
Addressing the light-soaking issue in inverted organic solar cells using chemical bath deposited fluorinated TiOx electron transport layer†
Fang Jeng
Lim
ab,
Ying Ting
Set
b,
Ananthanarayanan
Krishnamoorthy
b,
Jianyong
Ouyang
bc,
Joachim
Luther
d and
Ghim Wei
Ho
*a
aDepartment of Electrical and Computer Engineering, National University of Singapore, Blk EA #06-10, 9 Engineering Drive 1, 117575 Singapore, Singapore. E-mail: elehgw@nus.edu.sg; Fax: +65 6775 4710; Tel: +65 6516 8121
bSolar Energy Research Institute of Singapore (SERIS), National University of Singapore, 7 Engineering Drive 1, 117574 Singapore, Singapore
cDepartment of Material Science and Engineering, National University of Singapore, Blk E4 #01-01, 9 Engineering Drive 1, 119260 Singapore, Singapore
dFraunhofer Institute for Solar Energy Systems, Heidenhofstr. 2, 79110 Freiburg, Germany
First published on 24th October 2014
Abstract
The device lifetime of an inverted organic solar cell (IOSC) is significantly better compared to standard-architecture OSC under ambient conditions. However, various studies have shown that when an n-type oxide is used as a selective electron transport layer (ETL) in the IOSC, a reversible light-soaking treatment is required. This reversible treatment largely hampers the practicality of the device, especially in outdoor applications, in which the light-soaking time may take hours every morning. In this work, fluorinated TiOx (F-TiOx), prepared by low-temperature solution-processed chemical bath deposition technique, was used as the ETL to significantly reduce the light-soaking time for a P3HT:PCBM based IOSC. Without affecting the device efficiency, more than ten-fold reduction in light-soaking time was observed for fluorinated TiOx (F-TiOx) when compared with conventional sol–gel TiOx. Ultraviolet photoelectron spectroscopy (UPS) and UV photoconductivity measurements were used to understand the light-soaking time reducing mechanism. From the perspective of ITO/TiOx interface, shift in work function was observed in F-TiOx due to the partial filling of its defective sites by fluorine atoms. Consequently, this process reduces its intrinsic trap state density compared to sol–gel TiOx even before the light-soaking treatment. As a result, the trap filling action can be completed in a shorter time upon illumination, and thus significantly reduce the duration of the necessary light-soaking.
1 Introduction
Impressive progress in the forms of superior air-stability and increasing device efficiency in the development of inverted organic solar cells (IOSCs) has recently been reported.1–4 However, there is an inherent issue, i.e., the necessity of a mandatory “light-soaking” treatment, in which the device needs to be exposed to light with UV components to improve the device characteristics.5 This phenomenon is commonly observed in IOSCs with n-type oxide semiconductors (TiOx, ZnO) as a selective electron transport layer (ETL). The as-prepared device initially shows s-shaped current density–voltage characteristics, a low short circuit current and low fill factor.5–8 After prolonged illumination (∼10 min under AM 1.5G irradiation), the device efficiency gradually improves and saturates at the highest value. The light-soaking behavior in the IOSC appears to originate from the necessary filling of the defective trap sites in the oxide semiconductor layer by the photoelectrons generated by the UV component (λ ≤ 400 nm) in the AM 1.5G illumination (1000 W m−2).5,9 Such trap filling action would consequently lower the work function of the ETL and improve its electron selectivity.5,10 It is important to note that the light-soaking effect is reversible.11–13 As a result, the device would have to be light soaked repeatedly at each light–dark cycle (every morning if used for outdoor applications), and this requirement largely hinders the device practicality. In laboratory testing (AM 1.5G, 1000 W m−2), typical IOSC devices require at least 10 min to complete the light-soaking;3,5,14 in outdoor applications, the same device would require at least an hour of light-soaking every morning. Therefore, it would be highly beneficial to significantly reduce the light-soaking time in inverted organic solar cells. Strontium- and barium-doped ZnO and Al-doped ZnO on TiOx (AZO–TiOx) have already been used as the ETL in attempts to address this issue with the IOSC.7,10We hereby report on the use of fluorinated TiOx (F-TiOx), deposited by chemical bath deposition (CBD), as a functionalized electron transport layer in the IOSC to address the light-soaking issue. This material has been applied extensively in various applications, such as electrochromic applications,15 the anti-corrosion of metal,16 photocatalytic applications17 and solar cells.18 The presence of fluorine in TiOx could significantly reduce its dangling bonds by passivation.15 In CdS and CdSe quantum dot solar cells (QDSC), F-TiOx was used to enhance cell efficiency.18 Chemical bath deposition (CBD) is a routinely employed coating technique to fabricate metal oxide thin films.19,20 It is a facile, scalable, low temperature, solution processed and low-cost deposition method. Oxide thin films can be deposited by immersing a substrate in a chemical bath of the precursor solutions.21 Furthermore, a straightforward thickness control in the range of 20 nm to several microns can be achieved.22–25 This technique is normally adopted for the fabrication of transparent conducting oxide (TCO)20 and active layer in thin film transistors.26 In solar cell applications, CBD has been used to fabricate well-controlled TiOx layers.22,27 This technique can also be utilised to deposit a uniform TiOx layer on larger TCO substrates with relative ease compared with the conventional spin coating technique. The incorporation of fluorine into TiOx can also be intrinsically performed by CBD onto TCO substrates.15,16 To the best of our knowledge, this material has not been previously used in OSC applications, and there has been no report on addressing the light-soaking issue in IOSC devices using a chemical bath deposited ETL.
In this work, we demonstrate the use of the as-deposited (CBD) fluorinated TiOx (F-TiOx) as the electron transport layer in a P3HT:PCBM based inverted organic solar cell. Without affecting the device efficiency, its light-soaking time was reduced by more than ten-fold compared to IOSCs with conventional sol–gel coated TiOx as electron transport layer. A model from the perspective of the ITO/TiOx interface was suggested to explain the possible cause of reduction in light-soaking time.
2 Results and discussions
The inverted organic solar cells with F-TiOx interlayer were fabricated as described in detail in the Experimental section. The device architecture was ITO/F-TiOx/P3HT:PCBM/PEDOT:PSS-CFS31/Ag, as shown in Fig. 1. The corresponding cross section SEM image of the device is attached to the schematics in the same figure. An IOSC device with architecture ITO/sol–gel TiOx/P3HT:PCBM/PEDOT:PSS-CFS31/Ag, which was reported earlier,3 was used as a control device in this work.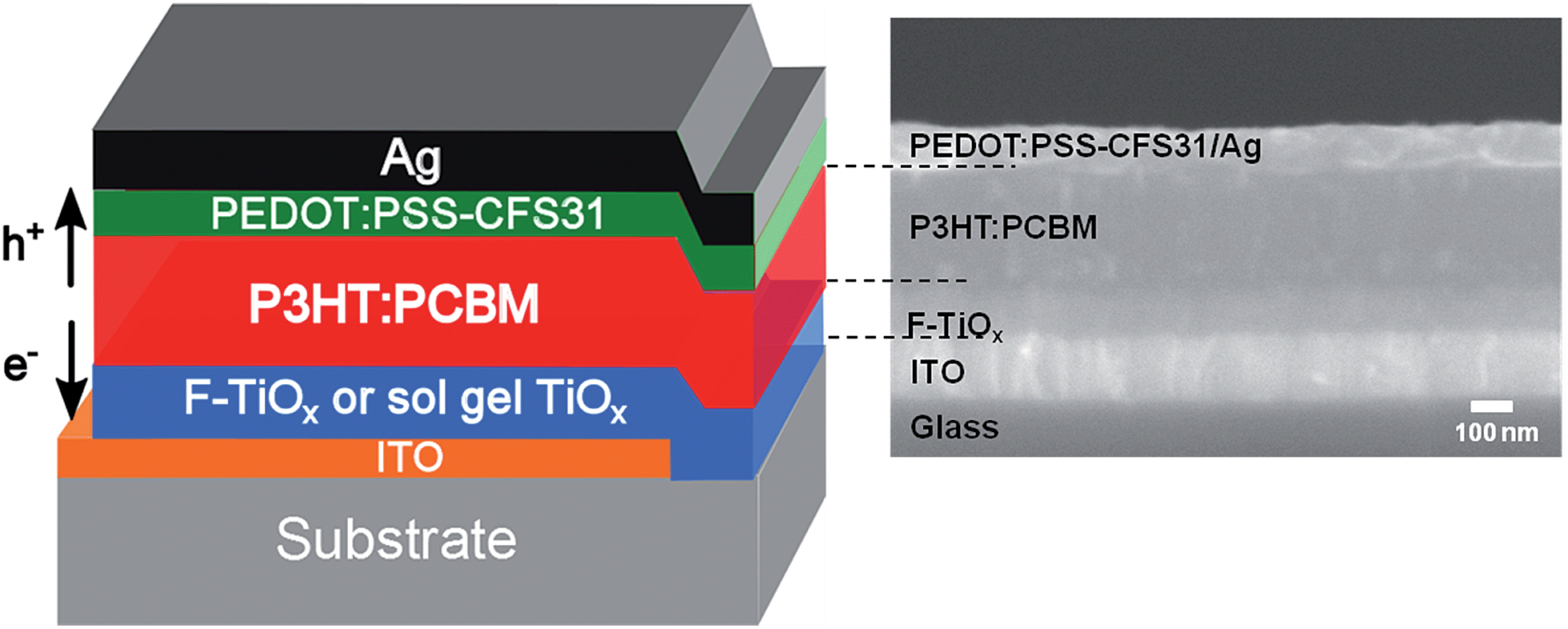 | ||
| Fig. 1 Illustration of the device architecture for inverted organic solar cells investigated in this study (left) and the corresponding cross section SEM image (right). | ||
Metal oxide electron transport layer characterization
The deposition reaction in CBD consists of hydrolysis equilibrium reaction of a metal–fluoro complex ((NH4)2TiF6) and the consumption reaction of F− from its ion scavenger (H3BO3). For the hydrolysis reaction, Deki et al. proposed the following reaction scheme:28| [TiF6]2− + nOH− → [TiF6−n(OH)n]2−n + nF− | (a) |
The addition of H3BO3 readily reacts with the F− ion produced in reaction (a) to form the more stable complex ion BF4−, as shown in reaction (b). According to the law of mass action, the equilibrium of reaction (a) will shift to the right due to the consumption of fluoride ions. According to their studies, Deki et al. have shown that apart from reacting with free fluoride ions, BO33− also reacts with the ions in the [TiF6−n(OH)n]2−. This reaction triggers the formation of [Ti(OH)6]2−, which forms TiOx after dehydration (reaction (c)).28 The nucleation of TiOx occurs in the chemical bath, causing the solution to become cloudy. Because of the environment of the chemical bath, fluorine ions readily adsorb onto the TiOx layers, thus forming fluorinated TiOx (F-TiOx).
| BO33− + 4F− + 6H+ ↔ BF4− + 3H2O | (b) |
 | (c) |
Because the deposition of F-TiOx as electron transport layer for organic solar cell applications was not previously reported, the optimal precursor concentration and the optimal F-TiOx thickness for maximum efficiency (η) were determined in this investigation. We determined that, in various concentrations of (NH4)2TiF6, the deposition rate can be varied from 35 nm h−1 to 65 nm h−1 (see ESI† for details). This variation in deposition rate was determined to affect the resulting F-TiOx film morphology.
The film morphology of the ETL is critical, as it determines whether a proper selective contact can be established with the adjacent photoactive layer. It was determined that at different (NH4)2TiF6 concentration regimes, islands, cracks or smooth TiOx film were observed. At a low concentration (0.02 M and 0.03 M, Fig. 2(a) and (b)) of (NH4)2TiF6, F-TiOx islands were formed and these islands could cause an increase in series resistance because of improper contact between the ETL with the photoactive layer. Thus, the presence of islands is detrimental for device performance. Cracks were formed in the F-TiOx when the deposition rate was relatively high (0.05 M (NH4)2TiF6), as clearly observed in Fig. 2(c). When the formation of TiOx accelerates, stress is introduced on the film and eventually forms cracks on the surface. These cracks form the recombination centers and deteriorate the electronic coupling at the TiOx/organic interface, increasing the loss due to recombination.29 Therefore, the presence of cracks is not beneficial for IOSCs. In the region of 0.1 M (NH4)2TiF6 and 0.2 M H3BO3, the morphology of the film is least defective, without the presence of islands and cracks at a thickness of 100 nm (Fig. 2(d)). The deposited F-TiOx film also has decent coverage over the ITO substrate. Such coverage would help in minimizing the likelihood of highly resistant and improper contact with the photoactive layer. Hence, by correlating the deposition rate and the F-TiOx film morphology, the precursor concentrations were set at 0.1 M (NH4)2TiF6 and 0.2 M H3BO3.
 | ||
| Fig. 2 SEM topographic images showing film morphology of F-TiOx on ITO. The samples were prepared with (a) 0.02 M, (b) 0.03 M, (c) 0.05 M, and (d) 0.1 M (NH4)2TiF6 at fixed 0.2 M H3BO3. The thickness of the reported F-TiOx layers are 100 nm, except for (a), for which the film growth stops at 80 nm. | ||
Fig. 3(a) and (b) show the summary of the thickness of the resulting F-TiOx film at various (NH4)2TiF6 concentration with 0.2 M H3BO3 and the deposition rate as a function of the two precursor concentrations. The red contour lines in Fig. 3(a) and (b) indicate the speculated optimal region for a relatively efficient IOSC. For validation, IOSC devices with F-TiOx deposited at various precursor concentrations were fabricated. The efficiency (η) and the fill factor of the devices under AM 1.5G illumination are shown in Fig. 3(c). The film grown with 0.05 M (NH4)2TiF6 expectedly gives a lower η (2.5%), because of a low fill factor (54%), indicating an improper contact between the ETL and the P3HT:PCBM layer because of the presence of islands and cracks. As the precursor concentration increases, the fill factor gradually increases and remains constant at 60%. The F-TiOx deposited at 0.1 M (NH4)2TiF6 gives the highest efficiency (3%), with a high fill factor (60%). At this concentration, the optimal F-TiOx thickness was determined to be 80 nm, which gives the highest η of 3% (Fig. 3(d)). Hence, the F-TiOx film with a thickness of 80 nm, deposited with 0.1 M (NH4)2TiF6 and 0.2 M H3BO3 was adopted for further studies in this work.
 | ||
Fig. 3 Color contour representation of (a) F-TiOx thickness over time at various (NH4)2TiF6 concentrations (fixed 0.2 M H3BO3). (b) Deposition rate of F-TiOx at various precursor concentrations. The red contour lines indicate the regions of desired thickness and film morphology for the F-TiOx ETL. (c) Efficiency ( ) and fill factor ( ) and fill factor ( ) of inverted organic solar cells with F-TiOx ETL deposited using various concentrations of (NH4)2TiF6 and 0.2 M H3BO3. (d) Efficiency at various F-TiOx thicknesses in inverted OSC device. All the data points were averaged for at least 5 reproducible devices. The respective error bars represent the standard deviation of each type of device. ) of inverted organic solar cells with F-TiOx ETL deposited using various concentrations of (NH4)2TiF6 and 0.2 M H3BO3. (d) Efficiency at various F-TiOx thicknesses in inverted OSC device. All the data points were averaged for at least 5 reproducible devices. The respective error bars represent the standard deviation of each type of device. | ||
It has been experimentally demonstrated that NaOH is able to remove fluorine atoms adsorbed onto a TiOx surface by replacing them with hydroxyl groups.30 This treatment can also convert the physically adsorbed F into Ti-bonded F.17 Wang et al. reported that if fluorinated TiOx is washed with NaOH and water, the concentration of Ti-bonded F can be increased.31 Thus, this method was also used in the present study to alter the fluorine content in the samples. X-ray photoelectron spectroscopy (XPS) was used to determine the fluorine atomic concentration on the TiOx surface. Fig. 4(a) shows the F 1s spectra for the sol–gel TiOx and F-TiOx subjected to various post treatments. It was determined that, in addition to the sol–gel TiOx, a peak at 688.6 eV was observed for all F-TiOx samples. This peak is attributed to Ti-bonded F in TiOx and not the adsorbed fluorine, for which a peak around 684.4 eV is generally observed.32 Elemental analysis shows that the as-prepared F-TiOx contains 1.3 at% of fluorine. As shown in Fig. 4(b), the amount of fluorine increases to 1.5 at% and 1.7 at% after the samples were subjected to NaOH and water post treatments, respectively. To validate the results, analysis of the Ti 2p and O 1s spectra (not shown in the figure) for the samples, to obtain the ratio of atomic concentrations between Ti–O and OH (hydroxyl) bonds, was carried out. The Ti–O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OH ratio of various samples in Fig. 4(b) shows that after the post treatments, the concentration of the OH group increases. This result correlates well with the increase in Ti-bonded F concentration, suggesting that the post treatments remove the surface F and replace them by OH groups, which is in line with earlier reports.31 Then, the removed fluorine can fill in the incomplete Ti–O–Ti matrix, which leads to increase in fluorine concentration in F-TiOx. For detailed studies in the next section, a device with sol–gel TiOx (0 at% fluorine) as ETL is used as the control.
:OH ratio of various samples in Fig. 4(b) shows that after the post treatments, the concentration of the OH group increases. This result correlates well with the increase in Ti-bonded F concentration, suggesting that the post treatments remove the surface F and replace them by OH groups, which is in line with earlier reports.31 Then, the removed fluorine can fill in the incomplete Ti–O–Ti matrix, which leads to increase in fluorine concentration in F-TiOx. For detailed studies in the next section, a device with sol–gel TiOx (0 at% fluorine) as ETL is used as the control.
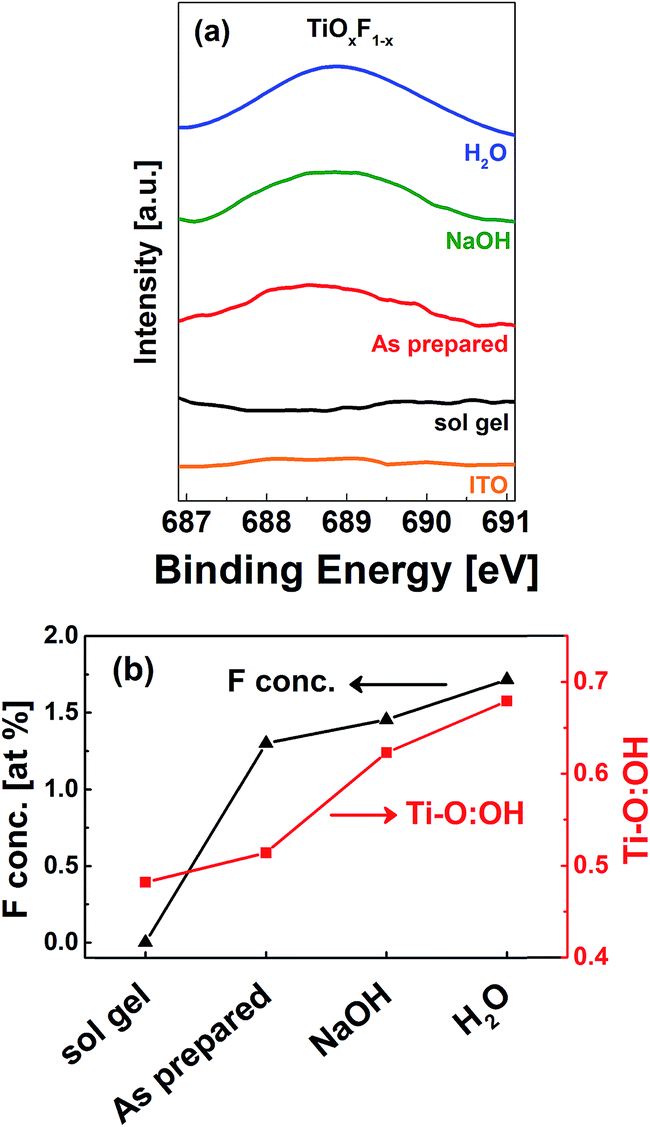 | ||
| Fig. 4 (a) F 1s photoelectron spectra for various TiOx samples on ITO substrate. The F 1s XPS spectra indicates the presence of adsorbed fluorine (∼688.6 eV) in all CBD F-TiOx devices. (b) Elemental analysis showing the atomic concentration of F and Ti–O:OH ratio on the sample surfaces corresponds to sol gel TiOx and the as-prepared, NaOH treated and H2O treated F-TiOx. | ||
Device performance
Table 1 shows the solar cell characteristics of devices with TiOx ETL at various fluorine atomic concentrations. While the open circuit voltages (Voc) of all the devices were similar, the short circuit current density (jsc) of the sol–gel TiOx, as-prepared F-TiOx and NaOH treated F-TiOx are similar (7.5 mA cm−2), while the H2O treated F-TiOx sample gives a slightly higher jsc (8 mA cm−2). However, this effect was traded off with a decrease in fill factor (62%) compared to the as-prepared F-TiOx. As a result, the cell efficiencies were not altered by the presence of fluorine in TiOx (3.0%).| Process | F at% | V oc [±17 mV] | j sc [±0.3 mA cm−2] | FF [±4%] | η [±0.3%] | τ soak [s] |
|---|---|---|---|---|---|---|
| Sol–gel | 0 | 628 | 7.5 | 62 | 2.9 | 450 |
| CBD (as-prepared) | 1.3 | 617 | 7.6 | 63 | 3.0 | 35 |
| CBD (NaOH treated) | 1.5 | 623 | 7.5 | 62 | 2.9 | 520 |
| CBD (H2O treated) | 1.7 | 615 | 8.0 | 62 | 3.0 | 110 |
However, the devices exhibited a significant difference when they were subjected to light-soaking, as shown in Fig. 5. Fig. 5(a) and (b) show the j–V characteristics for sol–gel TiOx and the as-prepared F-TiOx based IOSC devices, respectively, under AM 1.5G conditions (light-soaking treatments). S-shape characteristics were observed at the initial stage of illumination. The s-shape is a typical effect, which occurs due to the suppression in electron transport in TiOx, which results in a high resistance.33 It is noteworthy that the F-TiOx device reaches its maximum efficiency (50 s), considerably faster compared to the sol–gel TiOx device (350 s), after light-soaking. Fig. 5(c) shows the time evolution of device efficiency with various F atomic concentrations under AM 1.5G illumination in N2 atmosphere. The experimental data was fitted with a logistic function (see eqn (S1) and Table S2† for fitting function and parameters). By defining the light-soaking time (τsoak) as the time required for the efficiency to reach 95% of its maximum value (η(τsoak) = 0.95ηmax), the light-soaking effect for various devices was quantified, and are shown in Table 1. Devices with 0 at% fluorination (sol–gel TiOx) require approximately 450 seconds for the η to saturate, close to the values in previous reports.6,12 It is noteworthy that 450 s (8 min) of light soaking time in indoor laboratory conditions would require at least 1 h of light-soaking at outdoor conditions every morning (see Fig. S1† for rough calculations). For the as-prepared CBD F-TiOx devices (1.3 F at%), the η saturates in approximately 35 s of illumination. However, for NaOH treated F-TiOx (1.5 F at%), τsoak increases to approximately 520 s and H2O treated F-TiOx (1.7 F at%) shows 110 s for τsoak. All the post treated CBD F-TiOx samples, although having higher amounts of fluorine concentration, did not have shorter soaking time compared with 0 F at% TiOx. This result may suggest that OH groups on the TiOx surface could be the cause of trap formation, which leads to the necessity of light-soaking treatment. Nevertheless, the as-prepared F-TiOx, without further post treatments, can be used to address the light-soaking effect in inverted organic solar cells to significantly reduce the light-soaking time by a factor of 13 (450 s to 35 s) compared to devices with sol–gel TiOx ETL. As a result, a comparison of the as-prepared F-TiOx with sol–gel TiOx was performed in order to further understand the underlying reason for this light-soaking enhancement.
 | ||
| Fig. 5 Current density–voltage (j–V) characteristics of inverted organic solar cells with (a) sol–gel TiOx and (b) as prepared F-TiOx (1.3 F at%) as electron transport layer when subjected to light-soaking treatments, in which the illumination times are shown (guided by arrows). (c) Light-soaking effect on the efficiency of fresh devices with TiOx layers upon AM 1.5G illumination. The symbols represent experimental data, while the solid lines are fitted data by logistic function. The indicated time represents the corresponding light-soaking time (τsoak) for each sample, as defined in the main text. | ||
The device stability of the as-prepared IOSC with F-TiOx ETL was investigated by storing the sample in dark, at ambient conditions and monitoring its efficiency (η) over time, in accordance with the International Summit on Organic Photovoltaic Stability (ISOS) D-1 shelf scheme.34Fig. 6 shows the normalized open circuit voltage (Voc), short circuit current density (jsc), fill factor and η of the as-prepared CBD F-TiOx over a period of 1000 h, stored in a dark environment without encapsulation. The device shows superior stability over the non-encapsulated standard architecture OSC (inset), which was determined to be fully degraded in less than 24 h. The as-prepared F-TiOx device was able to retain 60% of its initial device performance even after 1000 h. The main cause of degradation is associated with the short circuit current density, as shown in Fig. 6 (red line). During the 70 h in presence of air, the device efficiency slightly increases compared to the freshly prepared cells. This initial increase was because of the increase in Voc. This phenomenon can be attributed to the oxidation of silver to silver oxide when stored at ambient condition. The work function is increased, which in turn increases the Voc.35,36jsc decrease may be due to the ingress of oxygen from the silver contact and moisture from the PEDOT:PSS to the photoactive layer, which may cause both chemical and physical changes in the photoactive layer.34 Overall, this result reveals that the incorporation of fluorine into TiOx has no detrimental effect on the photoactive layer, P3HT:PCBM, and the IOSC device performance.
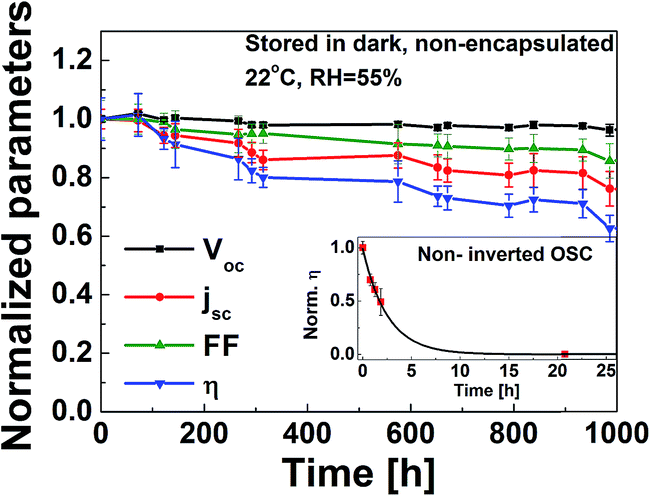 | ||
| Fig. 6 Normalized Voc, jsc, fill factor, η of ITO/CBD F-TiOx/P3HT:PCBM/PEDOT:PSS/Ag (without encapsulation) over 1000 h subjected to ISOS-D-1 scheme (stored in dark shelf, open circuit condition at 22 °C, R.H. 55%). The inset shows the degradation of the non-encapsulated standard architecture OSC (ITO/PEDOT:PSS/P3HT:PCBM/LiF/Al) under the same conditions. The error bar corresponds to the standard deviation of each parameter for 10 reproducible devices. | ||
Physical studies on the light-soaking characteristics
Several reports have suggested that the light-soaking behavior of an IOSC device with metal oxide ETL originates from at least two perspectives. Firstly, the light-soaking originates from the filling of trap states, which decreases the work function of the ETL and enables the electron extraction through the ITO/metal oxide interface.5 Secondly, the induced interfacial dipole between the metal oxide/organic interface may also determine the formation of an extraction barrier for the dissociated excitons in the device.10 In view of the new material (F-TiOx) proposed in this work, we could not rule out the effects by any one of them. Thus, we have conducted a control light-soaking experiment with various ETLs, such as sol–gel TiOx (80 nm), F-TiOx (80 nm), bilayer sol–gel TiOx (40 nm)/F-TiOx (40 nm) and F-TiOx (40 nm)/sol–gel TiOx (40 nm), to investigate which interface contributes to the light-soaking behavior (see Experimental section for film preparation). The results in Fig. 7 show that the F-TiOx based device has the shortest light-soaking time (∼30 s), followed by the sol–gel TiOx/F-TiOx (∼150 s), F-TiOx/sol gel TiOx (∼170 s) and sol–gel TiOx (∼450 s). This observation suggests that both ITO/TiOx and TiOx/organic interfaces contribute to the light-soaking behavior, as seen from the intermediate timings compared to that of pure sol–gel TiOx. In this work, we first examined the effect of trap-filling at the ITO/TiOx interface on the light-soaking behavior.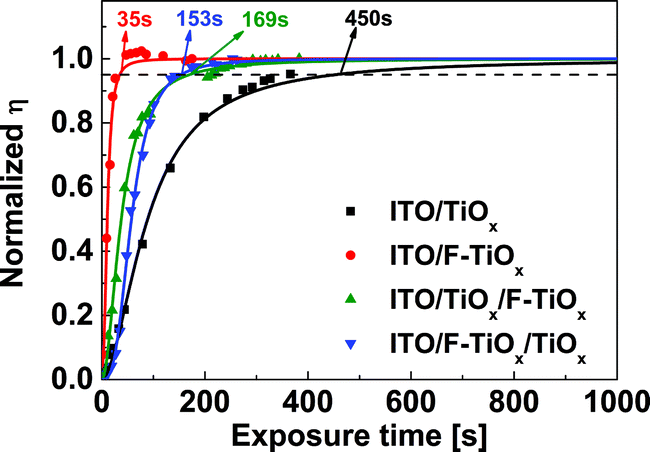 | ||
| Fig. 7 Light-soaking control experiment of fresh inverted organic solar cells with sol–gel TiOx (80 nm), F-TiOx (80 nm), sol–gel TiOx (40 nm)/F-TiOx (40 nm) and F-TiOx (40 nm)/sol–gel TiOx (40 nm) as electron transport layers upon AM 1.5G illumination. The symbols represent experimental data, while the solid lines are fitted data by logistic function. The indicated time represents the corresponding light-soaking time (τsoak) for each sample, as defined in the main text. | ||
The necessity of light-soaking seems to originate from the active unfilled electron traps caused by dangling bonds.5,7,10 These dangling bonds are susceptible to the adsorption of oxygen from air to form active trap sites, which could then be removed by UV photons.9 The presence of active traps affects the position of the quasi Fermi level for the electrons and the trap state density in the material. These two dominant parameters can be measured via ultraviolet photoelectron spectroscopy (UPS) and transient UV photoconductivity measurements, respectively. Through these techniques, the role of fluorine atoms on the light-soaking behavior of these amorphous TiOx thin films (see Fig. S3† for XRD spectra) was studied.
UPS technique was used to investigate the effect of fluorine atoms in TiOx on the light-soaking time from the energetics perspective. It was carried out by measuring the vacuum level shifts of sol–gel TiOx and F-TiOx films with respect to that of the ITO before light-soaking. Fig. 8(a) shows the region near the secondary electron cutoff and the emission onset for ITO, ITO/sol–gel TiOx (0 F at%) and ITO/F-TiOx (1.3–1.7 F at%) before light-soaking. A vacuum level shift (ΔEvac) of 0.41 eV between the ITO and sol–gel TiOx interface was observed. In the presence of fluorine, a total ΔEvac of 0.51 eV (0.41 + 0.10 eV) was observed. Therefore, F-TiOx has a lower work function (4.4 eV) than sol–gel TiOx (4.5 eV), as summarized in Fig. 8(b). Because all the films were subjected to the same heat treatment (180 °C), we can associate the change in work function to be largely dependent on the presence of fluorine incorporation into TiOx. These UPS measurements suggest a shift of Fermi level at the ITO/TiOx interface.
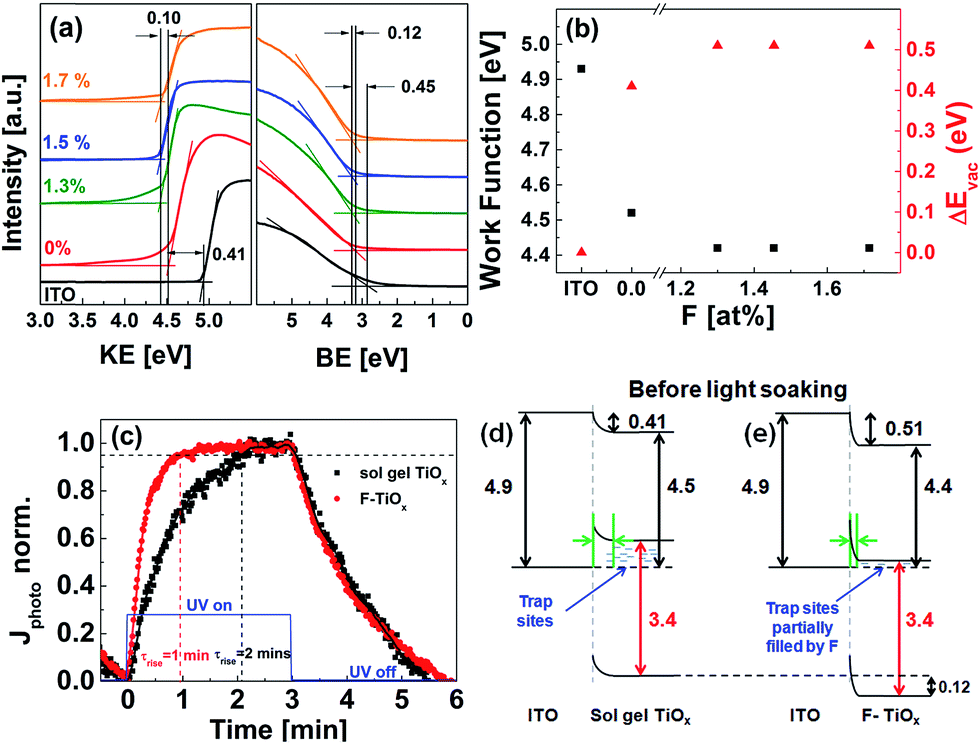 | ||
Fig. 8 (a) Ultraviolet photoelectron spectroscopy (UPS) secondary cut-off spectra of ITO and ITO/TiOx samples of various fluorine concentrations before light-soaking. The binding energy (BE) was set to zero at the Fermi level. (b) Work function values ( ) and vacuum level shift (ΔEvac, ) and vacuum level shift (ΔEvac,  ) at ITO/TiOx interface. (c) Normalized photocurrent of sol–gel TiOx (0 at%)/Ag ( ) at ITO/TiOx interface. (c) Normalized photocurrent of sol–gel TiOx (0 at%)/Ag ( ) and F-TiOx (1.3 F at%)/Ag ( ) and F-TiOx (1.3 F at%)/Ag ( ) under UV illumination. The blue line ( ) under UV illumination. The blue line ( ) indicates UV light ON/OFF state. (d and e) Resulting energy band diagrams at ITO/TiOx interface before light-soaking treatment. All values were obtained from UPS measurements, the band gaps were estimated by their respective absorption spectra (see Fig. S4†), and the green arrows at the ITO/TiOx interface represent the resulting barrier width due to Fermi level realignment. ) indicates UV light ON/OFF state. (d and e) Resulting energy band diagrams at ITO/TiOx interface before light-soaking treatment. All values were obtained from UPS measurements, the band gaps were estimated by their respective absorption spectra (see Fig. S4†), and the green arrows at the ITO/TiOx interface represent the resulting barrier width due to Fermi level realignment. | ||
TiOx is known to exhibit transient UV photoconductivity with long charging and relaxation times, due to the large amount of surface trap states.5,37 These trap states cause a decrease in the overall mobile carrier concentration of the material, and thus its conductivity. Transient UV photoconductivity measurements of sol–gel TiOx and F-TiOx (1.3 at%), before and after UV illumination, were carried out to investigate the existence of the traps and the filling characteristics of the traps. Fig. 8(c) shows the normalized photocurrent of sol–gel TiOx/Ag and the as-prepared CBD F-TiOx/Ag devices under UV illumination. At initial illumination, the trap states were gradually filled by photogenerated electrons in TiOx, increasing the overall mobile charge carrier concentration, and hence the photocurrent. For a detailed analysis, we define photocurrent rise time (τrise) as the time required for the photocurrent to reach 95% of its maximum value, indicating the time required for a complete trap filling. It is noteworthy that τrise of F-TiOx (1.3 at%) (1 minute) is twice as fast compared with that of sol–gel TiOx (2 min). Because the saturation of the photocurrent at constant illumination implies equilibrium between the trapping action and charge recombination,5 a shorter τrise would suggest that the material has a lower amount of trap states. Hence, we may conclude that the trap state density in the F-TiOx (1.3 at%) layer is considerably lower than that in sol–gel TiOx.
By combining the findings of the UPS spectra and the UV photoconductivity response, we suggest an explanation for the improvement in the light-soaking characteristics of the device with fluorinated TiOx electron transport layer from the perspective of the ITO/TiOx interface. Fig. 8(d) and (e) show the resulting energy level diagrams of the interfaces for sol–gel TiOx and F-TiOx before light-soaking. In sol–gel TiOx, due to the lower ΔEvac (0.41 eV) and higher work function (4.5 eV), the Fermi level (EF) is farther away from the conduction band. Because there are more trap states in sol–gel TiOx, the shift of EF suggests that the material has a lower overall carrier concentration due to these unfilled trap states. This phenomenon would influence its barrier width at the ITO/TiOx interface, which is related to Debye length (LD), a characteristic length scale for charge screening.10LD scales with the dielectric constant (ε) and the carrier concentration (n) (LD ∝ (ε/n)0.5). A lower carrier concentration would result in a large LD, and thus forms a larger barrier width (green arrow) between ITO and sol–gel TiOx (Fig. 8(d)).
In F-TiOx, due to the higher ΔEvac (0.51 eV) and the lower work function (4.4 eV), with an observable shift of 0.12 eV of the valence band to higher binding energy (Fig. 8(a)), EF shifts closer to the conduction band compared with that of sol–gel TiOx. Because the trap state density of F-TiOx is lower compared to sol–gel TiOx, the shift in EF suggests an increase in overall carrier concentration, probably due to partial filling of the electron trap sites by fluorine atoms in the TiOx. The increased carrier concentration decreases the LD and narrows the barrier width (green arrow) between ITO and F-TiOx (Fig. 8(e)). This result also corroborates with a recent study that shows a significant increase in a carrier concentration of TiO2 after doping with fluorine.38 It is noteworthy that all these phenomena are observed at the ITO/TiOx interface before light-soaking treatment.
When the device is illuminated, the ΔEvac of both TiOx and F-TiOx samples increases, and it is highly possible that they will have similar values because of their identical cell efficiencies (3.0%). As a result, their ΔEvac values would converge towards about 0.6 eV after light-soaking, as observed by Kim et al.5 Given that the initial ΔEvac value of F-TiOx (0.5 eV) is nearer to that of the light-soaked condition (0.6 eV), the time taken to fully fill the trap sites in F-TiOx is significantly shorter compared to sol–gel TiOx, as observed in Fig. 5(c). Hence, these findings provide a possible explanation for the significant reduction in the light-soaking time in F-TiOx inverted organic solar cells.
Given the same reduced work function, the NaOH (1.5 at%) and H2O (1.7 at%) treated F-TiOx samples show inferior light-soaking effect. This phenomenon leads to the idea that the ITO/TiOx interface may not be the only factor in influencing the light-soaking effect. The modification of the TiOx surface by these treatments suggests that there is an induced dipole at the TiOx/organic interface, which may also play a certain role in the light-soaking effect. An extended study from this perspective will be performed in future.
3 Conclusion
The application of fluorinated TiOx (F-TiOx) as the electron transport layer (ETL) was used to address the light-soaking issue in inverted organic solar cells (IOSC). The ETL was fabricated by a solution processed chemical bath deposition method. The presence of fluorine in TiOx seems to reduce the trap state density in the ETL by partially filling the states and causing a decrease in its work function before light-soaking is carried out. The effect significantly shortens the time required for trap filling when the device is illuminated. Therefore, the use of F-TiOx, without further post treatments, can significantly decrease the necessary light-soaking time by a factor of 13 (from 450 s to 35 s) in an air-stable inverted organic solar cell (IOSC) compared with the time for its sol–gel TiOx counterpart without affecting device efficiency. This material can significantly increase the practicality of an air-stable, efficient and solution-processed inverted organic solar cell.4 Experimental
Materials
Titanium isopropoxide (TTIP, 97%, Sigma-Aldrich), acetylacetone (AA, Sigma-Aldrich) and isopropanol (IPA, reagent grade, Aik Moh Paints & Chemical Pte Ltd.) were used to prepare the sol–gel TiOx electron transport layer. For the chemical bath deposited F-TiOx, ammonium hexafluorotitanate ((NH4)2TiF6, 99.99%, Sigma-Aldrich) and boric acid (H3BO3, ≥99.99%, Sigma-Aldrich) were used. Regioregular (>98%) poly(3-hexylthiophene) (P3HT, product code M101, average Mw 65500, Ossila), phenyl-C61-butyric acid methyl ester (PC61BM – 99.5% purity, product code Nano-CPCBM-BF, Nano-C) and poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS – Heraeus Clevios P VP AI 4083, product code M121, Ossila) were used as donor, acceptor and hole transport layer, respectively. 1,2-Dichlorobenzene (DCB, Sigma-Aldrich) was used as the solvent for donor and acceptor. Capstone® FS-31 (Dupont) was added into PEDOT:PSS prior to spin coating. Silver (Ag) metal was purchased from K. J. Kurt Lesker & Co. All the abovementioned materials were used as received.
Solution preparation
The sol–gel TiOx precursor solution was prepared by adding an appropriate amount of acetylacetone dropwise to TTIP under continuous stirring. Then, isopropanol was added to the mixture as solvent. The volume ratio of TTIP:AA:IPA was set to 1:0.5:10. Then, the solution was stirred for at least 2 h prior to spin coating onto the substrate. The as-prepared solution exhibits an orange-yellow color, indicating the d–d absorption originating from the titanium complex ion formed by the reaction between titanium isopropoxide and the stabilizer acetylacetone.39 The precursor solutions for the F-TiOx films were prepared by mixing the appropriate concentrations of ammonium hexafluorotitanate ((NH4)2TiF6, 99.99%, Aldrich) and boric acid (H3BO3). Both the solutions were separately stirred for at least 10 min at room temperature before mixing. The stirred solutions were mixed and placed in a pre-heated bath with temperature at 40 °C before immersing ITO substrates into the bath for F-TiOx deposition. P3HT:PCBM blend was made in the ratio 1:0.8 (the concentration of P3HT was 15 mg ml−1, whereas PCBM was 12 mg ml−1) in 1 ml of dichlorobenzene (DCB). The mixed solution was stirred and heated at 60 °C overnight in a N2 filled glove box prior to spin coating.
Device fabrication
Fluorinated TiOx (F-TiOx) was prepared via chemical bath deposition. The F-TiOx film was deposited by immersing the ITO substrate into a chemical bath comprised of 0.1 M ammonium hexafluorotitanate ((NH4)2TiF6) and 0.2 M boric acid (H3BO3) at 40 °C for 90 min to obtain a film thickness of 80 nm. To control the fluorine concentration, the F-TiOx samples were immersed in NaOH or ultrasonicated in de-ionised water for 30 min. Then, the films were rinsed with DI water after each treatment. Finally, the films were subjected to annealing at 180 °C for 1 h before proceeding to device fabrication. Inverted bulk heterojunction organic solar cells based on P3HT:PCBM were prepared on pre-patterned tin-doped indium oxide (ITO, Xinyan Technology Ltd.) coated glass substrates. The sheet resistance of the ITO (thickness: 90 ± 10 nm) was determined to be around 15–20 Ω □−1. The substrates were pre-cleaned using a detergent solution, followed by successive ultrasonication in de-ionized water, isopropanol and acetone for 15 min each. Then, the substrates were dried in an oven at 60 °C for two hours. Then, 80 nm of fluorinated TiOx (F-TiOx) was deposited onto ITO via chemical bath deposition. For sol–gel TiOx samples, the sol–gel precursor was spun on top of ITO substrates and hydrolyzed in a controlled environment (relative humidity 40%) for 2 h. All TiOx films were annealed at 180 °C for 1 h in air before transferring to a N2-filled glove box. For sol–gel TiOx/F-TiOx samples, the 40 nm of sol–gel precursor was first coated on ITO, hydrolyzed for 2 h and annealed at 180 °C for 1 h in air. After that, 40 nm of F-TiOx was chemical bath deposited onto the sol–gel TiOx in the abovementioned chemical bath and annealed at 180 °C for 1 h before transferring to a N2-filled glove box. For F-TiOx/sol–gel TiOx samples, the 40 nm of F-TiOx was chemical bath deposited onto the sol–gel TiOx in the abovementioned chemical bath and annealed at 180 °C for 1 h. After that, 40 nm of sol–gel precursor was first coated on ITO, hydrolyzed for 2 h and annealed at 180 °C for 1 h before transferring to a N2-filled glove box. P3HT:PCBM blend was spun onto the electron transport layer at 800 rpm for 30 s, and the resultant wet film was annealed on a hot plate at 140 °C for 1 min. The thickness of the photoactive layer was approximately 200 nm. Then, 70 nm of PEDOT:PSS blended with CFS-31 (PEDOT:PSS-CFS-31) with volume ratio of 5.5% was coated onto the active layer in air through a 0.45 μm cellulose filter and annealed at 140 °C for 1 min in the N2 filled glove box. Then, Ag metal (100 nm) was thermally evaporated by a pre-designed shadow mask, resulting in an active device with an area of 9 mm2. The vacuum condition during the evaporation was around 4 × 10−6 mbar. The completed device architecture is ITO/TiOx/P3HT:PCBM/PEDOT:CFS-31/Ag. Cell degradation study was carried out by storing the device in a dark cabinet in open circuit conditions at 22 °C and relative humidity of 55%, in accordance with the ISOS-D-1 shelf scheme.34Material characterization
The current density–voltage (j–V) measurements were obtained under 1 sun illumination (ABET technologies Sun 2000 Solar Simulator) and a Keithley 2400 sourcemeter. The light-soaking treatment was done using the same lamp source, in which the devices were continuously illuminated and data were periodically acquired. The thicknesses of all layers were measured using Veeco DEKTAK 150 Stylus Profilometer. The electron microscopic images were obtained using JEOL FEG JSM 6700F field emission SEM operating at 15 keV. All the fabrication steps, except coating of TiOx and PEDOT:PSS, were performed inside a glove box of N2 atmosphere (Charslton Technologies, ≤1 ppm moisture and O2). X-ray photoelectron spectroscopy (XPS) measurements were performed using monochromatized Al-Kα at 1486.71 eV on a Kratos AXIS UltraDLD system. All the spectra were calibrated with respect to the C 1s spectrum (C–C bond) at 284.5 eV, and the peaks were fitted using Voigt (Gaussian and Lorentzian) function. Ultraviolet photoelectron spectroscopy (UPS) was performed with the same equipment, but with a He I (21.22 eV) radiation (VG Scientific ESCALab Mark 2) and applied bias of 5 V to separate the signals from samples and detector. The binding energies were set zero at the Fermi level. UV photocurrent response measurements were carried out using a Zahner impedance analyzer, equipped with a calibrated 365 nm UV light emitting diode (LED) source in ambient conditions. The light intensity of the LED, which was regulated by a photosensor in a control loop, was set at 50 W m−2. The UV light source was time-programmed accordingly for periodic power on and off. TiOx/Ag with two parallel Ag contacts were used as the measurement devices. They were biased at 0.01 V in all measurements.Acknowledgements
SERIS is sponsored by the National University of Singapore (NUS) and Singapore's National Research Foundation (NRF) through the Singapore Economic Development Board (EDB). Fang Jeng Lim would also like to express his sincere gratitude to Singapore National Research Foundation (Energy Innovation Programme Office) for providing a PhD scholarship. We gratefully acknowledge Dupont™ through MegaChem Limited, Singapore for supplying Capstone® FS-31.References
- A. K. K. Kyaw, D. H. Wang, V. Gupta, J. Zhang, S. Chand, G. C. Bazan and A. J. Heeger, Adv. Mater., 2013, 25(17), 2397–2402 CrossRef CAS PubMed.
- M. Song, J. W. Kang, D. H. Kim, J. D. Kwon, S. G. Park, S. Nam, S. Jo, S. Y. Ryu and C. S. Kim, Appl. Phys. Lett., 2013, 102(14), 143303 CrossRef PubMed.
- F. J. Lim, A. Krishnamoorthy, J. Luther and G. W. Ho, J. Mater. Chem., 2012, 22(48), 25057–25064 RSC.
- S. Woo, W. H. Kim, H. Kim, Y. Yi, H. K. Lyu and Y. Kim, Adv. Energy Mater., 2014, 4, 1301692 Search PubMed.
- J. Kim, G. Kim, Y. Choi, J. Lee, S. H. Park and K. Lee, J. Appl. Phys., 2012, 111(11), 114511 CrossRef PubMed.
- T. Kuwabara, C. Iwata, T. Yamaguchi and K. Takahashi, ACS Appl. Mater. Interfaces, 2010, 2(8), 2254–2260 CAS.
- O. Pachoumi, C. Li, Y. Vaynzof, K. K. Banger and H. Sirringhaus, Adv. Energy Mater., 2013, 3(11), 1428–1436 CrossRef CAS.
- W. Wei, C. Zhang, D. Chen, Z. Wang, C. Zhu, J. Zhang, X. Lu and Y. Hao, ACS Appl. Mater. Interfaces, 2013, 5(24), 13318–13324 CAS.
- F. C. Krebs, T. Tromholt and M. Jørgensen, Nanoscale, 2010, 2(6), 873–886 RSC.
- S. Trost, K. Zilberberg, A. Behrendt, A. Polywka, P. Görrn, P. Reckers, J. Maibach, T. Mayer and T. Riedl, Adv. Energy Mater., 2013, 3(11), 1437–1444 CrossRef CAS.
- M. R. Lilliedal, A. J. Medford, M. V. Madsen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2010, 94(12), 2018–2031 CrossRef CAS PubMed.
- Z. Lin and J. Zhang, ACS Appl. Mater. Interfaces, 2013, 5(3), 713–718 CAS.
- H. Schmidt, K. Zilberberg, S. Schmale, H. Flügge, T. Riedl and W. Kowalsky, Appl. Phys. Lett., 2010, 96(24), 243305 CrossRef PubMed.
- J. C. Wang, C. Y. Lu, J. L. Hsu, M. K. Lee, Y. R. Hong, T. P. Perng, S. F. Horng and H. F. Meng, J. Mater. Chem., 2011, 21(15), 5723–5728 RSC.
- M. K. Lee, C. H. Fan, H. C. Wang and C. Y. Chang, J. Electrochem. Soc., 2011, 158(8), D511–D514 CrossRef CAS PubMed.
- C. Lei, H. Zhou, Z. Feng, Y. Zhu and R. Du, J. Alloys Compd., 2012, 513(5), 552–558 CrossRef CAS PubMed.
- Y. Wang, H. Zhang, P. Liu, T. Sun, Y. Li, H. Yang, X. Yao and H. Zhao, J. Mater. Chem. A, 2013, 1(41), 12948–12953 CAS.
- M. Samadpour and J. Bisquert, J. Phys. Chem. C, 2011, 115(29), 14400–14407 CAS.
- S. Pawar, B. Pawar, J. Kim, O. S. Joo and C. Lokhande, Curr. Appl. Phys., 2011, 11(2), 117–161 CrossRef PubMed.
- H. Khallaf, C. T. Chen, L. B. Chang, O. Lupan, A. Dutta, H. Heinrich, F. Haque, E. D. Barco and L. Chow, Appl. Surf. Sci., 2012, 258(16), 6069–6074 CrossRef CAS PubMed.
- C. Lokhande, Mater. Chem. Phys., 1991, 27(1), 1–43 CrossRef CAS.
- T. Kuwabara, H. Sugiyama, M. Kuzuba, T. Yamaguchi and K. Takahashi, Org. Electron., 2010, 11(6), 1136–1140 CrossRef CAS PubMed.
- Y. Masuda and K. Kato, Thin Solid Films, 2008, 516(9), 2547–2552 CrossRef CAS PubMed.
- S. Supothina and M. De Guire, Thin Solid Films, 2000, 371(1), 1–9 CrossRef CAS.
- A. More, T. Gujar, J. Gunjakar, C. Lokhande and O. S. Joo, Appl. Surf. Sci., 2008, 255(5), 2682–2687 CrossRef CAS PubMed.
- H. C. Cheng, C. F. Chen and C. C. Lee, Thin Solid Films, 2006, 498(1–2), 142–145 CrossRef CAS PubMed.
- H. Han, P. Sudhagar, T. Song, Y. Jeon, I. Mora-Sero, F. Fabregat-Santiago, J. Bisquert, Y. S. Kang and U. Paik, Phys. Chem. Chem. Phys., 2013, 49(27), 2810–2812 CAS.
- S. Deki, Y. Aoi, Y. Asaoka, A. Kajinami and M. Mizuhata, J. Mater. Chem., 1997, 733–736 RSC.
- L. M. Chen, Z. Xu, Z. Hong and Y. Yang, J. Mater. Chem., 2010, 20, 2575–2598 RSC.
- S. Liu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132(34), 11914–11916 CrossRef CAS PubMed.
- Q. Wang, W. Ma and J. Zhao, Langmuir, 2008, 24(14), 7338–7345 CrossRef CAS PubMed.
- K. Lv, B. Cheng, J. Yu and G. Liu, Phys. Chem. Chem. Phys., 2012, 14(16), 5349–5362 RSC.
- B. Ecker, H. J. Egelhaaf, R. Steim, J. Parisi and E. V. Hauff, J. Phys. Chem. C, 2012, 116(31), 16333–16337 CAS.
- M. Reese, S. Gevorgyan, M. Jørgensen, E. Bundgaard, S. Kurtz, D. Ginley, D. Olson, M. Lloyd, P. Morvillo and E. Katz, Sol. Energy Mater. Sol. Cells, 2011, 95(5), 1253–1267 CrossRef CAS PubMed.
- M. T. Lloyd, D. C. Olson, P. Lu, E. Fang, D. L. Moore, M. S. White, M. O. Reese, D. S. Ginley and J. W. P. Hsu, Phys. Chem. Chem. Phys., 2009, 19(41), 7638–7642 CAS.
- J. Kim, C. Kim, Y. Kim and Y. Loo, Appl. Phys. Lett., 2009, 95, 183301 CrossRef PubMed.
- K. Pomoni, A. Vomvas and C. Trapalis, Thin Solid Films, 2005, 479(1–2), 160–165 CrossRef CAS PubMed.
- W. Q. Fang, X. L. Wang, H. Zhang, Y. Jia, Z. Huo, Z. Li, H. Zhao, H. G. Yang and X. Yao, J. Mater. Chem. A, 2014, 2(10), 3513–3520 CAS.
- T. Kuwabara, T. Nakayama, K. Uozumi, T. Yamaguchi and K. Takahashi, Sol. Energy Mater. Sol. Cells, 2008, 92(11), 1476–1482 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta05042h |
| This journal is © The Royal Society of Chemistry 2015 |