
Polymer aggregation control in polymer–fullerene bulk heterojunctions adapted from solution
Christian
Kästner
a,
Daniel A. M.
Egbe
b and
Harald
Hoppe
*a
aInstitute of Physics, Ilmenau University of Technology, Langewiesener Str. 22, 98693 Ilmenau, Germany. E-mail: harald.hoppe@tu-ilmenau.de
bLinz Institute for Organic Solar Cells, Johannes Kepler University Linz, Altenbergerstr. 69, 4040 Linz, Austria
First published on 18th November 2014
Abstract
It is common knowledge that the polymer conformation and its phase separation with fullerene derivatives are delicate issues crucially impacting on the photovoltaic parameters of polymer based solar cells. While strongly intermixed polymer–fullerene phases presumably provide a large interfacial area and consequently a high quantum efficiency of exciton dissociation, pristine and primarily ordered polymer and fullerene domains may support efficient charge transport and its percolation. To study the aggregation and phase separation behaviour in polymer solar cells we investigated the counterbalancing influences of the polymer solution concentration and its blending ratio with PCBM ([6,6]-phenyl-C61-butyric acid methyl ester) on the basis of a semi-crystalline anthracene-containing poly(p-phenylene-ethynylene)-alt-poly(p-phenylene-vinylene) (PPE-PPV) copolymer statistically bearing branched 2-ethylhexyloxy and linear octyloxy side-chains (AnE-PVstat). The polymer aggregation was semi-quantitatively evaluated on the basis of its optical fingerprints and varied with both, the solution and the PCBM concentration, yielding a specific maximum within the parameter range studied. Upon relating photovoltaic parameters to the order within the polymer phase, the counterbalancing effect between charge generation and transport for increasing polymer aggregation is demonstrated, in agreement with sound hypotheses.
Introduction
The continuously increasing research interest in polymer-based organic photovoltaics (polymer solar cells) over the last two decades1–4 has resulted in improved fundamental and technological knowledge concerning polymer solar cells as a flexible5–7 and semi-transparent8–10 option for harvesting solar radiation at potentially low cost.11–13 But there are still big efforts to be undertaken in order to match the requirements of future power installations similar to conventional, inorganic photovoltaics. In general highly efficient polymer solar cells are based on the bulk heterojunction (BHJ)14,15 concept – in which the intimate intermixing of electron donating polymers and electron acceptors provides an efficient ultra-fast charge transfer within this blend.16–18 Among the most suited acceptors for efficient bulk heterojunction solar cells are fullerene derivatives, most commonly PCBM.18,19Since charge separation of excitons takes place at the interface between the polymer and PCBM, an intimate mixture of both materials is required for successful splitting of the photogenerated excitons due to the large interfacial area, yielding high charge generation rates.16,17,20 In contrast, recombination rates are also increased as charge percolation is limited within homogeneously intermixed phases yielding losses in photocurrent.21 Hence a pristine polymer phase may reduce charge recombination, while the hole mobility is additionally controlled by the order within the polymer phase21–24 – π–π-stacking on the short-range and crystallinity on the long-range order25–28 as well as by phase purity.29 The electron transport capability is comparably high within the PCBM phase, already benefitting from a higher order easily obtained in aggregates of spherical fullerene derivatives.30,31 Furthermore, fullerene aggregation/crystallization promotes charge separation within bulk heterojunctions due to the multitude of energy levels present for charge transfer.20,32,33 Thus, large phase separation between the polymer and PCBM improves the charge extraction from the bulk, but leads to a loss of interfacial area and thus potentially photocurrent. Contrarily, a strong intermixing leads to a large interfacial area and thus charge generation, but a loss in charge percolation pathways and thus to increased charge recombination. Furthermore, excitons generated within the pristine bulk material have to reach the interface between the polymer and PCBM for dissociation. But, the limited exciton life-time and consequently its diffusion length of approximately 10–20 nm require pristine domains of limited size, which still allow excitons to reach the interface.34,35 Conclusively, charge generation, recombination and percolation counterbalance each other and they are controlled by the morphology of the bulk heterojunction.36–39 Thus, a fine-tuned blend morphology is required to maximize charge generation and minimize charge recombination due to improved charge transport and extraction.
Several approaches were pursued to control the nanomorphology of organic bulk heterojunctions. For example, the so-called micro-phase separation between the donor and acceptor could be obtained by utilization of block copolymers consisting of alternating well-aligned donor and acceptor blocks fulfilling the requirements of suitable phase separation.40,41 In the case of binary donor–acceptor systems, like the well-known semi-crystalline poly(3-hexylthiophene) (P3HT), post-production treatments such as thermal annealing42,43 or slow drying44 improved the solar cell performance by enhanced structural ordering, and thus the controlled crystallization of P3HT.45 Several approaches for controlling the P3HT aggregation already within solutions were pursued in order to yield an improved morphology control for the evolving bulk heterojunction.46–50 The formation of semi-crystalline P3HT fibers was for example achieved by aggregation induced by the use of non-solvents such as additives within the P3HT-solution.49,51 Amorphous polymer based systems could be improved by increased phase separation between PCBM and an intercalated polymer–PCBM mix-phase52 utilizing solvent blends or additives.53–55 However, those systems still lack performance due to the amorphous nature of the polymer and limited hole percolation.22,56–58
In this study we present an approach to precisely control the structural order of the polymer as well as phase separation between the polymer and PCBM. The herein used copolymer AnE-PVstat is semi-crystalline as obtained from wide-angle X-ray scattering experiments.25 Previous studies revealed the degree of aggregation of semi-crystalline AnE-PV to be improved by the presence of PCBM.59,60 Furthermore, the degree of phase separation between AnE-PV copolymers and PCBM can be controlled by the solvent composition of chloroform and chlorobenzene and the PCBM concentration within the common solution. An optimized phase separation of AnE-PV copolymers with PCBM was obtained for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of chloroform and chlorobenzene based solutions.61 Based on its ability to aggregate and phase separate with PCBM, AnE-PVstat constitutes a perfect candidate to study the influence of structural order and phase separation on the solar cell device operation. In the following, the blend morphology was precisely controlled by solution concentration and PCBM weight fraction. The quantification of polymeric structural order in this material system is achieved by the introduction of a parameter of combined structural order, derived from the relative changes in the inter-chain and intra-chain order of polymer aggregates, probed with optical steady-state spectroscopy.
:1 ratio of chloroform and chlorobenzene based solutions.61 Based on its ability to aggregate and phase separate with PCBM, AnE-PVstat constitutes a perfect candidate to study the influence of structural order and phase separation on the solar cell device operation. In the following, the blend morphology was precisely controlled by solution concentration and PCBM weight fraction. The quantification of polymeric structural order in this material system is achieved by the introduction of a parameter of combined structural order, derived from the relative changes in the inter-chain and intra-chain order of polymer aggregates, probed with optical steady-state spectroscopy.
Theoretical background
The analytical model used in our studies for quantifying the polymeric order is based on the assumptions for H- and J-aggregation of polymers as proposed by Spano et al., whereas absorption spectroscopy generally reveals information about the inter-chain order and photoluminescence spectroscopy about the intra-chain order, compare with Fig. 1.62–64 The optical absorption and emission of J-aggregates, described by the HJ-aggregate model, is explicitly linked with the polymeric order: the ratio of the 0–0 to 0–1 transition of the absorption spectrum, A0–0/A0–1, increases with increasing order as well as the ratio of the polymer photoluminescence emission E0–0/E0–1 at room temperature.63 Apart from that, it is also well known that the near-field order, generated by the π–π-stacking of the polymer backbones, is parental for the origination of the 0–0 peak transition in the polymer absorption spectra.65,66 Furthermore, the absorption spectra also contain information about the degree of conjugation.67,68 J-aggregation has extensive consequences on the electrical properties: the intra-chain coupling is much stronger than the inter-chain coupling and leads therefore to large free charge carrier mobilities,69 but comparably low exciton mobilities along the polymer backbone.70 | ||
| Fig. 1 Correlation between emission/absorption transitions, E0–0/E0–1 and A0–0/A0–1, and intra-chain/inter-chain coupling for J-aggregated AnE-PV. | ||
To globally define the polymer order within the system, we linked the inter-chain order, represented by the π–π-stacking of polymer backbones, to the intra-chain order, represented by torsion-free planarization of polymer backbones. Hence the product of the A0–0/A0–1 polymer absorption peak ratio and the E0–0/E0–1 polymer emission peak ratio can be defined as the parameter of combined structural order, PCSO, valid for J-aggregates and under the assumption that inter- and intra-chain order must be strongly correlated. As within aggregates both conditions, intra-chain and inter-chain order, have to be fulfilled, the combined structural order parameter is introduced as a product of both individual order parameters. An arithmetic average, i.e. the weighted sum of both parameters, constitutes a weaker and compromising condition and is therefore discarded.
Next to the intra-chain order of polymer backbones, photoluminescence spectroscopy allows semi-quantitative insights into the degree of phase separation between the polymer and PCBM.71 Usually, the degree of polymer aggregation is accompanied by the degree of phase separation between the polymer and PCBM due to the fact that the undisturbed pristine polymer phase is more prone to reorganize within an ordered structure by free energy minimization.72 We have already shown that semi-crystalline AnE-PV tends to phase separate strongly from PCBM.60 An earlier comparison between the domain size and photoluminescence yield of thin AnE-PV:PCBM blend films yielded good agreement with the X-ray diffraction results.73 Whilst strongly phase separated systems showed remaining photoluminescence from both materials, strong intermixing led to substantial quenching of the polymer photoluminescence signal, and to the occurrence of interfacial charge transfer photoluminescence (CT-PL) signals.60,74 Thus photoluminescence provides potentially a lot of information about the scale of phase separation within bulk heterojunction blends.
Experimental
Scheme 1 displays the chemical structures of AnE-PVstat and PCBM, which were used within this study. AnE-PVstat was synthesized as reported earlier.59 As an electron acceptor, PCBM was used as obtained from the supplier (Nano-C, USA). | ||
| Scheme 1 Molecular structure of AnE-PVstat (C8H17 = octyl and/or 2-ethylhexyl) and PCBM. | ||
Thin films of AnE-PVstat:PCBM blends were spin cast onto glass substrates using 1:1 mixtures of chloroform–chlorobenzene based solutions under a nitrogen atmosphere. Thin film absorption spectra were recorded with a Varian Cary 5000 UV/Vis spectrophotometer under 2-beam VW-setup conditions to determine the sample absorption via transmission and reflection measurements. Thin film photoluminescence (PL) spectra were recorded with an Avantes avaspec 2048 fiber spectrometer within a range from 500 to 1100 nm and were normalized to the film absorption at the laser excitation wavelength of 445 nm. All optical characterizations were executed at room temperature.
Solar cell device preparation on glass involved partly etching the ITO-layer for selectively contacting the back electrode, followed by spin coating of PEDOT:PSS (Clevios PH, Heraeus). The PEDOT:PSS layers were annealed at 170 °C for 15 minutes to release water moieties and were afterwards transferred to a nitrogen (N2) glovebox for further processing of the photoactive layers. The photoactive layers were spin cast from AnE-PVstat:PCBM solutions with blend ratios varying from 1:1 to 1:2 by weight (polymer–fullerene) and a solution concentration varying from 0.4 to 0.6 wt% of the polymer part. Spin frequencies were varied from 500 to 1600 rpm to evaluate the optimum layer thickness for every blend. The top aluminum electrode was deposited by physical vapor deposition. Current–voltage (I–V) measurements of solar cell devices exhibiting an active area of 0.5 cm2 were recorded with a Keithley 2400 Source-Measure-Unit using a class A solar simulator. The external quantum efficiency spectra were recorded using bias illumination to resemble current densities typical under one sun illumination. Neither thin films prepared for the optical investigations nor those prepared for solar cell devices were annealed.
Results and discussion
To elucidate the influence of solution concentration and AnE-PVstat:PCBM blend ratio on the π–π-stacking of AnE-PVstat, absorption spectra of thin films were recorded. Fig. 2 depicts the thin film absorption spectra of AnE-PVstat blended with different amounts of PCBM at various solution concentrations. Spectra were normalized to the 0–1 transition peak height to highlight the relative change with respect to the 0–0 transition. Fig. 2a, b and c show the absorption of the AnE-PVstat:PCBM blend films over the full measurement range and Fig. 2d depict the zoom-in spectrum, spanning over the absorption edge with the 0–0 and 0–1 transition to highlight the degree of π–π-stacking induced order.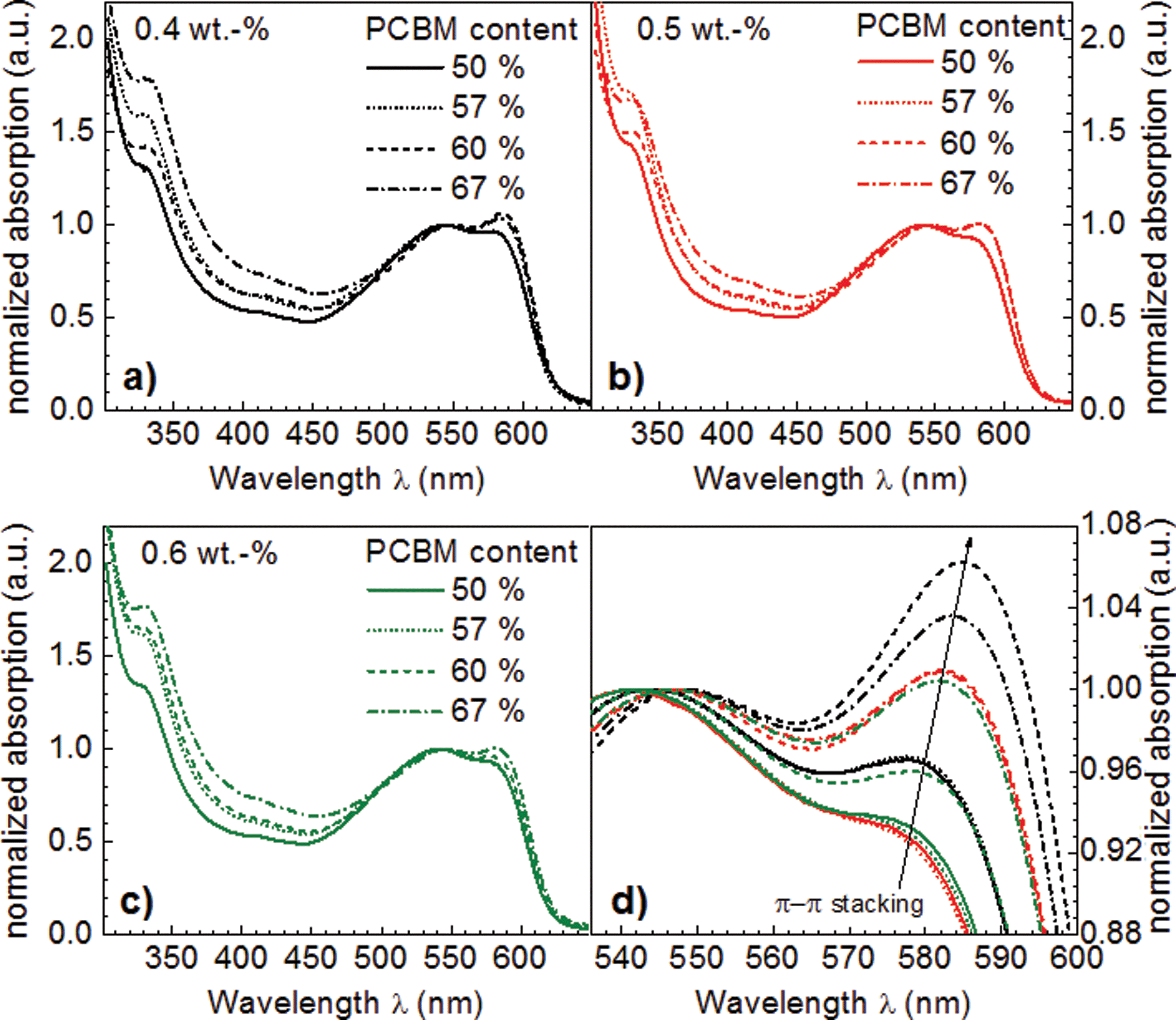 | ||
| Fig. 2 Thin film absorption spectra of AnE-PVstat:PCBM films, normalized to the 0–1 transition at around 545 nm to highlight the evolution of the 0–0 transition at around 585 nm, as a function of the AnE-PVstat solution concentration and the PCBM weight fraction. The full spectra are shown in (a), (b) and (c); the zoom-in spectra, highlighting the occurrence of the AnE-PVstat π–π-stacking, are shown in (d). | ||
The polymer–fullerene blend ratio and the polymer solution concentration imposed a strong impact on the aggregation behavior of AnE-PVstat which was related to the red-shift of the polymer absorption accompanied by the typical 0–0 transition at the absorption edge of the polymer. The reduced probability for the entanglement of polymers within diluted solutions seems to promote the formation of aggregates. Hence the larger degree of freedom enables aggregation, whereas higher solution concentrations may hinder the aggregate formation due to entanglement. Generally, the peak height of the 0–0 transition and thus the order increased with an increasing amount of PCBM at a certain solution concentration as well as with decreasing solution concentration at a certain AnE-PVstat:PCBM blend ratio. The induced polymer aggregation upon addition of PCBM was already discovered previously for the semi-crystalline analogue AnE-PVab.59,60 It has been demonstrated that upon blending with PCBM the polymer aggregation generally increased. However, maximum polymer aggregation was not found for the largest PCBM concentration at the lowest solution concentration, but instead for a AnE-PVstat:PCBM blending ratio of 2:3 for 0.4 wt% solution concentration of AnE-PVstat. This can be understood as with further increasing the PCBM content the diffusion rate of PCBM into the polymer domains grows by the progressive concentration gradient. In contrast to PBTTT, the PCBM does not form interdigitated nano-crystallites with AnE-PVstat.75–77 Thus higher volume fractions and thus concentrations of incorporated PCBM molecules tended to distort the polymeric order, which resulted in a slight reduction of the 0–0 transition oscillator strength. In conclusion, both parameters, PCBM volume fraction and polymer solution concentration, may counterbalance the degree of polymer aggregation and presumably the pristine domain sizes.
For a semi-quantitative analysis, the ratio between the 0–0 peak and 0–1 peak heights was taken as a measure for the degree of polymer aggregation.62–64 To visualize the degree of polymer aggregation – appointed to the π–π-stacking of AnE-PVstat – the normalized peak height ratios are plotted as a function of the processing parameters AnE-PVstat concentration and AnE-PVstat:PCBM blend ratio in Fig. 5a. It should be noted that even lower solution concentrations might lead to stronger polymer aggregation, as the observed maximum of the polymer aggregation is located at the edge of the investigated range. However, too low solution concentrations led to processing difficulties i.e. unacceptable film inhomogeneities, so that the required active layer thicknesses for solar cell application were not obtained anymore. Thus, lower solution concentrations than 0.4 wt% were irrelevant and not further considered.
To elucidate the influence of solution concentration and AnE-PVstat:PCBM blend ratio on the backbone planarization of AnE-PVstat, photoluminescence spectra of the identical thin films were recorded. Fig. 3 depicts the obtained thin film photoluminescence spectra of AnE-PVstat blended with different amounts of PCBM at various solution concentrations. Fig. 3a–c show the fully recorded wavelength range photoluminescence spectra of the AnE-PVstat:PCBM blend thin films whilst Fig. 3d shows the photoluminescence normalized to the 0–0 emission of the polymer within a zoom-in range, highlighting the typical photoluminescence contributions of AnE-PVstat and PCBM.
 | ||
| Fig. 3 Thin film photoluminescence spectra of AnE-PVstat:PCBM films normalized to the thin film absorption at 445 nm laser excitation wavelength as a function of the AnE-PVstat solution concentration and the PCBM weight fraction. Full range PL-spectra are shown in (a), (b) and (c); the 0–0 transition normalized zoom-in spectra of all samples, enabling a comparison concerning aggregation of AnE-PVstat and domain size evolution of PCBM, are shown in (d). The evolution of the relative PCBM PL peak strengths (for highlighting, spectra are normalized to the PL at 690 nm) are depicted in (e). | ||
The overall polymer photoluminescence signal strength (Fig. 4a) is directly correlated with the volume of pristine polymer phases in the film, in which the excitons are not able to reach an interface with PCBM during their lifetime. As the PL-signal intensity varied much more strongly over the entire processing parameter range than the increase of the volume fraction of ordered polymer phases, which is estimated by the relative increase in inter-chain order (compare with Fig. 4a), it is indicated that the domain size is varied 3-dimensionally. For PCBM a similarly large variation in PL-signal intensity was found and larger PCBM domains are conclusively found for the largest PCBM concentrations (i.e. 1:2 AnE-PVstat:PCBM blend ratios).78
 | ||
| Fig. 4 Interpolated contour plots of the photoluminescence intensity of (a) polymer and (b) PCBM as a function of the processing parameters. | ||
Overall the maximum polymer PL-signal – involving the largest polymer domain size – is observed for 0.4 wt% AnE-PVstat solution concentration and a 2:3 blending ratio of AnE-PVstat:PCBM. This is in accordance with the observations from the absorption measurements – the maximum polymer aggregation was found for the same concentration and blend ratio.
Fig. 5 summarizes the normalized parameters for inter- and intra-chain order. The strongest influence on both order parameters is imposed by the PCBM concentration, whereas the polymer solution concentration showed a stronger influence on the inter-chain as compared to the intra-chain order. For further considerations and as a compromise, the two order parameters were unified into a single combined structural order parameter, PCSO, as defined above (Fig. 5c).
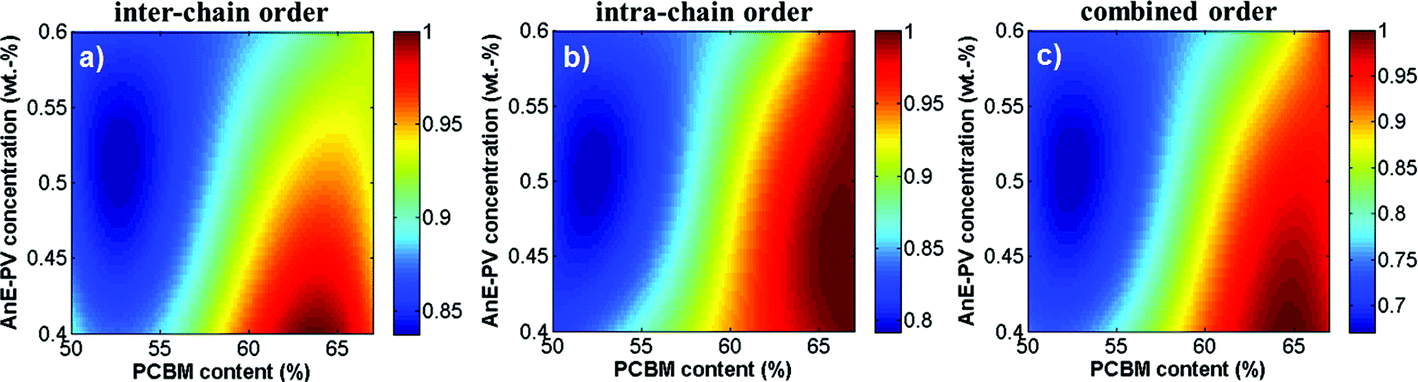 | ||
| Fig. 5 Interpolated contour plots of (a) A0–0/A0–1 ratio of the polymer transition peak heights from absorption as a measure for the inter-chain order (compare with Fig. 2), and for (b) E0–0/E0–1 polymer emission peak ratio obtained from photoluminescence as a measure of the intra-chain order (compare with Fig. 3d) and (c) of the product of both order parameters as a measure for combined structural order (PCSO) as a function of the AnE-PVstat solution concentration and the PCBM weight fraction. | ||
In summary, the optical characterization revealed that AnE-PVstat forms J-aggregates, since for absorption and photoluminescence the 0–0 peak could raise above the 0–1 peak. Coupling the intra-chain order and inter-chain order to a common parameter of combined structural order allowed to quantify the general degree of order in polymer aggregates. Finally, both the polymer order and the blend phase separation were controlled by the solution concentration and dominantly by the PCBM content.
To gain additional insight into the blend film morphology atomic force microscopy (AFM) measurements were carried out in tapping mode. The obtained topography images of 2.5 μm × 2.5 μm scans of the blend films are shown in Fig. 6. At lower PCBM contents a fine-scale structure is observed. With increasing PCBM content larger domains evolve, presumably originating from increasing PCBM inclusions.71 On top of these fullerene aggregates polymer aggregates of AnE-PVstat are clearly visible. In good agreement with the photoluminescence data, the PCBM aggregates increased with increasing PCBM concentration and lowered polymer solution concentration, which is also reflected in the surface roughness contour plot in Fig. 7. Thus the pronounced self-aggregation of AnE-PVstat in more diluted solutions seems to support the evolution of larger PCBM domains.
 | ||
| Fig. 6 Tapping mode 2.5 μm × 2.5 μm topography images (20 nm height scale) of AnE-PVstat:PCBM blend thin films as a function of AnE-PVstat solution concentration and PCBM content. | ||
 | ||
| Fig. 7 Normalized mean-square surface roughness, taken from AFM images, as a function of PCBM content and AnE-PV solution concentration. For visualization, the data points were entered into a matrix that was zero-filled and linearly interpolated between the experimental data points. | ||
To gain insight into the influence of polymer aggregation and phase separation between AnE-PVstat and PCBM on the opto-electronic properties of bulk heterojunctions, solar cells were fabricated, spanning over the same range of processing parameters, and characterized. The current density–voltage (J–V) characteristics and EQE spectra of all photovoltaic devices are shown in Fig. 8. Indeed, the EQE spectra confirmed the short-circuit current densities obtained from the J–V characteristics. Table 1 summarizes the photovoltaic parameters of these solar cells with optimized film thicknesses.
 | ||
| Fig. 8 (a) Current density–voltage characteristics under one sun illumination intensity of film thickness optimized solar cells for different polymer solution concentrations and PCBM weight fractions and (b) the corresponding EQE spectra recorded under one sun bias light illumination. | ||
| Concentration (wt%) | PCBM content (%) | J SC (mA cm−2) | V OC (mV) | FF (%) | PCE (%) | R S (Ω) | R P (kΩ) |
|---|---|---|---|---|---|---|---|
| 0.4 | 50 | 7.57 | 853 | 63.1 | 4.07 | 13.1 | 2.1 |
| 57 | 7.52 | 843 | 68.2 | 4.33 | 10.1 | 3.0 | |
| 60 | 7.03 | 848 | 69.8 | 4.16 | 9.5 | 4.8 | |
| 67 | 7.05 | 834 | 68.8 | 4.04 | 9.4 | 2.5 | |
| 0.5 | 50 | 7.62 | 858 | 59.7 | 3.91 | 14.2 | 2.3 |
| 57 | 7.48 | 842 | 62.9 | 3.96 | 11.5 | 2.1 | |
| 60 | 7.38 | 838 | 66.0 | 4.08 | 12.0 | 3.2 | |
| 67 | 7.18 | 821 | 67.0 | 3.95 | 10.4 | 3.7 | |
| 0.6 | 50 | 7.17 | 861 | 56.9 | 3.52 | 14.9 | 2.1 |
| 57 | 7.27 | 825 | 60.7 | 3.64 | 14.7 | 2.3 | |
| 60 | 7.61 | 835 | 64.4 | 4.09 | 11.7 | 5.8 | |
| 67 | 7.36 | 820 | 66.6 | 4.02 | 10.3 | 2.4 |
The first general observation is that the variation in all photovoltaic parameters remained relatively small and power conversion efficiencies varied around 4%, typical for AnE-PVstat.79 The first conclusion may therefore be, that upon small perturbations of the beforehand optimized donor–acceptor system only gradual changes within the bulk heterojunction blend morphology occurred. In order to visualize the variation in PV-parameters with respect to the processing parameters, the data were interpolated and plotted as a function of AnE-PVstat solution concentration and AnE-PVstat:PCBM blending ratio (Fig. 9). At the first glance it is obvious that the dependence of many PV-parameters is stronger on the PCBM concentration, and thus the blend ratio, than on the polymer solution concentration. This is especially valid for the short-circuit current density (JSC) and the parallel resistance (RP) and less strong for the open-circuit voltage (VOC). Overall the dependence of fill factor (FF) on the processing parameters is the most reminiscing of the development of the polymer order as displayed in Fig. 3 and 5 above.
 | ||
| Fig. 9 Contour plots of the determined mean-values of all PV-parameters of AnE-PVstat:PCBM based BHJ solar cells as a function of the AnE-PVstat solution concentration and the PCBM weight fraction: (a) short-circuit current density JSC (mA cm−2), (b) open-circuit voltage VOC (mV), (c) fill factor FF (%), (d) power conversion efficiency PCE (%), (e) series resistance RS (Ω) and (f) parallel resistance RP (Ω). | ||
For better comparison of the photovoltaic parameters with the underlying polymeric order, the data were replotted with respect to just one parameter of combined structural order (PCSO). The resulting plots are depicted in Fig. 10. The graphs show the mean-values with standard deviations as obtained from all solar cells (black squares with error bars) and the corresponding linear fits (red lines) with respect to the PCSO. Indeed the fill factor is most strongly and positively correlated with the order of the polymer. Vitarisi et al. found the same result for small molecule based organic solar cells: the fill factor increased with the degree of phase separation accompanying pristine phase order, indicating reduced recombination losses.80 Similarly the bulk resistance, expressed by the series resistance (RS), shows a strong anti-correlation with the increasing order of the polymer. Both parameters thus show that an improved polymeric order yields improved charge extraction properties, which are generally provided by an increased mobility-lifetime product. On the other hand, the short-circuit photocurrent JSC exhibits an anti-correlation with the polymeric order, which is in agreement with polymer domain coarsening yielding a reduction in the interfacial area between the polymer and fullerene derivative. This decrease in photocurrent is furthermore in good agreement with the observed increase in polymer photoluminescence, as depicted in Fig. 3 and 4. The open-circuit voltage is slightly anti-correlated with the combined structural order parameter, which is in good agreement with the lowering of the polymer band-gap due to ordering.20,32,33 Independent of the device film thickness, the highest values of the open-circuit voltage were found for less aggregated, more disordered polymer phases. On the other hand, the less ordered regions of both materials, polymer and PCBM, yield a lower effective HOMO of the polymer81,82 and a higher effective LUMO of PCBM,33 altogether yielding larger open-circuit voltages.
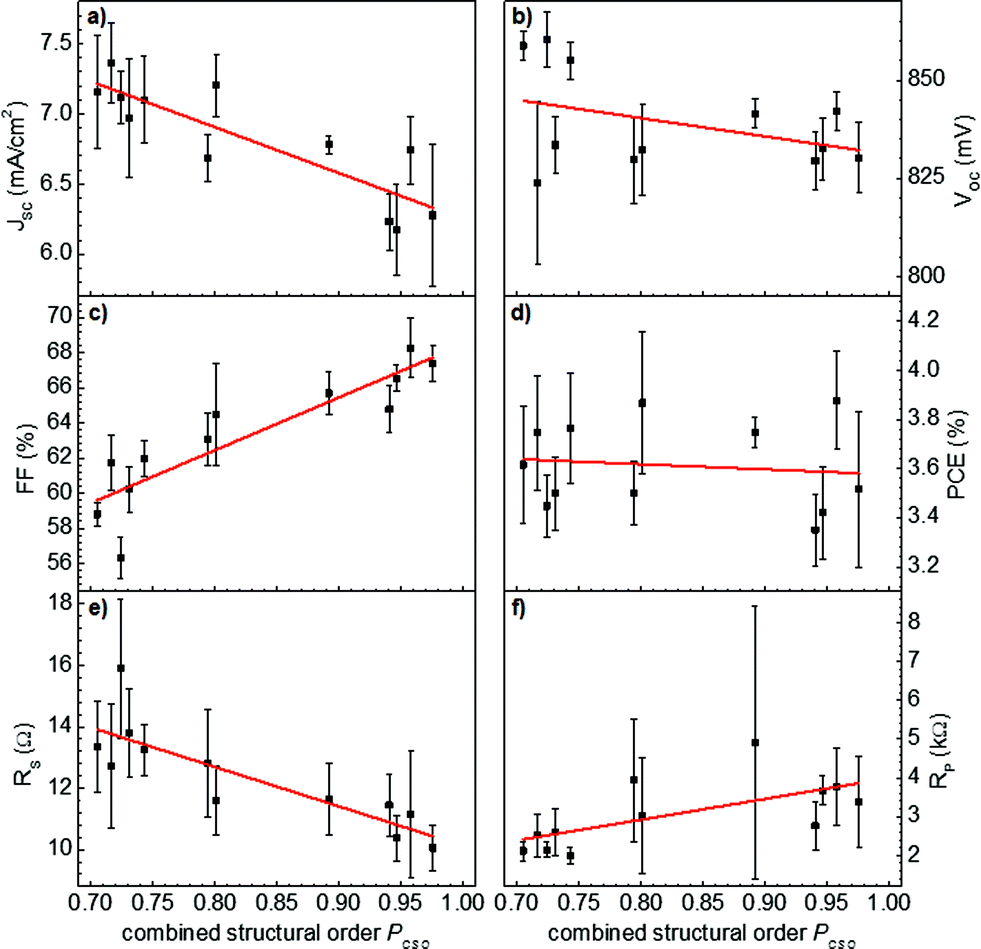 | ||
| Fig. 10 All photovoltaic parameters ((a) JSC, (b) VOC, (c) FF, (d) PCE, (e) RS, and (f) RP) replotted as a function of the combined structural order parameter PCSO. The black squares correspond to the mean-value of all solar cells investigated, whereas the standard deviation is depicted as range. The red line is a linear fit to the statistical data with respect to the PCSO. | ||
Conclusion
The aggregation of the semi-crystalline polymer AnE-PVstat was controlled in polymer–fullerene bulk heterojunction blends with PCBM by variation of the processing parameters – polymer solution concentration and PCBM content. The optical analysis via absorption and photoluminescence spectroscopy revealed that AnE-PVstat forms J-aggregates. Whereas the 0–0 to 0–1 peak ratio in absorption indicated the extent of inter-chain order, and thus π–π-stacking, the 0–0 to 0–1 peak ratio of the photoluminescence provided information about the intra-chain order, and thus the planarity of the polymer. In our semi-quantitative approach we normalized these peak ratios to the obtained maximum and unified both into a single combined structural order parameter PCSO. By analyzing and relating all photovoltaic parameters to the combined structural order, we find convincing evidence, that polymer aggregation- supports charge extraction – as confirmed by the increased fill factor and reduced series resistance,
- reduces photocurrent generation – presumably due to reduction in the interfacial area, and
- slightly reduces photovoltage – pointing out that aggregation yields energetic relaxation.
Overall the variation in polymer aggregation did not have any remarkable impact on the power conversion efficiency, as the above mentioned effect was balanced out, which indicates the present material system to be optimized at its maximum performance within the parameter range studied. In conclusion, with the model material system applied in this study, a number of hypotheses concerning the effect of subtle morphological changes in terms of phase separation and domain ordering could be verified. Future studies will be focused on precisely quantifying the extent of phase separation, the volume fraction of ordered polymer phases and the exciton and charge carrier dynamics.
Acknowledgements
The authors are grateful for financial support from DFG within the framework of SPP 1355. C.K. is grateful to the Thüringer Landesgraduiertenschule für Photovoltaik (PhotoGrad) for financial support.References
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- P. Kumar and S. Chand, Prog. Photovoltaics, 2012, 20, 377–415 CAS.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- Y. Wang, J. Sol. Energy Eng., 2012, 134, 011017–011025 CrossRef.
- T. T. Larsen-Olsen, F. Machui, B. Lechene, S. Berny, D. Angmo, R. Soslashndergaard, N. Blouin, W. Mitchell, S. Tierney, T. Cull, P. Tiwana, F. Meyer, M. Carrasco-Orozco, A. Scheel, W. Loumlvenich, R. de Bettignies, C. J. Brabec and F. C. Krebs, Adv. Energy Mater., 2012, 2, 1091–1094 CrossRef CAS.
- F. C. Krebs, S. A. Gevorgyan and J. Alstrup, J. Mater. Chem., 2009, 19, 5442–5451 RSC.
- F. C. Krebs, M. Jorgensen, K. Norrman, O. Hagemann, J. Alstrup, T. D. Nielsen, J. Fyenbo, K. Larsen and J. Kristensen, Sol. Energy Mater. Sol. Cells, 2009, 93, 422–441 CrossRef CAS PubMed.
- G. M. Ng, E. L. Kietzke, T. Kietzke, L. W. Tan, P. K. Liew and F. R. Zhu, Appl. Phys. Lett., 2007, 90, 103505-1–103505-3 Search PubMed.
- T. Ameri, G. Dennler, C. Waldauf, H. Azimi, A. Seemann, K. Forberich, J. Hauch, M. Scharber, K. Hingerl and C. J. Brabec, Adv. Funct. Mater., 2010, 20, 1592–1598 CrossRef CAS.
- D. Han, H. Kim, S. Lee, M. Seo and S. Yoo, Opt. Express, 2010, 18, A513–A521 CrossRef CAS PubMed.
- S. E. Shaheen, D. S. Ginley and G. E. Jabbour, MRS Bull., 2005, 30, 10–19 CrossRef CAS.
- E. Ahlswede, W. Muhleisen, M. W. B. M. Wahi, J. Hanisch and M. Powalla, Appl. Phys. Lett., 2008, 92, 143307–143309 CrossRef PubMed.
- S.-I. Na, B.-K. Yu, S.-S. Kim, D. Vak, T.-S. Kim, J.-S. Yeo and D.-Y. Kim, Sol. Energy Mater. Sol. Cells, 2010, 94, 1333–1337 CrossRef CAS PubMed.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Synth. Met., 1993, 59, 333–352 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CAS.
- B. Kraabel, C. H. Lee, D. McBranch, D. Moses, N. S. Sariciftci and A. J. Heeger, Chem. Phys. Lett., 1993, 213, 389–394 CrossRef CAS.
- R. A. J. Janssen, J. C. Hummelen, K. Lee, K. Pakbaz, N. S. Sariciftci, A. J. Heeger and F. Wudl, J. Chem. Phys., 1995, 103, 788–793 CrossRef CAS PubMed.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- B. M. Savoie, A. Rao, A. A. Bakulin, S. Gelinas, B. Movaghar, R. H. Friend, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 2014, 136, 2876–2884 CrossRef CAS PubMed.
- A. Pivrikas, N. S. Sariciftci, G. Juška and R. Österbacka, Prog. Photovoltaics, 2007, 15, 677–696 CAS.
- M. T. Dang, L. Hirsch, G. Wantz and J. D. Wuest, Chem. Rev., 2013, 3734–3765 CrossRef CAS PubMed.
- C. R. Singh, G. Gupta, R. Lohwasser, S. Engmann, J. Balko, M. Thelakkat, T. Thurn-Albrecht and H. Hoppe, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 943–951 CrossRef CAS.
- D. T. Duong, V. Ho, Z. Shang, S. Mollinger, S. C. B. Mannsfeld, J. Dacuña, M. F. Toney, R. Segalman and A. Salleo, Adv. Funct. Mater., 2014, 4515–4521 CrossRef CAS.
- S. Rathgeber, D. B. de Toledo, E. Birckner, H. Hoppe and D. A. M. Egbe, Macromolecules, 2010, 43, 306–315 CrossRef CAS.
- S. Rathgeber, J. Perlich, F. Kuhnlenz, S. Turk, D. A. M. Egbe, H. Hoppe and R. Gehrke, Polymer, 2011, 52, 3819–3826 CrossRef CAS PubMed.
- E. J. W. Crossland, K. Tremel, F. Fischer, K. Rahimi, G. Reiter, U. Steiner and S. Ludwigs, Adv. Mater., 2012, 24, 839–844 CrossRef CAS PubMed.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nature Mater., 2013, 12, 1037–1043 CrossRef PubMed.
- C. Kästner, X. Jiao, D. A. M. Egbe, H. W. Ade and H. Hoppe, Proc. SPIE, 2014, 9184, 91840Z-1–91840Z-6 CrossRef PubMed , Organic Photovoltaics XV.
- C. Waldauf, P. Schilinsky, M. Perisutti, J. Hauch and C. J. Brabec, Adv. Mater., 2003, 15, 2084–2088 CrossRef CAS.
- T. B. Singh, N. Marjanović, G. J. Matt, S. Günes, N. S. Sariciftci, A. Montaigne Ramil, A. Andreev, H. Sitter, R. Schwödiauer and S. Bauer, Org. Electron., 2005, 6, 105–110 CrossRef CAS PubMed.
- S. Gelinas, A. Rao, A. Kumar, S. L. Smith, A. W. Chin, J. Clark, T. S. van der Poll, G. C. Bazan and R. H. Friend, Science, 2014, 343, 512–516 CrossRef CAS PubMed.
- F. C. Jamieson, E. B. Domingo, T. McCarthy-Ward, M. Heeney, N. Stingelin and J. R. Durrant, Chem. Sci., 2012, 3, 485–492 RSC.
- S. M. Menke and R. J. Holmes, Energy Environ. Sci., 2014, 7, 499–512 CAS.
- I. W. Hwang, D. Moses and A. J. Heeger, J. Phys. Chem. C, 2008, 112, 4350–4354 CAS.
- J. Nelson, Mater. Today, 2011, 14, 462–470 CrossRef CAS.
- M. A. Brady, G. M. Su and M. L. Chabinyc, Soft Matter, 2011, 7, 11065–11077 RSC.
- F. Liu, Y. Gu, J. W. Jung, W. H. Jo and T. P. Russell, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1018–1044 CrossRef CAS.
- N. D. Treat and M. L. Chabinyc, Annu. Rev. Phys. Chem., 2014, 65, 59–81 CrossRef CAS PubMed.
- S.-S. Sun, Sol. Energy Mater. Sol. Cells, 2003, 79, 257–264 CrossRef CAS.
- S. M. Lindner and M. Thelakkat, Macromolecules, 2004, 37, 8832–8835 CrossRef CAS.
- F. Padinger, R. S. Rittberger and N. S. Sariciftci, Adv. Funct. Mater., 2003, 13, 85–88 CrossRef CAS.
- W. L. Ma, C. Y. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- G. Li, V. Shrotriya, J. S. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- X. N. Yang, J. Loos, S. C. Veenstra, W. J. H. Verhees, M. M. Wienk, J. M. Kroon, M. A. J. Michels and R. A. J. Janssen, Nano Lett., 2005, 5, 579–583 CrossRef CAS PubMed.
- N. Kiriy, E. Jahne, H. J. Adler, M. Schneider, A. Kiriy, G. Gorodyska, S. Minko, D. Jehnichen, P. Simon, A. A. Fokin and M. Stamm, Nano Lett., 2003, 3, 707–712 CrossRef CAS.
- W. Y. Huang, P. T. Huang, Y. K. Han, C. C. Lee, T. L. Hsieh and M. Y. Chang, Macromolecules, 2008, 41, 7485–7489 CrossRef CAS.
- A. J. Moule and K. Meerholz, Adv. Mater., 2008, 20, 240–245 CrossRef CAS.
- S. Y. Sun, T. Salim, L. H. Wong, Y. L. Foo, F. Boey and Y. M. Lam, J. Mater. Chem., 2011, 21, 377–386 RSC.
- M. J. Sobkowicz, R. L. Jones, R. J. Kline and D. M. DeLongchamp, Macromolecules, 2012, 45, 1046–1055 CrossRef CAS.
- H. Yan, Y. Yan, Z. Yu and Z. Wei, J. Phys. Chem. C, 2011, 115, 3257–3262 CAS.
- K. Vakhshouri, D. R. Kozub, C. Wang, A. Salleo and E. D. Gomez, Phys. Rev. Lett., 2012, 108, 026601-1–026601-5 CrossRef.
- F. C. Chen, H. C. Tseng and C. J. Ko, Appl. Phys. Lett., 2008, 92, 103316-1–103316-3 Search PubMed.
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619–3623 CrossRef CAS PubMed.
- Y. Yao, J. Hou, Z. Xu, G. Li and Y. Yang, Adv. Funct. Mater., 2008, 18, 1783–1789 CrossRef CAS.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS PubMed.
- H. Hoppe, T. Glatzel, M. Niggemann, W. Schwinger, F. Schaeffler, A. Hinsch, M. C. Lux-Steiner and N. S. Sariciftci, Thin Solid Films, 2006, 511–512, 587–592 CrossRef CAS PubMed.
- N. C. Cates, R. Gysel, J. E. P. Dahl, A. Sellinger and M. D. McGehee, Chem. Mat., 2010, 22, 3543–3548 CrossRef CAS.
- D. A. M. Egbe, S. Turk, S. Rathgeber, F. Kuhnlenz, R. Jadhav, A. Wild, E. Birckner, G. Adam, A. Pivrikas, V. Cimrova, G. Knor, N. S. Sariciftci and H. Hoppe, Macromolecules, 2010, 43, 1261–1269 CrossRef CAS.
- C. Kastner, S. Rathgeber, D. A. M. Egbe and H. Hoppe, J. Mater. Chem. A, 2013, 1, 3961–3969 Search PubMed.
- C. Kästner, B. Muhsin, A. Wild, D. A. M. Egbe, S. Rathgeber and H. Hoppe, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 868–874 CrossRef.
- J. Clark, J. F. Chang, F. C. Spano, R. H. Friend and C. Silva, Appl. Phys. Lett., 2009, 163306-1–163306-3 Search PubMed.
- H. Yamagata and F. C. Spano, J. Chem. Phys., 2012, 137, 184901-1–184901-14 CrossRef PubMed.
- F. C. Spano, J. Clark, C. Silva and R. H. Friend, J. Chem. Phys., 2009, 130, 234701-1–234701-11 CrossRef PubMed.
- R. Osterbacka, C. P. An, X. M. Jiang and Z. V. Vardeny, Science, 2000, 287, 839–842 CrossRef CAS.
- S. R. Amrutha and M. Jayakannan, J. Phys. Chem. B, 2008, 112, 1119–1129 CrossRef CAS PubMed.
- D. Baeriswyl, D. Campbell and S. Mazumdar, in Conjugated Conducting Polymers, ed. H. Kiess, Springer, Berlin Heidelberg, 1992, pp. 7–133 Search PubMed.
- K. Pichler, D. A. Halliday, D. D. C. Bradley, P. L. Burn, R. H. Friend and A. B. Holmes, J. Phys.: Condens. Matter, 1993, 5, 7155–7172 CrossRef CAS.
- R. Hoofman, M. P. de Haas, L. D. A. Siebbeles and J. M. Warman, Nature, 1998, 392, 54–56 CrossRef CAS PubMed.
- B. J. Schwartz, T. Q. Nguyen, J. J. Wu and S. H. Tolbert, Synth. Met., 2001, 116, 35–40 CrossRef CAS.
- H. Hoppe, M. Niggemann, C. Winder, J. Kraut, R. Hiesgen, A. Hinsch, D. Meissner and N. S. Sariciftci, Adv. Funct. Mater., 2004, 14, 1005–1011 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1945, 13, 453–465 CrossRef CAS PubMed.
- C. Kästner, D. K. Susarova, R. Jadhav, C. Ulbricht, D. A. M. Egbe, S. Rathgeber, P. A. Troshin and H. Hoppe, J. Mater. Chem., 2012, 22, 15987–15997 RSC.
- M. Hallermann, I. Kriegel, E. Da Como, J. M. Berger, E. von Hauff and J. Feldmann, Adv. Funct. Mater., 2009, 19, 3662–3668 CrossRef CAS.
- N. C. Cates, R. Gysel, Z. Beiley, C. E. Miller, M. F. Toney, M. Heeney, I. McCulloch and M. D. McGehee, Nano Lett., 2009, 9, 4153–4157 CrossRef CAS PubMed.
- N. C. Miller, E. Cho, M. J. N. Junk, R. Gysel, C. Risko, D. Kim, S. Sweetnam, C. E. Miller, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, A. Amassian, D. Acevedo-Feliz, C. Knox, M. R. Hansen, D. Dudenko, B. F. Chmelka, M. F. Toney, J.-L. Brédas and M. D. McGehee, Adv. Mater., 2012, 24, 6071–6079 CrossRef CAS PubMed.
- N. C. Miller, E. Cho, R. Gysel, C. Risko, V. Coropceanu, C. E. Miller, S. Sweetnam, A. Sellinger, M. Heeney, I. McCulloch, J.-L. Brédas, M. F. Toney and M. D. McGehee, Adv. Mater., 2012, 2, 1208–1217 CAS.
- R. Koeppe and N. S. Sariciftci, Photochem. Photobiol. Sci., 2006, 5, 1122–1131 CAS.
- C. Kastner, C. Ulbricht, D. A. M. Egbe and H. Hoppe, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1562–1566 CrossRef.
- A. Viterisi, F. Gispert-Guirado, J. W. Ryan and E. Palomares, J. Mater. Chem., 2012, 22, 15175–15182 RSC.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch, C. S. Ha and M. Ree, Nat. Mater., 2006, 5, 197–203 CrossRef CAS.
- T. M. Clarke, A. M. Ballantyne, J. Nelson, D. D. C. Bradley and J. R. Durrant, Adv. Funct. Mater., 2008, 18, 4029–4035 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |