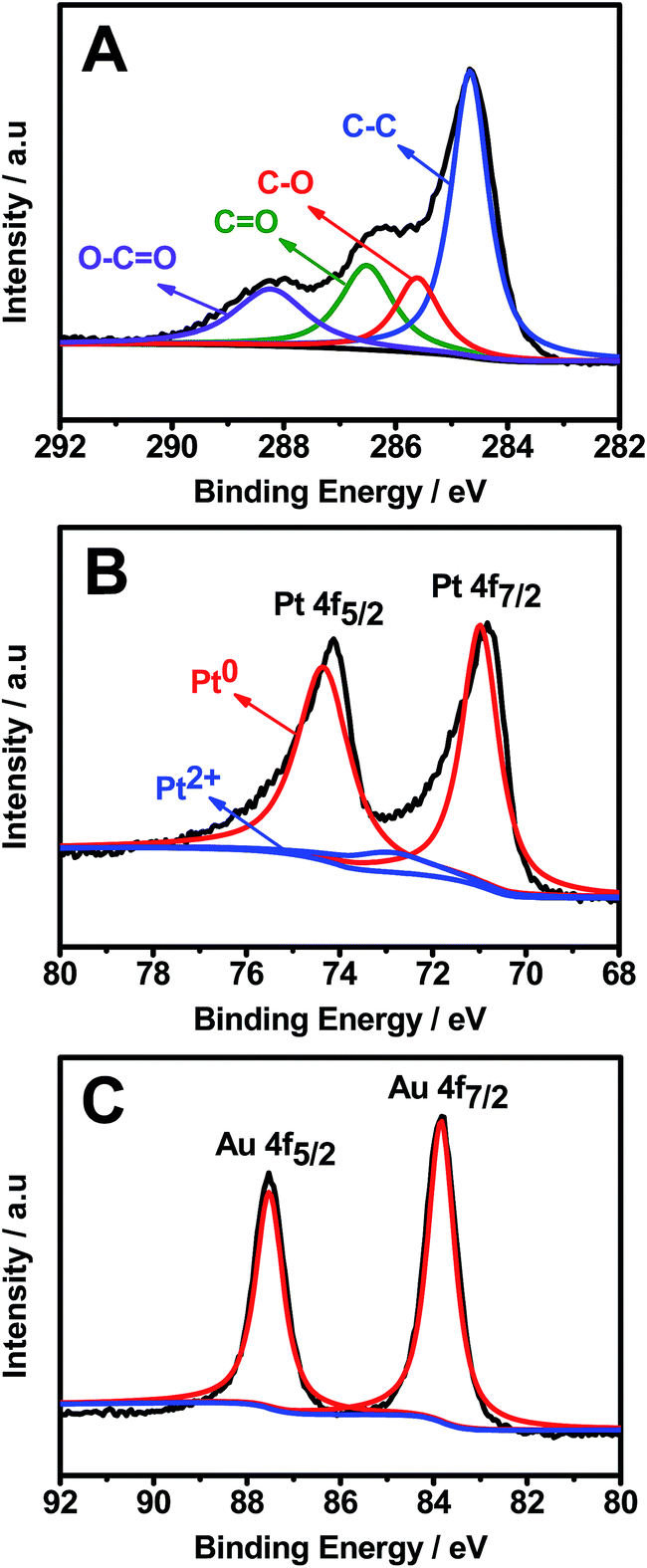 | ||
| Fig. 4 High-resolution C 1s (A), Pt 4f (B), and Au 4f (C) XPS spectra of Pt–Au pNDs/RGOs. | ||
The surface chemical states of Pt and Au were analyzed by the Pt 4f (Fig. 4B) and Au 4f (Fig. 4C) XPS spectra. The strong doublet peaks of Pt 4f emerging at 71.0 and 74.4 eV are ascribed to Pt0, whereas nearly no peaks are detected at 72.6 and 75.7 eV, showing the complete reduction of Pt2+ to Pt0 during the reaction.48 Meanwhile, the predominant Au0 4f5/2 and Au0 4f7/2 peaks illustrate that HAuCl4 is efficiently reduced to metallic Au0.49 These observations reveal that Pt0 and Au0 are the predominant species in Pt–Au pNDs/RGOs.
The amount of cytosine played a key role in the formation of Pt–Au nanocrystals (Fig. 5). The absence of cytosine yields numerous irregular nanoparticles with severe aggregation (Fig. 5A). A lower amount of cytosine (10 mM) results in the emergence of immature dendritic Pt–Au nanocrystals with a broad size distribution (Fig. 5B), and the best crystals are obtained using 25 mM cytosine (Fig. 1A and B). Nevertheless, excessive cytosine, such as 50 mM (Fig. 5C), induces the formation of irregular dendritic Pt–Au nanocrystals with low surface coverage and reduced particle size compared to 25 mM cytosine, demonstrating that cytosine acts as a structure-directing agent and weak stabilizing agent.
 | ||
| Fig. 5 TEM images of the products prepared without cytosine (A), and with 10 mM (B) and 50 mM (C) cytosine. | ||
Cytosine molecules selectively adsorbed on Pt and Au nuclei with different crystal planes.50 Specifically, the crystal growth was severely blocked on the high energy crystal planes, such as the (110) and (100) planes, which were bound by the adsorbed molecules.51 On the other hand, the growth on the (111) crystal planes with low energy was promoted according to the direct growth mechanism,20,36 as confirmed by the XRD pattern shown in Fig. 3A, resulting in the formation of dendritic nanostructures. Moreover, the presence of cytosine could also act as a physical barrier to hinder the aggregation of nanocrystals and thus improve their dispersity.52,53
Moreover, the absence of GOs leads to the formation of significantly aggregated Pt–Au dendritic products (Fig. S2†), indicating that GOs are responsible for the good distribution of Pt–Au products by acting as a support in the present synthesis. This is attributed to the fact that the many oxygenated functional groups on the surfaces of GOs control the reduction kinetics.49
The catalytic properties of Pt–Au pNDs/RGOs were examined via the reduction reaction of 4-NP to 4-AP in the presence of excess NaBH4, as depicted in Fig. 6. BH4− would adsorb onto the catalyst surface and transfer active hydrogen species to the particle surface to form a metal hydride complex.54 Meanwhile, 4-NP also adheres to the surfaces of Pt–Au pNDs/RGOs via chemical adsorption.55 As a result, the adsorbed hydrogen species would transfer to the nitro groups, inducing their reduction to amino groups; this hypothesis is strongly supported by previous work.56,57
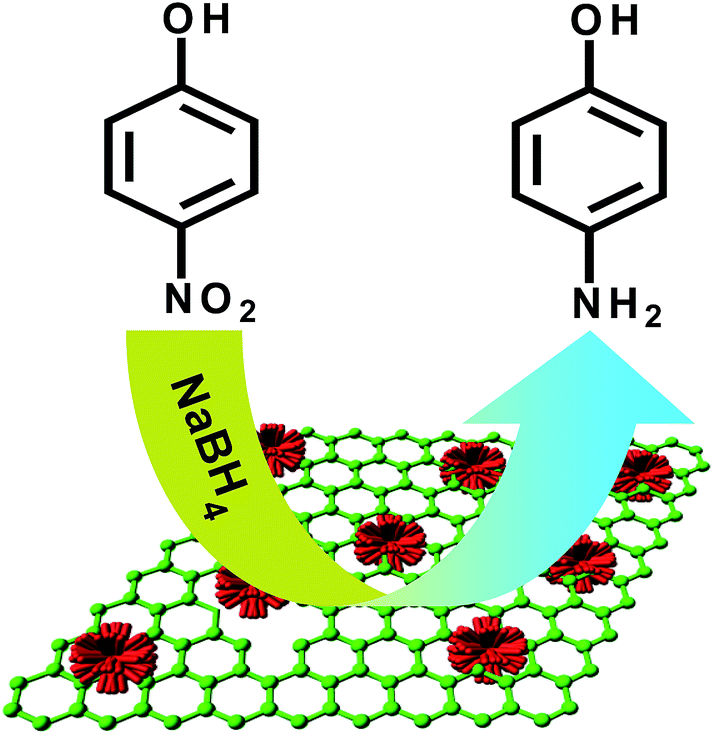 | ||
| Fig. 6 Schematic illustration for the reduction of 4-NP. | ||
The reduction process of 4-NP was monitored by UV-Vis spectroscopy at different time intervals and dosages, using Pt–Au pNDs/RGOs as a catalyst (Fig. 7A–C). It is known that this reaction has difficulty occurring without a catalyst.58 The absorption peak intensities at 400 nm were used to quantify the reduction concentration of 4-NP by extending the reaction time. After the addition of Pt–Au pNDs/RGOs to the mixed solution of 4-NP and NaBH4, the absorption peak at 400 nm significantly decreases as the reaction proceeds, owing to the reduction of 4-NP.7 Simultaneously, a new peak appears at 300 nm, accompanied with an increase of the peak intensity, which is ascribed to the formation of 4-AP.59 In addition, increasing the dosage of Pt–Au pNDs/RGOs facilitates the reduction reaction; the solution color changes from yellowish green to colorless within 10 min by using 50 μL Pt–Au pNDs/RGOs with a concentration of 0.1 mg mL−1 (Fig. 7C).
 | ||
| Fig. 7 Time-dependent UV-Vis spectra of 4-NP catalyzed by different volumes of Pt–Au pNDs/RGOs (0.1 mg mL−1): 10 μL (A), 25 μL (B), and 50 μL (C). (D) Conversion of 4-NP with reaction time in the presence of 0.005 mg Pt–Au pNDs/RGOs (curve a), 0.05 mg Pt black (curve b), and 0.05 mg Au NCs (curve c). | ||
For comparison, the catalytic performances of commercial Pt black (1 mg mL−1) and home-made Au NCs (1 mg mL−1) were investigated under the identical conditions. However, the reaction time is 1.0 h for Pt black (Fig. S3A†) and 2.5 h for home-made Au NCs (Fig. S3B†). With respect to the conversion of 4-NP (Fig. 7D), the catalytic activity is significantly improved for Pt–Au pNDs/RGOs compared to Pt black and Au NCs.
Fig. 8 shows the logarithm plot of the absorbance (−ln(A/A0)) with reaction time, using Pt black and Au NCs as references; this allows us to further examine the catalytic activity of Pt–Au pNDs/RGOs. The apparent kinetic rate constant (kapp) is proportional to the total surface area of the catalyst, which can be estimated based on the regression of the slope from the logarithm plot (−dCt/dt = kappCt).60 As listed in Table 1, the kapp value is 3.8 × 10−3 s−1 for Pt–Au pNDs/RGOs. This value is about 5-fold and 12-fold higher than those of Pt black (0.7 × 10−3 s−1) and Au NCs (0.3 × 10−3 s−1), respectively, evidencing the enhanced catalytic acitivity of Pt–Au pNDs/RGOs.
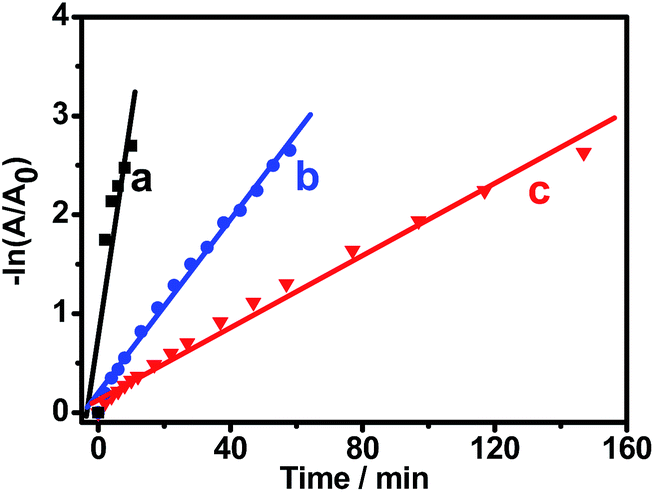 | ||
| Fig. 8 Plots of −ln(A/A0) against the reaction time for the reduction of 4-NP catalyzed by Pt–Au pNDs/RGOs (curve a), Pt black (curve b), and Au NCs (curve c). | ||
| Catalyst | k app (×10−3 s−1) | k nor (s−1 g−1) | Time (min) |
|---|---|---|---|
| Pt–Au pNDs/RGOs | 3.8 | 926 | 10 |
| Pt black | 0.7 | 15 | 58 |
| Au NCs | 0.3 | 6 | 147 |
Meanwhile, the normalized rate constant (knor) was also determined, which was associated with the amount of catalyst, i.e., knor = kapp/m. Herein, TGA analysis demonstrated that Pt and Au loading was 81.4 wt% in Pt–Au pNDs/RGOs, owing to the presence of RGOs (Fig. S4†).61 As shown in Table 1, the knor value of Pt–Au pNDs/RGOs (926 s−1 g−1) is much higher than those of Pt black (15 s−1 g−1) and Au NCs (6 s−1 g−1). At the same time, the kapp and knor values of Pt–Au pNDs/RGOs are comparable to catalysts reported in the literature (Table S1†), including Pt55Pd38Bi7 nanowires (knor = 286 s−1 g−1),62 AuPd nanocrystals (knor = 78 s−1 g−1),63 PtNi nanoparticles (knor = 483 s−1 g−1),64 and Fe@Au–ATPGO (knor = 400 s−1 g−1).65 These results strongly indicate the improved catalytic activity of Pt–Au pNDs/RGOs.
Interestingly, the dosage of Pt–Au pNDs/RGOs is 10 times less than the dosages of Pt black and Au NCs, while it needs a much shorter reaction time to achieve the same conversion quantity of 4-NP. The above results verify the enhanced catalytic performance of Pt–Au pNDs/RGOs, owing to the unique interconnected nanostructures of Pt–Au pNDs which provide more available active sites,58 the improved mass transport by using RGOs as a support,66 along with the synergistic effects between Pt and Au.67
4. Conclusions
In conclusion, we have prepared Pt–Au pNDs/RGOs through an efficient co-reduction wet-chemical route at room temperature. The co-existence of cytosine and GOs plays a crucial role in the growth of Pt–Au pNDs with high quality. The as-prepared Pt–Au pNDs/RGOs show improved catalytic activity for the reduction of 4-NP to 4-AP, compared with commercial Pt black and home-made Au NCs. It is believed that the as-prepared Pt–Au pNDs/RGOs will be used as a promising catalyst for application in organic synthesis or other fields.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21475118, 21175118, 21275130 and 21275131) and colleges in Zhejiang province through the young academic leaders of academic climbing project (pd2013055).References
- P. Deka, R. C. Deka and P. Bharali, New J. Chem., 2014, 38, 1789–1793 RSC.
- J. Li, C.-y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426–8430 RSC.
- T. R. Mandlimath and B. Gopal, J. Mol. Catal. A: Chem., 2011, 350, 9–15 CrossRef CAS PubMed.
- K.-L. Wu, X.-W. Wei, X.-M. Zhou, D.-H. Wu, X.-W. Liu, Y. Ye and Q. Wang, J. Phys. Chem. C, 2011, 115, 16268–16274 CAS.
- H. Lu, H. Yin, Y. Liu, T. Jiang and L. Yu, Catal. Commun., 2008, 10, 313–316 CrossRef CAS PubMed.
- R. Rahimi, S. Safalou Moghaddam and M. Rabbani, J. Sol-Gel Sci. Technol., 2012, 64, 17–26 CrossRef CAS PubMed.
- Y. Liu, Z. Fang, L. Kuai and B. Geng, Nanoscale, 2014, 6, 9791–9797 RSC.
- J. A. Johnson, J. J. Makis, K. A. Marvin, S. E. Rodenbusch and K. J. Stevenson, J. Phys. Chem. C, 2013, 117, 22644–22651 CAS.
- J. Zheng, Y. Dong, W. Wang, Y. Ma, J. Hu, X. Chen and X. Chen, Nanoscale, 2013, 5, 4894–4901 RSC.
- A. Gangula, R. Podila, R. M. L. Karanam, C. Janardhana and A. M. Rao, Langmuir, 2011, 27, 15268–15274 CrossRef PubMed.
- Z. Jin, M. Xiao, Z. Bao, P. Wang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 6406–6410 CrossRef CAS PubMed.
- S. Sahu, A. Samantara, A. Dash, R. R. Juluri, R. Sahu, B. K. Mishra and B. Jena, Nano Res., 2013, 6, 635–643 CrossRef CAS PubMed.
- A. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS PubMed.
- W. Si, J. Li, H. Li, S. Li, J. Yin, H. Xu, X. Guo, T. Zhang and Y. Song, Nano Res., 2013, 6, 720–725 CrossRef CAS.
- S. Guo, D. Li, H. Zhu, S. Zhang, N. M. Markovic, V. R. Stamenkovic and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3465–3468 CrossRef CAS PubMed.
- Y. Lu, Y. Jiang and W. Chen, Nano Energy, 2013, 2, 836–844 CrossRef CAS PubMed.
- F. Lu, Y. Zhang, L. Zhang, Y. Zhang, J. X. Wang, R. R. Adzic, E. A. Stach and O. Gang, J. Am. Chem. Soc., 2011, 133, 18074–18077 CrossRef CAS PubMed.
- B. Sadeghi, M. A. S. Sadjadi and R. A. R. Vahdati, Superlattices Microstruct., 2009, 46, 858–863 CrossRef CAS PubMed.
- B. Lim and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 76–85 CrossRef CAS PubMed.
- J. Xu, A. R. Wilson, A. R. Rathmell, J. Howe, M. Chi and B. J. Wiley, ACS Nano, 2011, 5, 6119–6127 CrossRef CAS PubMed.
- S. Guo, S. Dong and E. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed.
- W. He, X. Wu, J. Liu, X. Hu, K. Zhang, S. Hou, W. Zhou and S. Xie, Chem. Mater., 2010, 22, 2988–2994 CrossRef CAS.
- J.-N. Zheng, L.-L. He, F.-Y. Chen, A.-J. Wang, M.-W. Xue and J.-J. Feng, J. Mater. Chem. A, 2014, 2, 12899–12906 CAS.
- C. Koenigsmann, A. C. Santulli, K. Gong, M. B. Vukmirovic, W.-p. Zhou, E. Sutter, S. S. Wong and R. R. Adzic, J. Am. Chem. Soc., 2011, 133, 9783–9795 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- H. Zhang, M. Jin, J. Wang, W. Li, P. H. C. Camargo, M. J. Kim, D. Yang, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2011, 133, 6078–6089 CrossRef CAS PubMed.
- R. Ning, J. Tian, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, Langmuir, 2013, 29, 13146–13151 CrossRef CAS PubMed.
- J.-N. Zheng, J.-J. Lv, S.-S. Li, M.-W. Xue, A.-J. Wang and J.-J. Feng, J. Mater. Chem. A, 2014, 2, 3445–3451 CAS.
- Y. He, N. Zhang, L. Zhang, Q. Gong, M. Yi, W. Wang, H. Qiu and J. Gao, Mater. Res. Bull., 2014, 51, 397–401 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- Y.-Q. Dang, H.-W. Li, B. Wang, L. Li and Y. Wu, ACS Appl. Mater. Interfaces, 2009, 1, 1533–1538 CAS.
- L. Wang and Y. Yamauchi, Chem. Mater., 2009, 21, 3562–3569 CrossRef CAS.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS PubMed.
- W. Zhang, J. Yang and X. Lu, ACS Nano, 2012, 6, 7397–7405 CrossRef CAS PubMed.
- X. Huang, E. Zhu, Y. Chen, Y. Li, C.-Y. Chiu, Y. Xu, Z. Lin, X. Duan and Y. Huang, Adv. Mater., 2013, 25, 2974–2979 CrossRef CAS PubMed.
- Z.-C. Zhang, J.-F. Hui, Z.-G. Guo, Q.-Y. Yu, B. Xu, X. Zhang, Z.-C. Liu, C.-M. Xu, J.-S. Gao and X. Wang, Nanoscale, 2012, 4, 2633–2639 RSC.
- S. Zhang, Y. Shao, H.-g. Liao, J. Liu, I. A. Aksay, G. Yin and Y. Lin, Chem. Mater., 2011, 23, 1079–1081 CrossRef CAS.
- Y. Lu, Y. Jiang, H. Wu and W. Chen, J. Phys. Chem. C, 2013, 117, 2926–2938 CAS.
- Q.-L. Zhang, T.-Q. Xu, J. Wei, J.-R. Chen, A.-J. Wang and J.-J. Feng, Electrochim. Acta, 2013, 112, 127–132 CrossRef CAS PubMed.
- J. Li, W. Tang, J. Huang, J. Jin and J. Ma, Catal. Sci. Technol., 2013, 3, 3155–3162 CAS.
- S.-S. Li, J.-N. Zheng, X. Ma, Y.-Y. Hu, A.-J. Wang, J. Chen and J.-J. Feng, Nanoscale, 2014, 6, 5708–5713 RSC.
- M. Liu, Y. Lu and W. Chen, Adv. Funct. Mater., 2013, 23, 1289–1296 CrossRef CAS.
- J. Tian, S. Liu, Y. Zhang, H. Li, L. Wang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Inorg. Chem., 2012, 51, 4742–4746 CrossRef CAS PubMed.
- S. Liu, L. Wang, J. Tian, W. Lu, Y. Zhang, X. Wang and X. Sun, J. Nanopart. Res., 2011, 13, 4731–4737 CrossRef CAS.
- J.-J. Lv, S.-S. Li, A.-J. Wang, L.-P. Mei, J.-R. Chen and J.-J. Feng, Electrochim. Acta, 2014, 136, 521–528 CrossRef CAS PubMed.
- L. Li, M. Chen, G. Huang, N. Yang, L. Zhang, H. Wang, Y. Liu, W. Wang and J. Gao, J. Power Sources, 2014, 263, 13–21 CrossRef CAS PubMed.
- G. Fu, X. Jiang, M. Gong, Y. Chen, Y. Tang, J. Lin and T. Lu, Nanoscale, 2014, 6, 8226–8234 RSC.
- X.-R. Li, X.-L. Li, M.-C. Xu, J.-J. Xu and H.-Y. Chen, J. Mater. Chem. A, 2014, 2, 1697–1703 CAS.
- Z.-Y. Lv, A.-Q. Li, Y. Fei, Z. Li, J.-R. Chen, A.-J. Wang and J.-J. Feng, Electrochim. Acta, 2013, 109, 136–144 CrossRef CAS PubMed.
- M. Chen, B. Wu, J. Yang and N. Zheng, Adv. Mater., 2012, 24, 862–879 CrossRef CAS PubMed.
- G. Fu, K. Wu, J. Lin, Y. Tang, Y. Chen, Y. Zhou and T. Lu, J. Phys. Chem. C, 2013, 117, 9826–9834 CAS.
- Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
- H.-Z. Zhu, Y.-M. Lu, F.-J. Fan and S.-H. Yu, Nanoscale, 2013, 5, 7219–7223 RSC.
- Y. Choi, H. S. Bae, E. Seo, S. Jang, K. H. Park and B.-S. Kim, J. Mater. Chem., 2011, 21, 15431–15436 RSC.
- L. Ai, H. Yue and J. Jiang, J. Mater. Chem., 2012, 22, 23447–23453 RSC.
- W. Lu, R. Ning, X. Qin, Y. Zhang, G. Chang, S. Liu, Y. Luo and X. Sun, J. Hazard. Mater., 2011, 197, 320–326 CrossRef CAS PubMed.
- Q. Ji, J. P. Hill and K. Ariga, J. Mater. Chem. A, 2013, 1, 3600–3606 CAS.
- N. Zhang and Y.-J. Xu, Chem. Mater., 2013, 25, 1979–1988 CrossRef CAS.
- W. Ye, Y. Chen, F. Zhou, C. Wang and Y. Li, J. Mater. Chem., 2012, 22, 18327–18334 RSC.
- J.-N. Zheng, S.-S. Li, X. Ma, F.-Y. Chen, A.-J. Wang, J. Chen and J.-J. Feng, J. Mater. Chem. A, 2014, 2, 8386–8395 CAS.
- Y.-Y. Shen, Y. Sun, L.-N. Zhou, Y.-J. Li and E. S. Yeung, J. Mater. Chem. A, 2014, 2, 2977–2984 CAS.
- J. Zhang, C. Hou, H. Huang, L. Zhang, Z. Jiang, G. Chen, Y. Jia, Q. Kuang, Z. Xie and L. Zheng, Small, 2013, 9, 538–544 CrossRef CAS PubMed.
- S. K. Ghosh, M. Mandal, S. Kundu, S. Nath and T. Pal, Appl. Catal., A, 2004, 268, 61–66 CrossRef CAS PubMed.
- V. K. Gupta, N. Atar, M. L. Yola, Z. Üstündağ and L. Uzun, Water Res., 2014, 48, 210–217 CrossRef CAS PubMed.
- J.-J. Lv, J.-N. Zheng, L.-L. Chen, M. Lin, A.-J. Wang, J.-R. Chen and J.-J. Feng, Electrochim. Acta, 2014, 143, 36–43 CrossRef CAS PubMed.
- Y. Yamauchi, A. Tonegawa, M. Komatsu, H. Wang, L. Wang, Y. Nemoto, N. Suzuki and K. Kuroda, J. Am. Chem. Soc., 2012, 134, 5100–5109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta05034g |
| This journal is © The Royal Society of Chemistry 2015 |