
First principles guide to tune h-BN nanostructures as superior light-element-based hydrogen storage materials: role of the bond exchange spillover mechanism†
E. Mathan
Kumar
,
S.
Sinthika
and
Ranjit
Thapa
*
SRM Research Institute, SRM University, Kattankulathur 603203, Tamil Nadu, India. E-mail: ranjit.t@res.srmuniv.ac.in; Fax: +91-44-27456702; Tel: +91-44-27417918
First published on 28th October 2014
Abstract
We investigate the interaction of molecular hydrogen with light-element-based n-doped hexagonal boron nitride (h-BN) nanostructures and moreover explore the bond exchange mechanism for spillover of atomic hydrogen using dispersion-corrected density functional theory (DFT-D) calculations. A number of doped configurations were tested and it has been found that co-doping of C and O on h-BN sheet significantly increases the adsorption energy of molecular H2. The charge transfer from the n-doped h-BN surface to H2 is found to be the reason for the higher interactions that boosted the binding energy. In addition, the doped h-BN surfaces act as catalysts and dissociate the H2 molecule with a very low activation barrier, but the migration of the resulting H atoms on the surface requires high energy. In order to facilitate easy and fast migration of H atoms, we introduce the bond exchange mechanism using external mediators i.e. borane (BH3) and gallane (GaH3) molecules which serve as secondary catalysts and help in lowering the migration barrier, leading to the formation of a hydrogenated surface. The partially hydrogenated surface in turn can also act as a hydrogen storage material, with a higher propensity to adsorb hydrogen molecules when compared to the unhydrogenated surface. Hence the surface proposed in this work can be used to store a substantial quantity of hydrogen as an energy source with easy adsorption and desorption kinetics.
1 Introduction
Hydrogen as an energy source can be stored mainly in two forms, one as a hydrogen molecule (H2) adsorbed weakly on various surfaces (mainly by physical adsorption)1–3 and another as atomic hydrogen (H) bonded strongly with other elements (chemical bonding).4–6 H2 storage via physical adsorption is fully reversible and follows fast kinetics in comparison to the chemically bonded H atom, which usually needs high release temperatures. Due to concern for reversibility and stability, the H2 binding energy should be within a certain energy window i.e. about 0.2–0.7 eV.7 From various studies it has been observed that the adsorption energy of H2 on transition metal (TM) clusters supported on graphene and other 1D/2D nanostructures is in the desirable range.8–13 But the major problem with hydrogen storage using metal atoms/clusters via physical interaction is low gravimetric ratios at ambient conditions.14,15 So to store H2 with high gravimetric density, the host material should only consist of light elements such as Be, B, C, O, N etc.16–19 However storing H2 in the light-element-based materials with moderate binding energy remains a challenge. Overall, current research is mainly focused on designing efficient hydrogen storage materials to reach the Department of Energy (DOE) targets viz. gravimetric density of 9.0 wt%, volumetric density of 81 g L−1, operating temperature in the range of −40 to 85 °C and applied pressure in the range of 1–100 atm.There are several motivating works on carbon based nanomaterials (such as carbon nanotube (CNT), graphene, and fullerene) and metal organic frameworks (MOF) that illustrate them as possible materials for hydrogen storage.20–24 Recently Chen et al. reported that Mg doped graphene oxide (GO) enhances the storage capacity up to 5.6 wt% at a temperature of about 200 K. The binding energy of H2 with GO is found to be mainly dependent on the polarization interaction between Mg and O.22 Another study reported that boron doped graphene is a good medium for hydrogen storage when decorated with alkaline earth metals (AEM) like Mg and Ca. The H2 interaction on this medium shows a crossover between Kubas and multipole Coulomb effect depending on the ionic state of AEM and the number of adsorbed H2 molecules.23 Durgun et al. have found that substitutional doping of Be into single wall carbon nanotubes (SWNT) increased the H2 binding energy up to 0.40 eV, which is very high compared to the pure SWNT (60 meV). The charge transfer from Be (∼1.2e) to the nearest carbon atoms (∼0.4e to each neighbors) was established to be the cause of the enhanced binding strength.24 Hexagonal boron nitride (h-BN) nanostructures have also emerged as a good medium for hydrogen storage, when their electronic structure is perturbed.25–27 For instance, carbon doped boron nitride cages are proven to have promising potential as hydrogen storage materials with a storage capacity of 7.43 wt%. The activation energy for hydrogenation and dehydrogenation from the B11N12C surface is found to be about 2 eV at room temperature and 10 bar applied pressure, with the repulsion between the electrons of the hydrogen and π electron of B11N12C reducing the energy barrier for dehydrogenation.28 Also it was reported that Li-decorated boron nitride atomic chains (BNAC) show a high storage capacity of 25.4 wt% because of strong hybridization between the lithium 2p orbital and the nitrogen 2p orbital along with the polarization interaction between the H2 molecule and the Li decorated BNAC.29 On the other hand Han et al.30 explained that the spillover mechanism using a secondary catalyst can help in the formation of a partially hydrogenated graphene surface with very low barrier energy and the number of atomic hydrogens bound to the substrate is a predominant factor for tuning the magnitude of the H2 binding energy value.
In this work, we extensively study the interaction of H2 with pure, and C, O, 2C, C–O and 2C–O doped h-BN monolayers and boron nitride nanocages (B12N12). Our calculations reveal that among the entire doping configuration, C–O doping synergistically enhances the chemical activity of h-BN nanostructures, leading to a higher value of adsorption energy. The donation of electrons from the pz (of doped surface) to the 1σ* orbital of H2 is assigned to be the cause of the higher binding energy as compared to the pure h-BN case. We found that there is a probability of H2 dissociation on the doped h-BN surface with its low activation energy barrier. However, the migration of the dissociated hydrogen atom along the doped and pure h-BN surfaces is unfavorable. We propose that secondary mediators such as borane (BH3) and gallane (GaH3) can help in the migration of H atom along the surface with very low barrier energy via the bond exchange spillover mechanism. So during the H2 adsorption and desorption process it is possible to form a partially hydrogenated surface. Interestingly, it has been found that this partially hydrogenated h-BN monolayer has the tendency to bind the H2 molecule with a higher binding energy value, and also could be considered as a H2 storage surface.
2 Computational details
We performed the calculations using spin polarized density-functional theory as implemented in the Cambridge Serial Total Energy package (CASTEP).31 The Vienna ab-initio simulation package (VASP) has also been used for further verification.32 The generalized gradient approximation (GGA) was employed for the exchange and correlation effects at Perdew–Burke–Ernzerhof (PBE)33 and Perdew and Wang's 1991 (PW91) level.34 The potentials of the atoms were described by the ultrasoft pseudopotentials approach35 and the projected augmented wave (PAW) method.36 The plane-wave basis set was considered with an energy cutoff of 400 eV. Brillouin zone sampling was made with the Monkhorst Pack scheme and a K-point grid of 7 × 7 × 1. For long-range van der Waals interactions, the parameter-free Tkatchenko–Scheffler (TS)37 and Ortmann Bechstedt and Schmidt (OBS) method38 were used with PBE and PW91 functionals respectively (denoted as PBE + D or PBE + TS and PW91 + D or PW91 + OBS from here on). All the structures were optimized until the total energy converged to less than 10−5 eV per atom and the maximum force converged to lower than 0.01 eV Å−1. The complete linear and quadratic synchronous transit method (LST/QST) implemented in CASTEP was used to search for the transition states.39 The nudged elastic band (NEB) method was also employed to estimate the transition states for further verification. We used (4 × 4) and (5 × 5) supercells (along the a and b axis) and 20 Å vacuum along c-axis as the model geometry to explain our results. The binding energy (EB.E) between the H2 molecule and the doped surface is defined as| EB.E = E(doped h-BN + nH2) − E[doped h-BN + (n − 1)H2] − E(H2), | (1) |
3 Results and discussion
3.1 Single H2 adsorption
Hexagonal boron nitride monolayers and B12N12 nanocages were chemically modified by substitutional doping with carbon and oxygen atoms. We substituted the C and O atoms for B and N in h-BN surfaces correspondingly (an example of an n-doped system). As the formation energy of an O atom replacing a B atom of h-BN is very high, this structure is predicted to be highly unstable,40 and hence we did not investigate this case. In Table S1 and Fig. S1 of the ESI,† the formation energies of different configurations of doped h-BN sheets are summarized. Moreover, we have reported that doping enhances the activity of the h-BN monolayer.41 In order to test the storage capacity for hydrogen molecules on this class of surfaces, we first made a single H2 molecule interact with a monolayer of doped h-BN and a B12N12 nanocage. Since the interaction energy of nonpolar H2 molecules with the substrates containing light elements is mainly in the physisorption (London dispersion) range,42 we considered the van der Waals-interaction-corrected total energy term to get more accurate results. We mainly used the PBE + D and PW91 + D approaches to serve this purpose.43The optimized structures of a single H2 molecule adsorbed on various doped h-BN monolayers are shown in Fig. 1. The H2 binding energies and H2-surface distances (da) are shown in the inset of each subfigure of Fig. 1. First, we examined the role of the adsorption sites of different doped h-BN surfaces. Four different sites were tested, the top sites of the C, O, N and B atoms. We confirmed that for doped systems, B and C atoms anchor the H2 molecule with slightly higher binding energies as compared to O and N atoms, as shown in Fig. 1 and S2 of the ESI.† The adsorbed geometry of a H2 molecule on C doped h-BN (CBN) is shown in Fig. 1a. Using the PW91 + D functional the binding energy value was found to be about −0.16 eV, which is higher than the value obtained using the PBE + D functional (EB.E = −0.06 eV). Next we took into account the oxygen-doped h-BN (OBN) surface, shown in Fig. 1b for H2 adsorption. A similar trend was observed in this surface, the PW91 + D functional showing a higher binding energy value compared to the PBE + D functional. The higher binding energy of H2 on the OBN (EB.E = −0.18 eV using PW91 + D) as compared to the CBN surface indicates that O doping enhances the interaction energy of the h-BN system for capturing H2 molecules, which has been also experimentally observed by Lei et al.44 This may be attributed to the fact that, upon doping with oxygen, the h-BN surface undergoes structural modification and unequal amounts of charge are imparted to the surrounding boron atoms,41 which causes the B atom adjacent to the O dopant to become highly active. Next we assessed the H2 adsorption ability of the co-doped (C and O both) h-BN monolayer (COBN).40 We chose the para-configuration of C and O co-doped h-BN surface based on lowest formation energy (please see Fig. S1 in the ESI†). The H2 adsorption is even stronger on the COBN surface, with EB.E = −0.28 eV (optimized structure is shown in Fig. 1c) and is higher than the other surfaces. In this case the H2-surface distance is about 2.58 Å. For this system the binding energy value is larger than the required chemical potential of H2 to store at 30 °C and 100 bar pressure.30 Next we increased the number of C dopants to two (2CBN) (optimized structure shown in Fig. 1d). The binding energy of H2 on this system is higher than single-atom doping, but is less than the OBN and COBN systems. In order to increase the hydrogen storage ability of this surface, we doped an O atom onto the 2CBN system (2COBN) (Fig. S3†). Our results reveal that the binding energy of H2 on the 2CO doped h-BN surface is higher than the C, O and 2C doped h-BN systems, but is still less than that of the COBN system. These results indicate that the COBN structure is highly efficient in storing the hydrogen molecule as compared to all the other doping configurations studied.
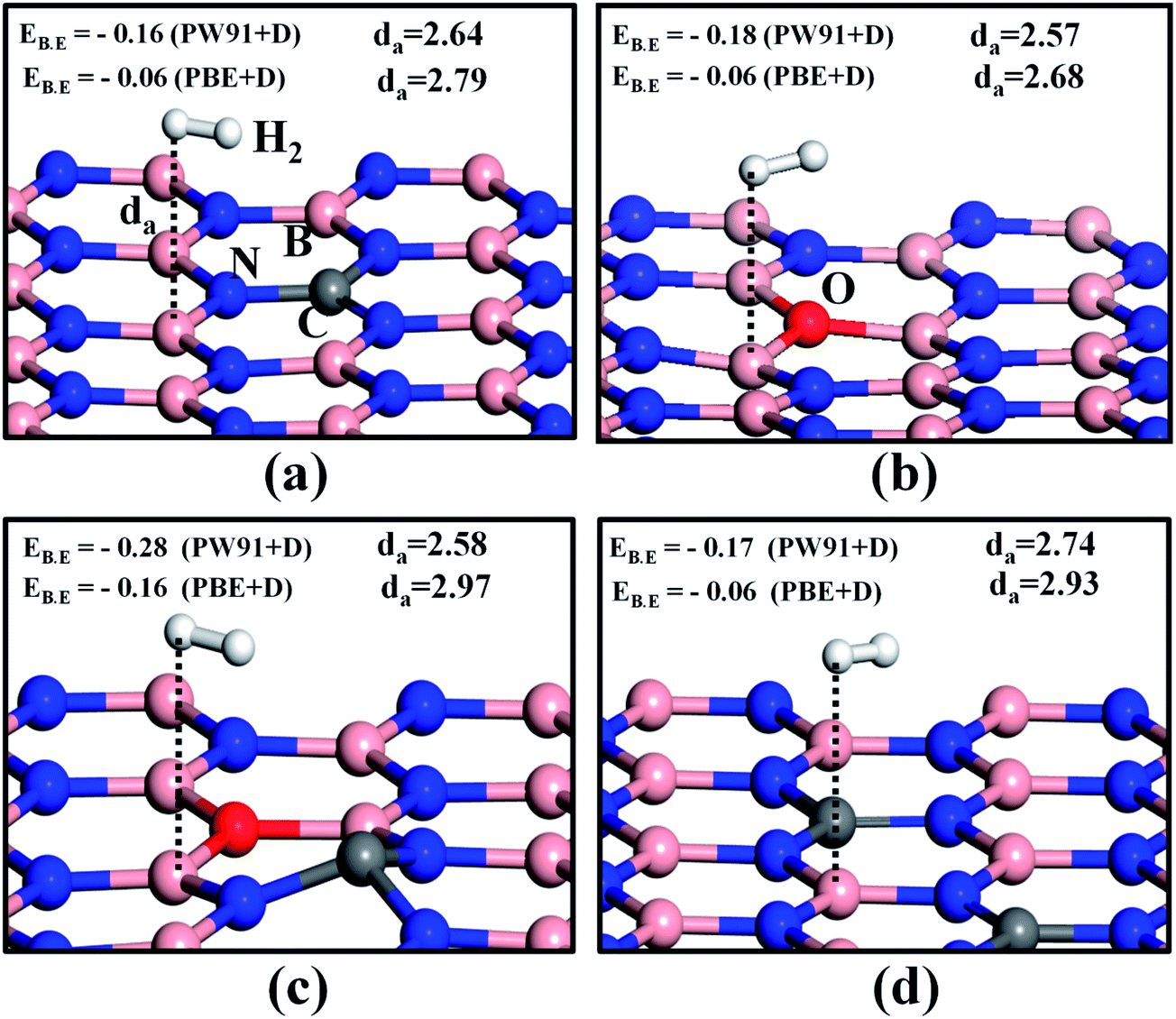 | ||
| Fig. 1 Optimized geometries for single H2 adsorption on (a) CBN (b) OBN (c) COBN and (d) 2CBN sheets. The red, gray, pink and blue spheres represent the oxygen, carbon, boron, nitrogen atoms. The hydrogen molecule is represented by white spheres. Binding energies (EB.E) are in eV and are calculated using two different functionals. H2-surface distances (da) are in Å. The black dotted line is a guide to the eye to show nearest active sites. | ||
In order to find out the role of the functionals on the H2-surface interaction energy value, single hydrogen molecule binding energies were calculated for all the types of configurations using the PBE, PBE + TS, PW91, PW91 + OBS and PBEsol functionals and are tabulated in Table 1. Examination of Table 1 reveals that the binding energy values estimated using the PW91 functional are mostly negative and higher than the EB.E using the PBE functional. On these surfaces the PBE functional underestimates the binding energy values, agreeing well with the reported results of Wang et al.29 Also Sun et al.45 emphasized that the PW91 functional can estimate the proper binding energy in the case of hydrogen adsorption. Similarly, considering the dispersion energy term, it has been found that the magnitudes of the binding energies are higher for the PW91 + OBS functional compared to the PBE + TS functional. Both the functionals follow the same trend in binding energy for different surfaces. It can be interpreted that the PW91 + OBS functional treats van der Waals forces more correctly in these cases.46 So we used the PW91 + OBS functional for the further studies and the functional-dependent binding energy of H2 on various doped h-BN configurations have been extensively discussed in the later section. From Table 1, it can also be inferred that H2 adsorption is strongly preferred on COBN and 2COBN systems (in view of all functionals), suggesting these surfaces to be efficient materials for hydrogen storage.
| System | E B.E (PBE) | E B.E (PBE + TS) | E B.E (PW91) | E B.E (PW91 + OBS) | E B.E (PBEsol) |
|---|---|---|---|---|---|
| h-BN | +0.04 | −0.04 | +0.01 | −0.15 | +0.01 |
| C-h-BN | +0.01 | −0.06 | −0.02 | −0.16 | −0.01 |
| O-h-BN | +0.04 | −0.06 | −0.04 | −0.18 | +0.01 |
| CO-h-BN | −0.05 | −0.16 | −0.045 | −0.28 | −0.11 |
| 2C-h-BN | +0.11 | −0.06 | −0.033 | −0.17 | −0.01 |
| 2CO-h-BN | −0.008 | −0.09 | −0.013 | −0.21 | −0.05 |
Now we consider one more example of a BN surface (BN-nanocage, B12N12) to find the trend of the H2 binding energy value as a function of the dopants. B12N12 is recognized to be a stable form of boron nitride,28 and its high surface to volume ratio can be employed to store a larger number of hydrogen molecules. Similarly to the monolayer case, the BN-nanocage was doped with C and O atoms, and the optimized structures are shown in Fig. 2. The binding energy of a single H2 adsorption on the C doped BN-nanocage (CB11N12, we use a similar notation from here on for all the cases) is EB.E = −0.12 eV (see Fig. 2a) (this value is similar to previous work28). In the case of OB12N11 the EB.E = −0.14 eV, the increment in binding energy can be attributed to the fact that O doping on B12N12 leads to a slight structural distortion of the host material, thereby increasing its ability to adsorb hydrogen molecules (see Fig. 2b). C–O co-doped on a BN-nanocage (COB11N11) is shown in Fig. 2c, here the EB.E = −0.15 eV, which is very low compared to COBN but is slightly higher than OB12N11 and CB11N12, and the distance between H2 and the surface is about 2.83 Å. Using the enthalpy ΔH (i.e. binding energy), the estimated free energy change (ΔG) during hydrogenation (H2 adsorption) on the (COB11N11) surface at room temperature and 10 bar pressure shows COB11N11 to be a better storage medium compared to the (CB11N12) surface.28 If we increase the number of carbon dopants to two in the BN-nanocage (2CB10N12), the binding energy is found to be −0.13 eV; the structure is shown in Fig. 2d. The CB11N12 and 2CB10N12 doping configurations of the nanocages exhibit similar adsorption energy values because of low charge transfer from the doped surface to the H2 molecule. In order to promote the interaction of H2 with the cages, we doped one more oxygen into the 2CBN cage (2COB10N11), and the binding energy increased to −0.16 eV with the optimized da = 2.410 Å (see Fig. S4†). 2COB10N11 can be regarded as the best storage medium among the doped BN cages considered here as its interaction with H2 surpasses the others.
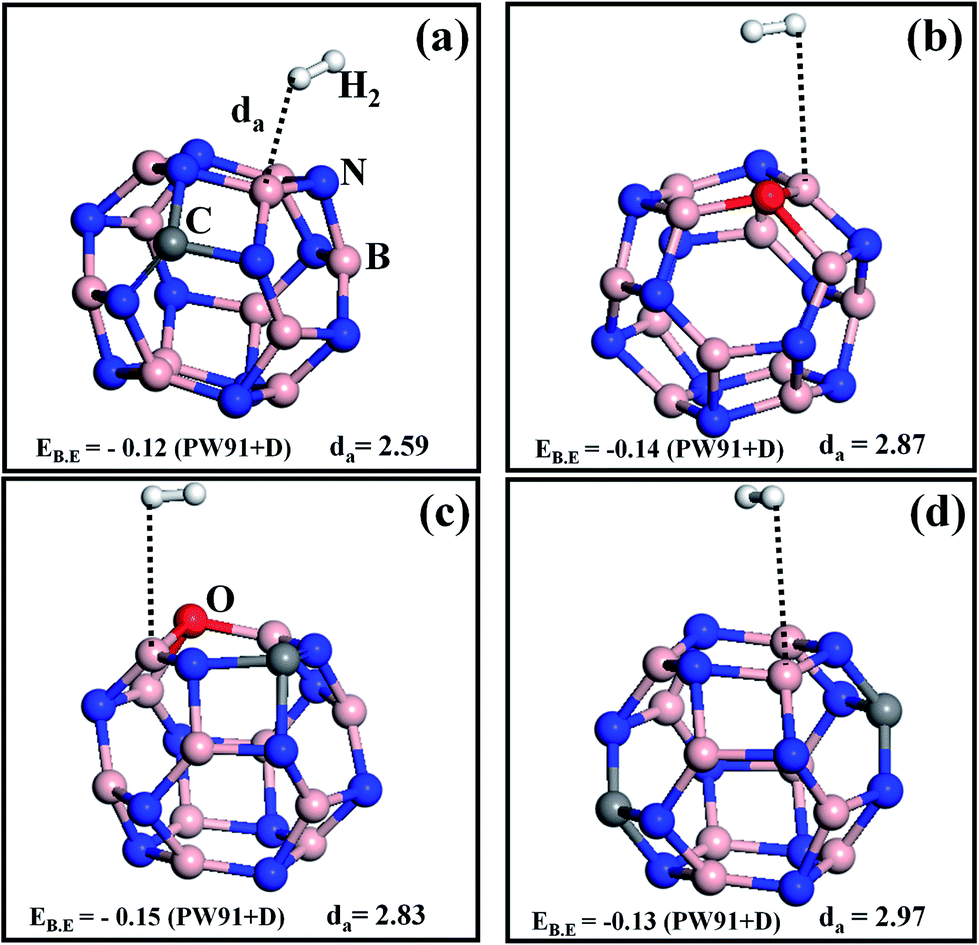 | ||
| Fig. 2 Single H2 molecule adsorption on different doped B12N12 nanocages (a) CB11N12, (b) OB12N11, (c) COB11N11, (d) 2CB10N12. All colors and symbols follow the same convention as those in Fig. 1. Binding energies (EB.E) are in eV and H2-surface distances (da) are in Å. | ||
3.2 Multi-H2 adsorption
Next we systematically investigated the interaction of more than one H2 molecule on the various doped h-BN monolayers (COBN, 2CBN, 2COBN). We used the PW91 + D functional for this purpose as it provides consistent energy values, described earlier in the text. In Fig. 3, the binding energy is presented as a function of the number of adsorbed H2 molecules. We added H2 molecules to the surface one by one, relaxing the structure each time (taking into account that H2 molecules adsorb on the surface one by one, during storage). Since lateral interactions between H2 molecules may contribute significantly to the binding energy (discussed in detail in a later section), we placed the H2 molecules at a distance of ∼3 Å from each other to ensure that H2–H2 interactions were minimal. The optimized structure of two hydrogen molecules on the COBN surface is shown in Fig. 3a. Here the binding energy per H2 on the COBN surface is about −0.23 eV, which is comparatively smaller than that of the single H2 adsorbed on the COBN surface. Three H2 molecules adsorbed on the COBN surface are shown in Fig. 3b, here the estimated EB.E per H2 is about −0.25 eV, which is slightly higher than in the case of two H2 molecules adsorbed on the COBN surface. Now upon addition of the fourth H2 molecule on COBN (see Fig. 3c) the EB.E per H2 increases to about −0.28 eV. Trends in the estimated results indicate that for the COBN surface the binding energy per H2 increases with the total number of H2 molecules per supercell, which is contrary to the case of metal-based systems wherein the binding energy per H2 decreases with number of H2 molecules.29,47 We also note that the bond length of a free H2 molecule is db = 0.75 Å which is elongated to 0.78 Å in the case of H2 (first molecule) adsorbed on the COBN system. Next, all the B and C sites of the COBN surface (only considering the single side of the COBN surface) were covered with H2 molecules (16H2) in order to assess the ultimate binding energy limit. In fact, our results reveal that the binding energy per H2 molecule for 16 H2 molecules adsorbed on the COBN surface increases to −0.42 eV. This increase in binding energy clearly indicates that the COBN surface is a good medium for H2 uptake in ambient conditions. In the same way, we followed the multi-hydrogen-molecule adsorption on 2CBN and 2COBN surfaces and plotted the binding energy of all these configurations as a function of the number of H2 molecules as shown in Fig. 3d. The trend of binding energies obtained is similar for all the configurations. We need additional studies to understand the nature of interaction among the H2 molecules and the surface. Also it should be noted that increasing the C–O co-doping concentration (e.g.: 2C2O, 3C3O per unit supercell are considered here) enhances the binding energy of the H2 molecule, which is an obvious phenomenon and also a simple way to tune the binding energy (for more details see Fig. S5†).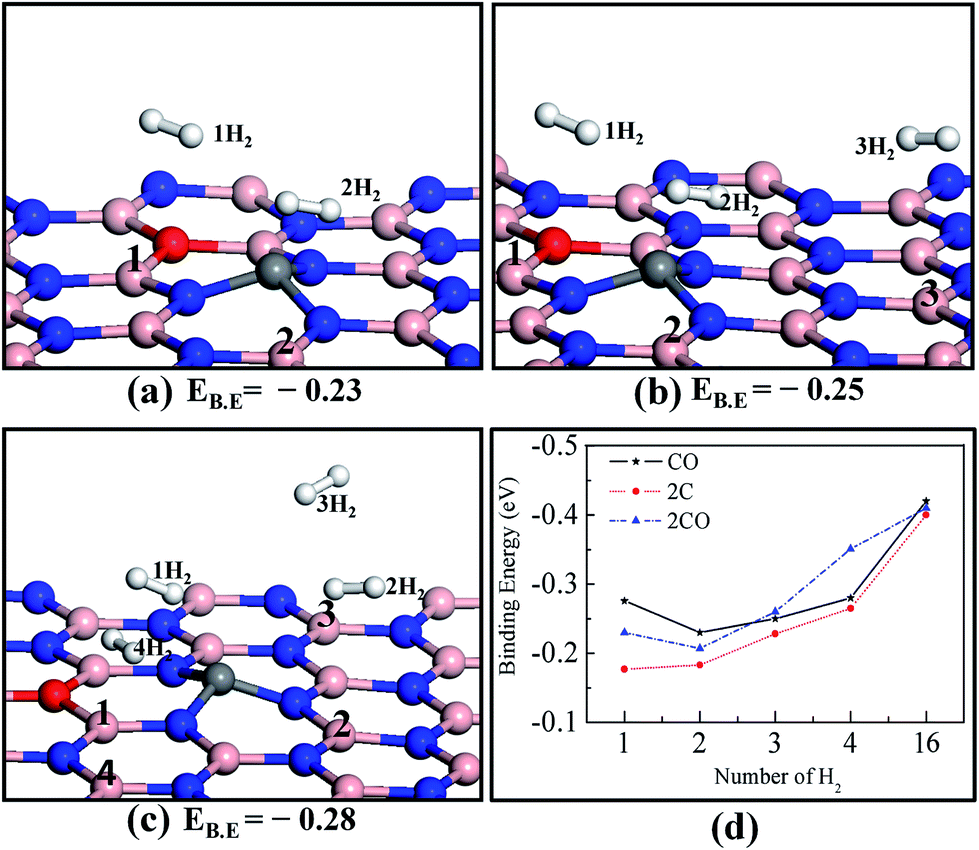 | ||
| Fig. 3 Optimized structures for (a) two, (b) three, and (c) four hydrogen molecules adsorbed on the COBN monolayer. The numbers preceding each H2 represent the order in which the H2 molecules are added, and the numbers on the surface denote the corresponding adsorption site of each molecule. (d) Graph demonstrating the binding energy as a function of the number of H2 molecules adsorbed on different surfaces. Binding energies (EB.E) are in eV. | ||
3.3 Interaction I: partial density of states (PDOS)
In order to gain a deeper insight into the cause of binding between the H2 molecules and the COBN monolayer, we plotted the spin polarized partial density of states (PDOS) of (one, two, three and sixteen) H2 adsorbed COBN systems, shown in Fig. 4. Here we considered the density of states only for C, O and B atoms (nearest to the H2 molecule) for simplicity (indicated by the solid line), and the PDOS of the H2 molecules are depicted using a filled area plot, for easier interpretation. The donor electronic state (of both minority and majority carriers) at about −0.8 eV below the Fermi level (Ef) of the COBN surface is a clear indication of two-electron doping. The non-magnetic ground state of C–O co-doped h-BN is more favorable as per the functional used in this case. Before adsorption, the 1σ* orbital of the free H2 molecule is empty and lies above the Fermi level. From the PDOS for the single H2 adsorbed on B site of COBN in Fig. 4a, it is clear that there is electron donation from the surface donor state to the 1σ* orbital of the H2 molecule, making it partially occupied. The inset shows a closer view of the PDOS near the Fermi level. The hybridization of states between the defect levels and the 1σ* orbital of H2 molecule can be inferred to be the cause of the increased binding energy of surface-H2 due to doping. Fig. 4b–d show the PDOS of two, three and sixteen H2 molecules adsorbed on the same surface (COBN). Fig. 4 clearly demonstrates that the partial occupancy of state of H2 molecules below the Fermi level is relatively increased as compared to the single H2 case. This phenomenon explains why the binding energy per H2 molecule on such surfaces remains similar even when the concentration of H2 increases. Also it should be noted that the partial occupancy of the 1σ* orbital of the H2 molecule increases in the case of a higher percentage C–O co-doping on the surface.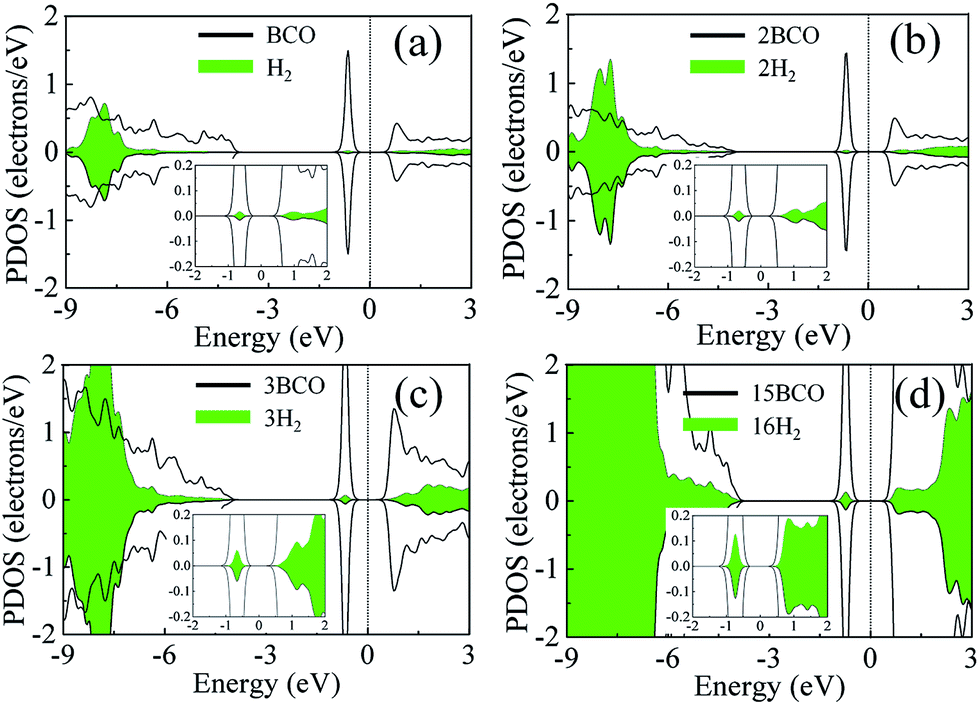 | ||
| Fig. 4 Partial density of states of the H2 molecules and the C, O and B atoms (adsorb sites) of (a) single H2 molecule adsorption on COBN system, (b) two H2 adsorption on COBN, (c) three H2 adsorption on COBN and (d) 16H2 adsorption on COBN. The insets are magnified views of PDOS near the Fermi level. | ||
3.4 Interaction II: isosurface and electron density difference
In order to analyze the nature of interactions that contribute to the total energy of the system, we made two models: in the first case, H2 molecules were adsorbed on four boron sites and were made to interact with each other with an average distance of 2 Å (henceforth named as 4H2-cluster) and in the second case, we removed a single H2 molecule from the most active B site and placed it far from the other 3H2 molecules at a distance of ∼4 Å (termed as far-H2–3H2 cluster). The binding energy per H2 molecule was calculated for the two cases. The binding energy per H2 molecule in the case of the 4H2-cluster is about −0.31 eV (the structure is shown in Fig. 5a). For the far-H2–3H2 cluster case we found that the binding energy per H2 decreases to about −0.22 eV (see Fig. 5b). This result clearly indicates that an active B site, near or far from C–O doping area, plays a vital role in determining the adsorption energy of H2. We believe that as H2 clustering is more favorable in this case, the H2–H2 self-interaction also contributes to the enhancement in binding energy (discussed later). Now we examine the origin of stronger binding of H2, particularly with the C–O co-doped h-BN monolayer and provide more insight into the cause of the difference in binding energy of the H2-cluster and the far-H2–3H2 cluster with the surface by performing isosurface and charge redistribution analysis. We studied mainly how the electronic charge density is modified upon adsorption of H2 molecule. More specifically, we plotted isosurfaces using the equation,| Δρ = ρ(COBN + H2) − ρ(COBN) − ρ(H2) | (2) |
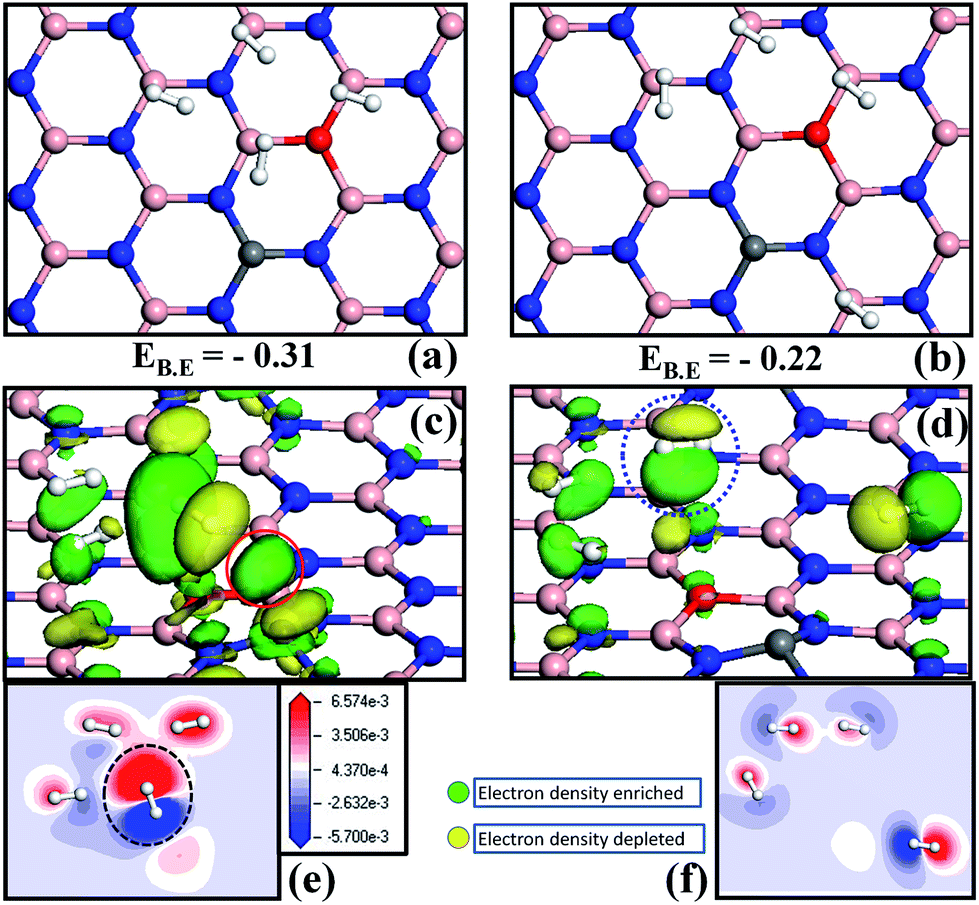 | ||
| Fig. 5 Energy values and optimized structures of (a) the 4H2 cluster, and (b) the far-H2–3H2 cluster on the COBN surface. Redistribution of electronic charge upon adsorption of (c) the 4H2 cluster, and (d) the far-H2–3H2 cluster on the COBN surface. Green and yellow lobes correspond to a gain and depletion of electronic charge, respectively. The isosurface value of 2 × 10−3 e Bohr−3 is considered for all the cases. Charge density difference value is the same for the two cases, a slice along the direction parallel to the COBN surface touching all four H2 molecules is demonstrated in (e) & (f). Gain of electron density is indicated by red color, loss of electron density is shown by blue color and white color indicates the region with very small change in the electron density. Binding energies (EB.E) are in eV. | ||
Charge density differences were also calculated for the same two cases (the 4H2-cluster and the far-H2–3H2 cluster) and are shown in Fig. 5e and f, respectively. A planar slice along the direction parallel to the COBN surface touches all the four H2 molecules, which helps in understanding the interaction between H2–H2 molecules (if any) and between the H2 molecule and the surface. The electron enrichment is indicated by red color, loss of electrons is shown by blue color and white color indicates a region with very small change in electron density. The dotted black circle around the H2 molecule indicates a large gain of electronic charge from the COBN surface. This is consistent with the insight obtained from the PDOS and isosurface analysis of the previous sections. The H2–H2 self-interaction is higher in the case of the 4H2-cluster compared to the far-H2–3H2 cluster, as can be inferred from Fig. 5e and f. This clearly suggests that the self-interaction energy also contributes to the binding energy value. For more insight we performed a test calculation of only the 4H2-cluster and the far-H2–3H2 cluster without considering the surface (COBN). In this case the total energy of 4H2-cluster is found to be more negative than the far-H2–3H2 cluster. This energy difference signifies that there is a contribution to the binding energy due to H2–H2 self-interaction along with partial charge transfer from the surface to H2 molecules. The results indicate that sharing of electrons of bonding orbitals of H2 is the cause of this phenomenon. Our results thus suggest that, as the binding value is in the physisorption range here, the H2–H2 self-interaction energy also needs to be considered while computing the binding energies. The details of H2 adsorption on the different surfaces and the various means used to explain the interaction are tabulated in Table 2.
| System | Functional | −EB.E (eV) | wt% | d a (Å) | d b (Å) | Interaction |
|---|---|---|---|---|---|---|
| Metal as adsorbent | ||||||
| Mg doped graphene oxide22 | PBE | 0.38 | 5.6 | 2.4 | 0.79 | Charge transfer from H2 to the metal ions, polarization and Kubas interaction |
| Mg and Ca doped graphene complexes23 | LDA & GGA | 0.31 (0.21) | 6.2 | 2.66 | 0.76 | Kubas and multipole Coulomb interaction |
| Be substituted SWNT24 | GGA | 0.40 | 2.4 | 2.0 | 0.77 | Charge transfer from Be to nearest carbon atom |
| Platinum doped BNNT25 | PBE | 0.365 | 4–8 | 1.88 | 0.82 | Charge transfer from surface to H2 and polarization interaction |
| Aluminum doped graphene47 | LDA | 0.260 | 5.13 | 2.7 | — | High charge transfer from Al to H2 |
| Clustering of Ti on C60 surface49 | PW91 | 0.38 | 2.85 | — | 0.75 | Charge polarization interaction |
| Sc or Ti decorated on B doped fragmented graphitic shells (FGS)50 | LDA and GGA | 0.3–0.5 | 4–8 | — | — | Charge transfer and Coulomb interaction |
| Metallo-carbohedrenes (M8C12) (M = Ti, Sc and V)51 | PBE | 0.15–0.46 | 10.96–15.06 | — | — | Charge transfer from metal to H2 |
| Ti(Sc) decorated boron–carbon-nitride sheet52 | GGA | 0.13–0.56 | 7.6–7.8 | 1.85 (2.21) | 0.91 (0.78) | Kubas and polarization interaction |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Low-elements as adsorbent | ||||||
| BN sheet53 | BLYP | 0.14 | 7.5 | — | 0.74 | Electric field induces polarization interaction |
| BNNT (stone-Wales defect)54 | PBE | 0.017 | — | — | 0.74 | Charge transfer from defect site to H2. |
| Charged fullerene {C283+ & C826+}55 | LDA | 0.32 | 8.0 | — | — | Charge transfer from metal atom and polarization interaction due to electric field |
| {Li12C60}56 | PW91 | 0.075 | 9.0 | 2.07 | 0.75 | Polarization interaction |
| COBN surface (current study) | PW91 + OBS | 0.28 | 7.4 | 2.58 | 0.78 | Charge transfer from donor state of the substrate to 1σ* orbital of H2 molecule |
| 2COB10N11 (current study) | PW91 + OBS | 0.16 | 7.36 | 2.41 | 0.76 | |
| Partially hydrogenated COBN (current study) | PW91 + OBS | 0.32 | 7.3 | 2.53 | 0.75 | |
3.5 Bond exchange mechanism
Here we investigate the possibility and limitation on hydrogenation of pure and doped h-BN monolayers and discuss how an external mediator can assist in the spillover of hydrogen atoms via the bond exchange mechanism. First we extensively studied the H2 interaction with partially hydrogenated surfaces taking into account that that the surfaces are highly stable and can be realized. It has been found that the H2 binding with partially hydrogenated COBN surface is high compared to all the other surfaces discussed earlier. The optimized structures of the H2 molecule on the partially hydrogenated COBN surface are shown in Fig. 6a. The binding energy value is −0.32 eV and the distance da is found to be 2.53 Å. A similar result was observed for the case of partially hydrogenated, undoped h-BN surface.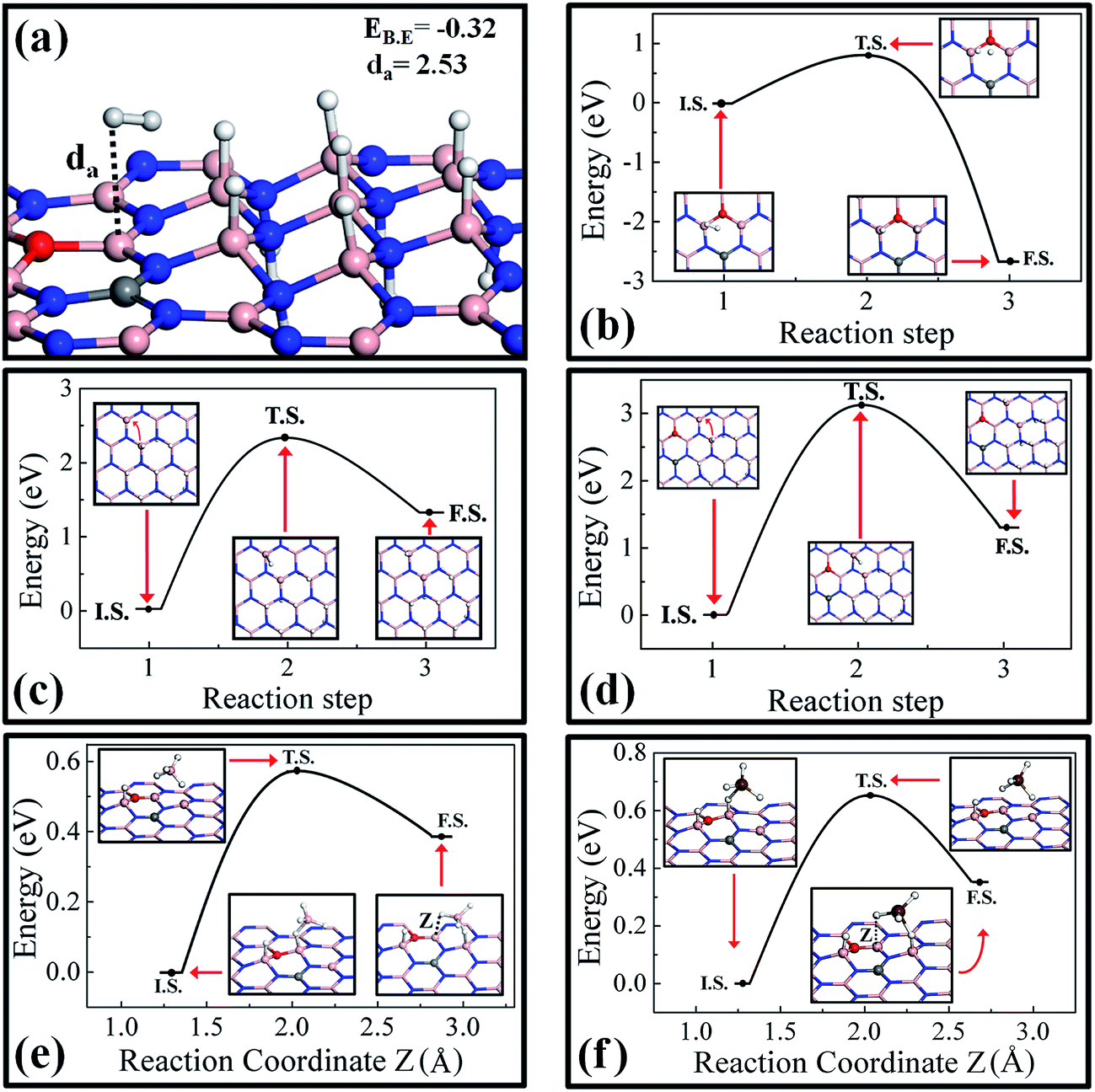 | ||
| Fig. 6 Optimized structure for (a) H2 adsorption on the partially hydrogenated COBN surface. The binding energy (eV) and H2-surface distance (Å) are written in the inset; (b) shows the barrier energy for the dissociation of H2 on COBN surface. The barrier energy curve during the migration of the H atom from the paired configuration on the pure h-BN and COBN surfaces are shown in (c) and (d) respectively. The energy barrier curve during the hydrogen spillover on the COBN surface via bond exchange mechanism using secondary mediators BH3 and GaH3 are shown in (e) and (f) respectively. In (b) to (f) the insets show the structure of the initial state (I.S.), transition state (T.S.) and final state (F.S.). The black lines connecting I.S., T.S. and F.S. are only to guide the eye. | ||
In order to achieve the hydrogenation, the first step is the dissociation of the H2 molecule. On the pure h-BN surface the energy required to dissociate the H2 molecule into two H atoms is more than 2.29 eV, suggesting that dissociation at ordinary temperatures is quite unlikely. A similar result has been observed on a pristine graphene surface, for which the dissociation barrier for H2 is quite high,30 but the dissociation of H2 and hopping of an H atom to the nearest carbon atom is feasible by using a metal cluster that acts as a catalyst and lowers the energy barrier (Ea) to about 0.65 eV.57 However, in our case the doped COBN system is itself acting as a catalyst.41 In order to calculate the dissociation barrier, we performed transition state search calculations considering a single H2 molecule adsorbed on the COBN surface as the initial state (I.S.) and two hydrogen atoms adsorbed on top of boron atoms as the final state (F.S.), the structures of which are shown in the inset of Fig. 6b. Our calculations reveal that the energy required for the bond breaking of H2 on the COBN surface is about 0.78 eV; the transition state (T.S.) structure is shown in the inset of Fig. 6b. The reaction energy in this case is about −2.75 eV, indicating the high stability of the H atom as compared to H2 molecule on COBN surface. Also it should be noted that the binding energy of the atomic hydrogen adsorbed on COBN is very high compared to on the pure h-BN system. These results clearly indicate that dissociation of H2 from the COBN surface is possible at low temperature, whereas on the pure h-BN surface it requires a very high temperature.
Now after dissociation, the migration of a hydrogen atom with a low energy barrier is required for easy and fast kinetics at low temperature to achieve the hydrogenation of the surface. Partially hydrogenated pure h-BN and COBN surfaces are considered for the purpose of finding the migration energy barriers. First we discuss the case of H atom migration on the pure h-BN surface. The I.S., F.S. and the T.S. structures are shown in the inset of Fig. 6c. In the inset of Fig. 6c (see the I.S. structure) the small, bent red arrow is used to depict the H migration direction, pointing to one of the closest and unoccupied B atoms to its I.S. on the surface . The dragging of the H atom from the paired chair like site to the nearest B site has a very high barrier energy of about Ea = 2.35 eV. The reaction energy in this case is highly endothermic, indicating that clustering of H atoms is more favorable.30 Similarly, considering the COBN surface, the energy barrier to migrate the H atom is Ea = 3.05 eV (the energy barrier is defined as the energy of T.S. in reference to the energy of I.S, shown in Fig. 6d), which is higher than of the pure hydrogenated case. In this case also the reaction energy is endothermic in nature. The corresponding T.S. structure is illustrated in the inset of Fig. 6d, in which the H atom is distant from the boron sites with an average B–H distance of about 2.4 Å. The barrier energy in both cases indicates an impractical migration of the hydrogen atom under ambient conditions. We also considered the single H migration just after the H2 dissociation near the active site. For this case the barrier energy is about 2.32 eV (the I.S., F.S and the calculated barrier energy are demonstrated in Fig. S6†). It can be concluded from this study that although dissociation from the COBN surface is possible, the migration of the H atom on its own requires a very high temperature (and thus does not look viable).
To solve the problem of enabling easy and fast migration of the H atom in ambient conditions, we proposed a new mechanism that can be termed as a bond exchange mechanism using external catalysts or agents. Here we introduce two examples of external catalysts, borane (BH3) and gallane (GaH3) molecules. A BH3 molecule near one of the dissociated H atoms adsorbed on COBN surface is considered as the I.S. (completely relaxed structure) and a BH3 molecule in close proximity with an H atom adsorbed on a neighboring B atom has been considered as the F.S. (structures are shown in the insets of Fig. 6e). So here we introduce the bond exchange spillover mechanism i.e. H atom exchange at the same ring via migration of a H atom using bond breaking (from surface) and swapping of one of the H atoms of BH3 in the surface; this is clearly seen from the structures of the I.S. and F.S. In a sense, the H atom (neutral form) spillover through the secondary catalyst via bond exchange is similar to the Grotthuss-proton exchange in the water network.39,58 The bond exchange barrier energy for migration of the H atom using the BH3 molecule is about Ea = 0.57 eV, which is very low compared to the energy required to desorb BH4 by breaking the surface H-bond far from the COBN surface (about 2.56 eV). The reaction energy is about 0.38 eV, indicating the route is slightly endothermic in nature. This is an obvious fact, as during bond exchange spillover, the H atom is moving away from the active site. It is also worth noting that while BH3 interacts quite strongly with a H atom that is adsorbed near the doping site, the interaction energy of BH3 is not so strong with a H atom adsorbed far from the dopant site. Hence we can infer that the complete dissociation of BH4 is less favorable compared to the H atom migration using the bond exchange mechanism. Similar results have been observed by considering GaH3 as a secondary catalyst. The structures of the reaction states (I.S., F.S. and T.S.) to explain the bond exchange mechanism using GaH3 as a secondary catalyst on the hydrogenated COBN surface are shown in the inset of Fig. 6f. In this case the maximum energy required for the bond exchange is about Ea = 0.65 eV, and the reaction energy is about 0.35 eV. Hence the bond exchange mechanism sufficiently decreases the activation barrier (Ea < 0.65 eV) for H migration on the substrate. We also considered NH3 and SiH4 as secondary catalysts, but found them not to be suitable candidates to initiate bond exchange spillover (see Fig. S7†).
4 Conclusions
In summary, first principles theory based DFT calculations explain significantly the increased binding strength of a H2 molecule with an n-doped h-BN surface as compared to the pristine form. To find the best storage medium, we checked various types of h-BN nanostructures and concluded that some particular systems like COBN, 2COBN, 2COB10N11 nanocage and partially hydrogenated COBN surfaces are superior media for the purpose. It has been observed that electron donation from the surface donor state to the 1σ* orbital of the H2 molecule is the governing reason for the stronger binding. 6.25% C–O doping is enough to store 7.4 wt% of H2 which is quite acceptable according to the DOE target. On the COBN surface the barrier energy to split the H–H bond is estimated to be about 0.78 eV. But the spillover of the H atom requires a very high temperature, which seems quite unfeasible for the hydrogenation of such a surface. We proposed that easy H atom migration along the surface is possible via a bond exchange mechanism using some external mediators (e.g.: GaH3, BH3). The hydrogen atom can migrate via bond exchange mechanism in the same hexagonal ring with an energy barrier of about 0.57–0.65 eV. Also it has been found that the partial hydrogenation on the surfaces can enhance the binding of molecular hydrogen. Our findings conclude that a metal-free light-element-based C and O doped h-BN nanostructure is an efficient medium to store hydrogen energy and that the bond exchange mechanism can be used as an effective approach for adsorption and desorption of hydrogen atoms.Acknowledgements
RT and SS thank the Science and Engineering Research Board (SERB), India for the financial support (Grant no: SB/FTP/PS028/2013). All authors thank SRM Research Institute, SRM University for providing a supercomputing facility and financial support.References
- S. Patchkovskii, J. S. Tse, S. N. Yurchenko, L. Zhechkov, T. Heine and G. Seifert, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10439–10444 CrossRef CAS PubMed.
- H. Si, L. J. Peng, J. R. Morris and B. C. Pan, J. Phys. Chem. C, 2011, 115, 9053–9058 CAS.
- R. E. Barajas-Barraza and R. A. Guirado-López, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155426 CrossRef.
- P. O. Krasnov, F. Ding, A. K. Singh and B. I. Yakobson, J. Phys. Chem. C, 2007, 111, 17977–17980 CAS.
- F. H. Yang and R. T. Yang, Carbon, 2002, 40, 437–444 CrossRef CAS.
- E. Yoo, L. Gao, T. Komatsu, N. Yagai, K. Arai, T. Yamazaki, K. Matsuishi, T. Matsumoto and J. Nakamura, J. Phys. Chem. B, 2004, 108, 18903–18907 CrossRef CAS.
- Y. Guo, K. Jiang, B. Xu, Y. Xia, J. Yin and Z. Liu, J. Phys. Chem. C, 2012, 116, 13837–13841 CAS.
- D. Sen, R. Thapa and K. K. Chattopadhyay, Int. J. Hydrogen Energy, 2013, 38, 3041–3049 CrossRef CAS PubMed.
- W. H. Shin, S. H. Yang, W. A. Goddard and J. K. Kang, Appl. Phys. Lett., 2006, 88, 053111 CrossRef PubMed.
- A. Sigal, M. I. Rojas and E. P. M. Leiva, Int. J. Hydrogen Energy, 2011, 36, 3537–3546 CrossRef CAS PubMed.
- H.-L. Park and Y.-C. Chung, Comput. Mater. Sci., 2010, 49, S297–S301 CrossRef PubMed.
- T. Yildirim and S. Ciraci, Phys. Rev. Lett., 2005, 94, 175501 CrossRef CAS.
- P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Nature, 2002, 420, 302–304 CrossRef CAS PubMed.
- L. Zhou, Renewable Sustainable Energy Rev., 2005, 9, 395–408 CrossRef CAS PubMed.
- Y. Ma, Y. Xia, M. Zhao, R. Wang and L. Mei, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 115422 CrossRef.
- Y.-H. Kim, Y. Zhao, A. Williamson, M. J. Heben and S. B. Zhang, Phys. Rev. Lett., 2006, 96, 016102 CrossRef.
- R. C. Lochan and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 1357–1370 RSC.
- Z. Zhang and K. Cho, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 75, 075420 CrossRef.
- M. Sankaran and B. Viswanathan, Carbon, 2006, 44, 2816–2821 CrossRef CAS PubMed.
- M. Suri, M. Dornfeld and E. Ganz, J. Chem. Phys., 2012, 116, 3661–3666 Search PubMed.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- C. Chen, J. Zhang, B. Zhang and H. M. Duan, J. Phys. Chem. C, 2013, 117, 4337–4344 CAS.
- G. Kim, S.-H. Jhi, S. Lim and N. Park, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 155437 CrossRef.
- E. Durgun, Y.-R. Jang and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 073413 CrossRef.
- X. Wu, J. L. Yang and X. C. Zeng, J. Chem. Phys., 2006, 125, 044704 CrossRef PubMed.
- Md. Shahzad Khan and M. Shahid Khan, Int. Nano Lett., 2011, 1, 103–110 Search PubMed.
- M. Chen, Y.-J. Zhao, J.-H. Liao and X.-B. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 045459 CrossRef.
- H. Y. Wu, X. F. Fan, J.-L. Kuo and W.-Q. Deng, Chem. Commun., 2010, 46, 883–885 RSC.
- Y. Wang, F. Wang, B. Xu, J. Zhang, Q. Sun and Y. Jia, J. Appl. Phys., 2013, 113, 064309 CrossRef PubMed.
- S. S. Han, H. Jung, D. H. Jung, S.-H. Choi and N. Park, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155408 CrossRef.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- Y. Wang and J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 13298 CrossRef.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- F. Ortmann, F. Bechstedt and W. G. Schmidt, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 205101 CrossRef.
- R. Thapa and N. Park, J. Phys. Chem. Lett., 2012, 3, 3065–3069 CrossRef CAS.
- O. L. Krivanek, M. F. Chisholm, V. Nicolosi, T. J. Pennycook, G. J. Corbin, N. Dellby, M. F. Murfitt, C. S. Own, Z. S. Szilagyi, M. P. Oxley, S. T. Pantelides and S. J. Pennycook, Nature, 2010, 464, 571–574 CrossRef CAS PubMed.
- S. Sinthika, E. Mathan Kumar and R. Thapa, J. Mater. Chem. A, 2014, 32, 12812–12820 Search PubMed.
- V. Tozzini and V. Pellegrini, Phys. Chem. Chem. Phys., 2013, 15, 80–89 RSC.
- D. Jiang and Z. Chen, Graphene Chemistry Theoretical Perspectives, John Wiley & Sons Ltd., 1st edn, 2013, ch. 16, p. 382 Search PubMed.
- W. Lei, H. Zhang, Y. Wu, B. Zhang, D. Liu, S. Qin, Z. Liu, L. Liu, Y. Ma and Y. Chen, Nano Energy, 2014, 6, 219–224 CrossRef CAS PubMed.
- Y. Y. Sun, K. Lee, L. Wang, Y.-H. Kim, W. Chen, Z. Chen and S. B. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 073401 CrossRef.
- J. Wang, J. Li, S.-S. Li and Y. Liu, J. Appl. Phys., 2013, 114, 124309 CrossRef PubMed.
- Z. M. Ao, Q. Jiang, R. Q. Zhang, T. T. Tan and S. Li, J. Appl. Phys., 2009, 105, 074307 CrossRef PubMed.
- K. Ulman, D. Bhaumik, B. C. Wood and S. Narasimhan, J. Chem. Phys., 2014, 140, 174708 CrossRef PubMed.
- Q. Sun, Q. Wang, P. Jena and Y. Kawazoe, J. Am. Chem. Soc., 2005, 127, 14582–14583 CrossRef CAS PubMed.
- S.-H. Jhi, G. Kim and N. Park, J. Korean Phys. Soc., 2008, 52, 1217–1220 CrossRef CAS.
- S. Banerjee, C. G. S. Pillai and C. Majumder, Appl. Phys. Lett., 2013, 102, 073901 CrossRef PubMed.
- N. Song, Y. Wang, Q. Sun and Y. Jia, Appl. Surf. Sci., 2012, 263, 182–186 CrossRef CAS PubMed.
- J. Zhou, Q. Wang, Q. Sun, P. Jena and X. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2801–2806 CrossRef CAS PubMed.
- W. An, X. Wu, J. L. Yang and X. C. Zeng, J. Phys. Chem. C, 2007, 111, 14105–14112 CAS.
- M. Yoon, S. Yang, E. Wang and Z. Zhang, Nano Lett., 2007, 7, 2578–2583 CrossRef CAS PubMed.
- Q. Sun, P. Jena, Q. Wang and M. Marquez, J. Am. Chem. Soc., 2006, 128, 9741–9745 CrossRef CAS PubMed.
- H.-F. Wu, X. Fan, J.-L. Kuo and W.-Q. Deng, J. Phys. Chem. C, 2011, 115, 9241–9249 CAS.
- Y. Zhao and T. Gennett, Phys. Rev. Lett., 2014, 112, 076101 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: H2 molecule interaction with additional doped surfaces, and also the case of a higher doping percentage has been considered. Explanation of the possibility of a bond exchange mechanism using ammonia (NH3) and silane (SiH4) molecules as an external mediator. See DOI: 10.1039/c4ta04706k |
| This journal is © The Royal Society of Chemistry 2015 |