
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable transition from hydration to monomer-supported lubrication in zwitterionic monolayers revealed by molecular dynamics simulation†
Christoph
Klein
ab,
Christopher R.
Iacovella
ab,
Clare
McCabe
abc and
Peter T.
Cummings
*ab
aDepartment of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, Tennessee 37235, USA. E-mail: peter.cummings@vanderbilt.edu
bVanderbilt Facility for Multiscale Modeling and Simulation (MuMS), Vanderbilt University, Nashville, Tennessee 37235, USA
cDepartment of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, USA
First published on 9th March 2015
Abstract
The tribology of 2-methacryloyloxyethyl phosphorylcholine monolayers in water is studied using molecular dynamics simulations. Our results show two distinct shear regimes where the first is dominated by hydration lubrication, exhibiting near zero friction coefficients, and the second by chain–chain interactions, resembling monomer-supported lubrication. These results provide insight into the hydration lubrication mechanism – a phenomena thought to underlie the extremely efficient lubrication provided by surfaces functionalized with polyzwitterionic polymer brushes and the mammalian synovial joint.
1 Introduction
Natural, mammalian synovial joints can sustain friction coefficients in the range of 0.001 (ref. 1) to 0.1 (ref. 2) while subject to physiological pressures. Surfaces functionalized with zwitterionic polymer brushes mimic aspects of a joint's composition3 and can yield friction coefficients as low or lower than those found in natural synovial joints.4–9 Such materials resist compression up to normal loads of several MPa while maintaining friction coefficients orders of magnitude lower than comparable neutral polymers in non-polar10 and aqueous solvents11 as well as non-grafted charged polymers in aqueous solution.12 Kyomoto et al. compared polymer brushes comprised of different neutral, charged and zwitterionic monomers and found that those grown from 2-methacryloyloxyethyl phosphorylcholine (MPC) exhibited the lowest coefficients of friction while enduring negligible wear over millions of cycles8 and proposed that these brushes could improve the performance of artificial hips.13 The mechanism by which such materials are thought to provide ultra-low friction coefficients has been termed “hydration lubrication”.14–17 Qualitatively, water is hypothesized to facilitate this mechanism by rapidly relaxing between hydration shells surrounding charged groups while simultaneously supporting high normal loads due to the tightly bound nature of these hydration shells. The rapid relaxation of water molecules allows systems to exhibit a fluid shear response. However, the precise nature of this mechanism and hence the validity of the hypothesized mechanism has yet to be determined.17 Additionally, the hydration lubrication mechanism itself presents but one pathway for dissipation of frictional forces in complex systems like zwitterionic polymer brushes.5,11,18 Molecular dynamics simulation, as used herein, provides a promising avenue for studying this mechanism and its role in more complex systems as it grants direct insight into the dynamic motion of charged groups and water molecules during shearing.Previous studies have demonstrated the utility of molecular simulation in elucidating tribological mechanisms19–23 but have not specifically focused on hydration lubrication. Other simulation studies have investigated both neutral24 and zwitterionic25–27 monolayer systems in aqueous environments. However, these simulation studies considered densely populated monolayers that do not compare to the environment existing within the experimentally studied brush systems thought to exhibit hydration lubrication. Moreover the foci of the studies considering zwitterionic monolayers were not centered directly on the behavior within zwitterionic brush films but rather on dense phospholipid and carboxybetaine coated surfaces and their interactions with solvated ions.
In this work, atomistic molecular dynamics simulations are used to study the frictional properties of sparse, silane-functionalized MPC (Fig. 1A) monolayers in an aqueous environment. More specifically, the aim is not to directly model the behavior of pMPC brush films but rather gain a deeper understanding of the molecular level environment surrounding individual monomers found in the brushes. Additionally, the monolayers studied herein differ from previously studied monolayers in two primary ways. First, the zwitterionic nature of the monomers clearly distinguishes them from neutral monolayers such as alkylsilanes. Second, in contrast to previously studied densely packed monolayers (both neutral and zwitterionic), our sparse monolayers are designed to mimic the aqueous environment found in experimentally studied pMPC brushes where water can interpenetrate the polymer film. Clearly the experimental brush systems are more complex due to the network of chain–chain interactions but the goal of this study is to clearly capture the MPC–water interactions, largely eliminating the chain–chain interactions to establish a baseline behavior.
 | ||
| Fig. 1 (A) Silane functionalized MPC monomer. (B) Schematic slice of solvated, unequilibrated systems. Silica substrates shown in red/yellow; chains in black/white; water in blue. Plus/minus symbols denote approximate locations of charged regions. The non-periodic z-dimension is defined as normal to the substrates; the x- and y-dimensions are both periodic and shearing is performed in the x-dimension. | ||
2 Methods
Monolayers were created on 4.1 × 4.7 nm (19.7 nm2) β-cristobalite silica substrates which provide an atomically smooth surface with well defined binding sites to enable examination of tribological phenomena under idealized conditions. Previous work has shown that this system size is sufficiently large to avoid any size effects related to periodic boundary conditions during tribological simulations.28 Silane-functionalized MPC monomers were randomly attached to oxygen atoms with open valencies on the silica surfaces via the monomers' terminal silane groups. Surface oxygen atoms not bound to a monomer were terminated as hydroxyl groups.Two systems were prepared with average chain surface densities of 1.02 and 2.03 chains per nm2 which corresponds to monomers attached to 20 and 40% of the under-coordinated surface oxygens, respectively. The chain arrangement for both systems is shown in the ESI (Fig. S1†) where opposing substrates were mirrored. The sparser chain density (1.02 chains per nm2) was chosen because monomers in the experimentally studied polymer brush systems are likely to possess higher mobility than would be granted by a densely packed monolayer on a flat substrate. The denser system (2.03 chains per nm2) compares to chain densities found in previous simulation studies of zwitterionic monolayers (1.98 chains per nm2)25 and thus provides a reference point. The previously studied monolayers, however, consisted of longer, more linear monomers than MPC and were formed in regular lattice arrangements that did not allow for water to penetrate into the monolayer. Additionally, observing similar results across two chain densities allows us to rule out anomalous behavior arising from one particular system. Both systems were then solvated in TIP3P water to create a density of ∼1.0 g cm−3 in the interstitial water layer post-compression, at the largest separation distances where friction simulations were performed. The separation distance, D, between opposing monolayers is defined as the distance between the outermost layers of silicon atoms in each substrate. A schematic overview of the system setup is given in Fig. 1B. The number of water molecules remains constant for all separation distances thus representing the edge case where compression happens fast enough to prevent significant squeeze out of water molecules during the elapsed simulation time. This model makes sense for a closed system where squeeze out occurs on a much longer timescale than the simulations performed here; e.g., we are considering a nanoscale segment of what would typically be a milli- to centimeter scale substrate where water may not be able to rapidly escape from the highly absorbent zwitterionic films. The other edge case, not considered in this work, would consider complete equilibration with bulk water outside of the pore at each separation distance.
Simulations were performed in LAMMPS29 using OPLS-aa parameters for both the MPC monomers30 and the substrate.31 To ensure charge neutrality of the system, partial charges were adjusted from their values for free chains, which by construction are essentially charge neutral; these adjusted charges were localized to the region around the attachment site to avoid significantly altering more relevant components of the original model. The partial charges used in this work are summarized in Table S1.† The rRESPA multi-timestep integrator32 was applied to the MPC chains and water molecules with timesteps of 0.25 fs for bonds, 0.5 fs for angles/dihedrals and 1 fs for non-bonded interactions. Periodic boundary conditions were applied in the x- and y-directions to simulate an infinite surface. Electrostatic interactions were modeled using the 2D particle–particle particle–mesh algorithm33 where the real space component is calculated within a 1 nm radius. Lennard–Jones interactions were calculated with a 1 nm cutoff radius. The temperature of the water and chain molecules was maintained at 300 K using the Nosé–Hoover thermostat.34,35
Two distinct sets of simulations were performed. First, compression runs were carried out by fixing the bottom substrate in space and displacing the top substrate down at 0.001 nm ps−1. Second, using configurations obtained from the compression runs as input, systems with fixed separation were examined under shear. Shearing simulations were performed at separation distances of 4.1–2.8 nm and 3.8–2.5 nm in increments of 0.05 nm for chain densities of 2.03 and 1.02 chains per nm2, respectively, for a total of 54 shearing simulations. Shear stresses were induced by displacing the top substrate at 10 m s−1 in the x-direction. From each independent shearing simulation, 15–30 ns of post-equilibration data were collected; systems with higher separation distances were sampled for longer periods of time due to larger fluctuations in shear forces. Normal and shear forces were calculated by summing the forces experienced by the bottom substrate plus chains in the z- and x-directions, respectively. These forces were sampled every 1 ps and block averaged across each simulation to yield the data points and standard errors shown in Fig. 4.
Although shearing speeds of 10 m s−1 are greater than those typically studied experimentally, they are frequently encountered in practical applications. For example, micro- and nanoelectromechanical devices routinely operate at speeds on the order of 10–20 m s−1 and natural joints can experience shearing on the order of 1 m s−1, e.g., when a kicking motion is made where angular velocities can reach over 20 rad s−1.36 Considering artificial joints or cartilage scaffolds as real world applications for materials lubricated via the mechanisms studied here, speeds experienced in knee joints are of obvious interest. Additionally, both experimental5 and simulation studies28 of shearing, functionalized surfaces show little dependence on shearing velocities across several orders of magnitude. This is not to suggest that there is no dependence but that the effects are minimal within the, respectively, accessible ranges of shearing speeds for experimental and simulation studies.
3 Results and discussion
3.1 Effects of compression
In Fig. 2 we present the normal force response for the aqueous MPC system as it is compressed for both surface densities of 1.02 and 2.03 chains per nm2. Both systems show nearly identical trends, although the less dense system (1.02 chains per nm2) appears to have its force curve shifted to smaller separations (∼0.22 nm lower) and a lower normal force, roughly proportional to surface coverage. This phenomenon is highlighted in the inset of Fig. 2 where the base line normal forces are shifted to zero and the separation distance of the denser system is shifted by 0.22 nm to show the overlap. The general trend shows that as separation distances between the substrates decrease, normal forces increase. However, there are noticeable deviations from the overall trend.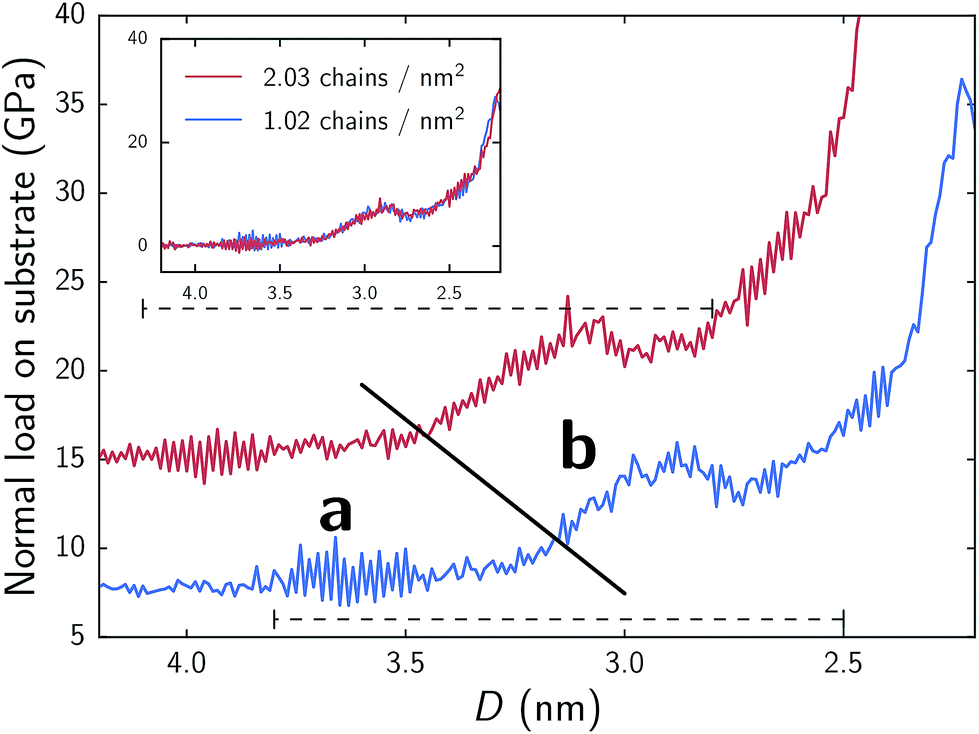 | ||
| Fig. 2 Normal force responses during compression of MPC systems. In main plot, shown forces are acting on substrate only in order to highlight the effects of chain density. Summing the forces acting on both the substrate and chains shifts the curves down the y-axis, yielding a base line normal force (at high separation) of approximately zero; additionally, shifting the separation distance by 0.22 nm for the denser system shows almost complete overlap in trend behavior for the two systems (shown in inset). The diagonal solid line approximately denotes the two regimes (a and b) discussed in text and highlighted in Fig. 3. The upper and lower horizontal dashed lines correspond to ranges of separations across which we performed shearing simulations for the respective chain densities. | ||
Two clear regimes are observed with respect to compression (hereinafter referred to as a and b). In systems of 2.03 and 1.02 chains per nm2, the transitions between these two regimes respectively occur at approximately 3.4 nm & 3.1 nm. This transition is consistent between the two surface densities, although, as noted, shifted to lower separations for the less dense monolayer. In regime a, as highlighted in Fig. 3, an interstitial water layer separates the opposing monolayers. The transition, from a to b, approximately corresponds to the first contact of terminal groups of chains attached to opposing substrates, rather than just a pure interstitial water layer (compare Fig. 3a and b). Throughout the transition and beyond, a large spike in normal force occurs. Examination of the total amount of water within the monolayer region as a function of separation reveals a similar trend to Fig. 2; relatively constant values within regime a and an increase as we cross over into regime b (see Fig. S2†). These phenomena suggests a high resistance to compression marked by swelling of the monolayer with water, which is qualitatively consistent with experimental studies of polymeric brushes built from the same monomers.4–8
 | ||
| Fig. 3 Snapshots of systems with 1.02 (left) and 2.03 (right) chains per nm2 at various separation distances. Regime (a) interstitial water layer separates surfaces. Regime (b) interstitial water layer no longer exists and chains from opposing substrates contact one another. Substrates are shown as black van der Waals spheres, chains as red van der Waals spheres and water is depicted as blue van der Waals spheres in the left image for every configuration and a surface map in the right image for the same configuration. All visualizations were performed using VMD.37 | ||
3.2 Effects of shearing
Fig. 4 shows shear stresses experienced by substrates at different normal forces that fall within regimes a and b. The normal force values on the x-axis correspond to the aforementioned separation distances and do not fluctuate significantly. At normal forces below ∼35 nN, which corresponds to regime a and approximately 1.8 GPa, shear stresses increase with a nearly linear trend. By performing linear least squares fitting of the data, the slope of Fsversus Fn was estimated which provides an estimate of the friction coefficient, μ. In regime a, we find friction coefficients of 0.014 for the sparser system and 0.016 for the denser system which are comparable to those exhibited by neutral, densely packed alkylsilane monolayers separated by water.24 However, the MPC monolayers sustain this coefficient of friction at normal loads nearly an order of magnitude higher. Unlike in dense hydrophobic monolayers, the sparse, flexible, zwitterionic monolayers enable water to penetrate into the monolayer instead of being restricted to a narrow regime bounded by the chains' terminal groups, providing similarly low friction coefficients at higher normal loads. Similarly, our sparse monolayers provide much lower friction coefficients when compared to densely packed phospholipid26 and carboxybetaine27 monolayers which range from ∼0.2–0.5 under the studied conditions. The friction coefficients found in regime a are, in fact, comparable to values reported for experimentally synthesized pMPC brush structures4,7,8 yet slightly above the lowest reported values.5,6 Since our system is a monolayer analogue to the experimentally studied one that features much larger polymer brushes, we cannot offer a direct comparison to these experimental results. However, the similarly low coefficients of friction at high normal loads observed here suggest that regime a, may be a good model for the local environment between brushes in the experimentally synthesized systems which are able to leverage the hydration lubrication mechanism to provide an efficient dissipation pathway for frictional forces. Here, chains attached to opposing surfaces or different polymer backbones, such as in the looped bottle brushes synthesized by Banquy et al.,9 come into close proximity but are still separated by a few water molecules that form the hydration shells around monomers and can rapidly exchange between shells.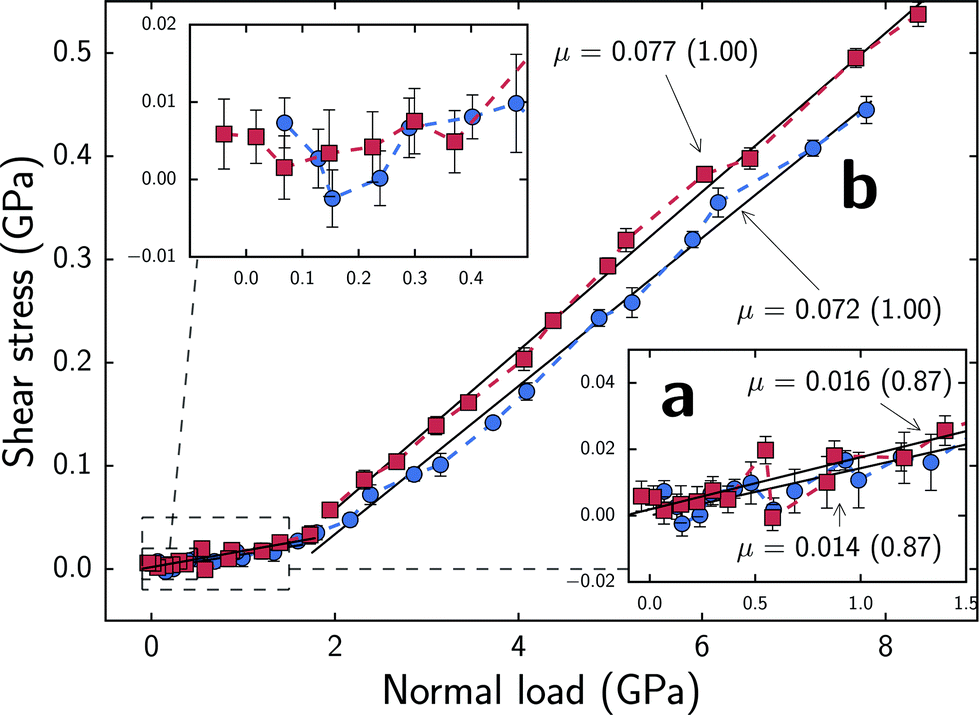 | ||
| Fig. 4 Block averaged shear stresses as a function of normal force per area. Colors correspond to Fig. 2. Error bars represent standard errors of the means; x-axis error bars are plotted but smaller than data markers for all points. Friction coefficients, μ, correspond to the slope of Fsversus Fn as determined by linear least squares fitting. R-values are shown in parentheses. The friction coefficients and fitted slopes near a and b respectively correspond to the regimes discussed in text and highlighted in Fig. 3. | ||
As normal loads trend towards zero, shown in the top left inset of Fig. 4, the average shear stresses fluctuate about zero. Portions of this regime correspond to the normal force fluctuations observed during compression beginning around 3.8 nm for the sparser system and 4.1 nm for the denser system (Fig. 2). We suspect that some of this behavior is caused by the transition from the attractive regime, where the two substrates are not yet in compressive contact, to the repulsive regime.
At normal forces exceeding ∼35 nN (1.8 GPa), which corresponds to regime b, shear stresses monotonically increase in a linear trend. In regime b, we find friction coefficients of 0.072 for the sparser system and 0.077 for the denser system. This magnitude of friction coefficients is comparable to those reported in previous experimental and simulation studies of neutral, densely packed monolayers without water38–42 but again at higher normal loads. Due to consistent chain–chain interactions between opposing monolayers, the shear response in this regime is likely more similar to that of systems without water which gives rise to the comparable friction coefficients. Just as in other studies without water, chains from opposing monolayers provide the primary normal load and shear stress response as opposed to the water molecules exchanging via the hydration lubrication mechanism. The chains are far less mobile than water molecules and are thus unable to provide a comparably fluid shear response yet they are able to provide a strong resistance to compression as shown in Fig. 2.
We also note that additional shearing simulations conducted at 1 m s−1 demonstrate the same trends; these results are summarized in Fig. S3.†
3.3 Water distribution during shearing
In the low shear stress regime, interactions between chains from opposing substrates are rare as is reflected in the velocity profiles shown in the top panels of Fig. 5. Here, all parts of the chains are moving at approximately the speed of the substrate as is a layer of water approximately as tall as the chains. There is also a distinct layer of water separating the two monolayers. Above ∼35 nN (1.8 GPa), opposing chains begin to consistently interact as shown in the bottom panels of Fig. 5. Opposing chains' terminal choline groups interpenetrate, drag each other and displace the interstitial water layer. The water velocity profiles in our systems differ from profiles of nanoconfined water43,44 and water confined between alkylsilane monolayers;24 where these systems exhibit a near linear slope from wall-to-wall or chain-to-chain with approximate boundary layers of less than 0.5 nm. The profiles in our systems assume an S-like shape with water layers over 1 nm thick within the sparse, zwitterionic monolayers. While monolayer thickness does decrease with decreasing separation distances (Fig. 6), the change in thickness is relatively minimal. A decrease in separation distance of 1.3 nm for both systems, yields only a decrease in film thickness of about 0.13 and 0.12 nm for 1.02 and 2.03 chains per nm2 respectively. This almost negligible change suggests that the monolayers swell with water as normal loads increase – a phenomenon also observed experimentally.8,13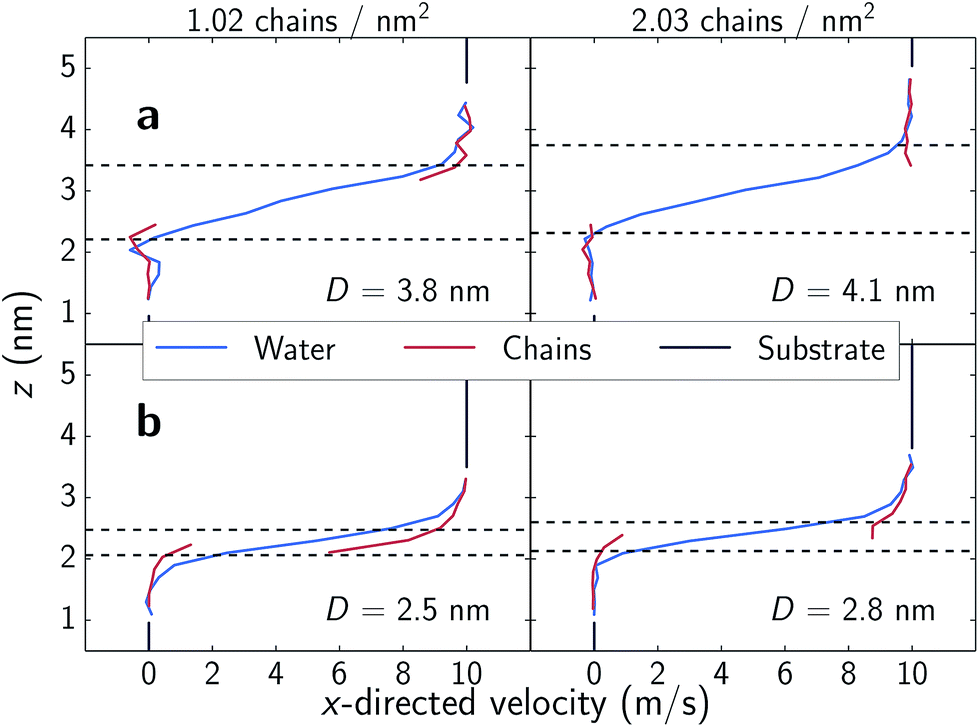 | ||
| Fig. 5 Velocity profiles of system components in the shear direction. The dashed lines denote the monolayer film thickness, wherein 90% of the monolayer atoms remain over the course of a simulation. Film thickness calculation is detailed in Fig. S4† and trends as a function of separation are shown in Fig. 6. Note that in the bottom two panels, interpenetration of chains from opposing monolayers occurs for portions outside the defined film thickness cutoff thus displacing the interstitial water layer. | ||
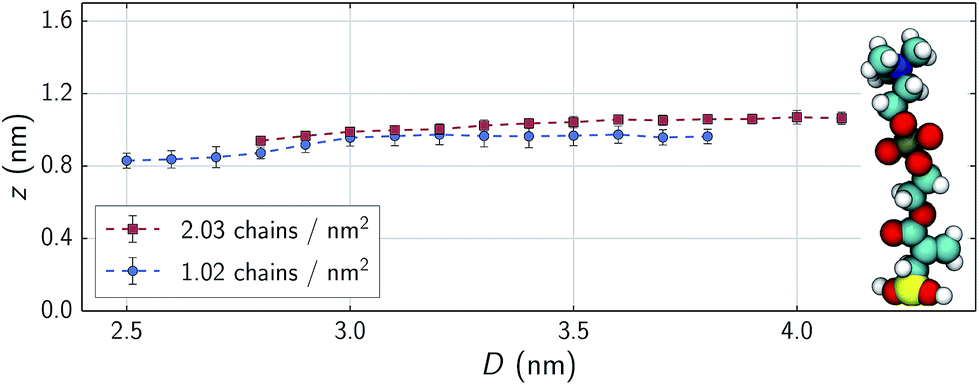 | ||
| Fig. 6 Dependence of monolayer thickness on separation distance during shearing. Equilibrated MPC chain in vacuo shown for reference (to scale of y-axis). Error bars correspond to standard deviations through time. | ||
Fig. 7 further details the distribution of water across the pores. The bulk of the water remains in the interstitial area but spreads into the outer parts of the monolayers (approximately denoted by the dashed lines) where it appears to bind near where the choline headgroups are located. As the systems are compressed and they transition from regime a to b the interstitial water does densify but water is also pushed into the monolayers as is particularly evident in the sparser system.
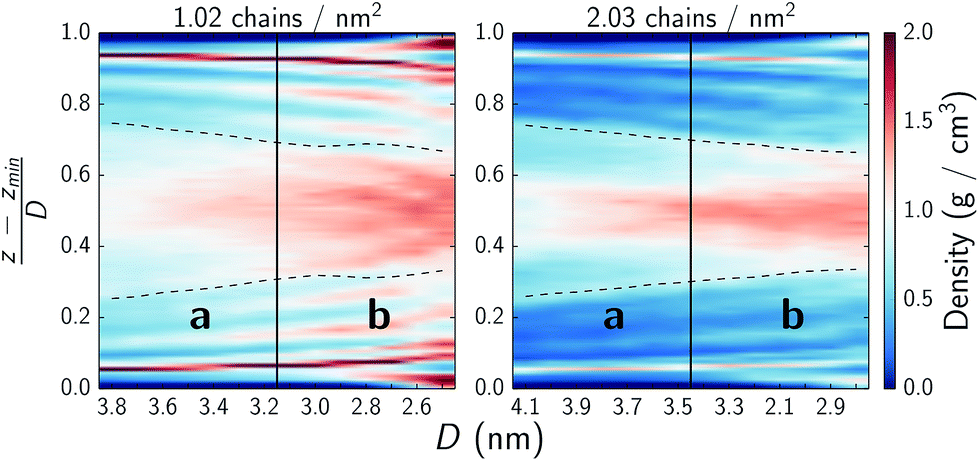 | ||
| Fig. 7 Water density across xy-planes as a function of normal force. The y-axis denotes the normalized position in the pore from the bottom substrate at 0 to the top substrate at 1. Solid vertical lines denote approximate transitions between regimes a and b as discussed in text and highlighted in Fig. 3. The dashed lines denote the monolayer film thickness, wherein 90% of the monolayer atoms remain over the course of a simulation and which the choline headgroups frequently protrude from. Note that these are local water densities which do not include the monomers. | ||
3.4 Water mobility during shearing
Experimental studies suggest that ultra-low shear stresses in hydration lubrication schemes arise due to rapid water exchange rates between hydration shells or hydration shells and a bulk-like fluid state.5,6,9,11,15–17 In Fig. 8, we quantify water mobility, by considering flux through xy-planes as a function of normal force. Low normal forces allow for rapid water exchange in the interstitial region as well as with water bound to the choline headgroups. Comparing to Fig. 4, at both chain densities, systems with the highest interstitial water flux correspond to those operating in regime a where fluxes are up to three times higher than in regime b and friction coefficients are up to an order of magnitude lower. Upon transitioning to regime b, where the hydration lubrication mechanism does not seem to be the dominant mechanism due to a lack of water mobility, the chains are still able to provide some form of lubrication while simultaneously resisting higher normal loads.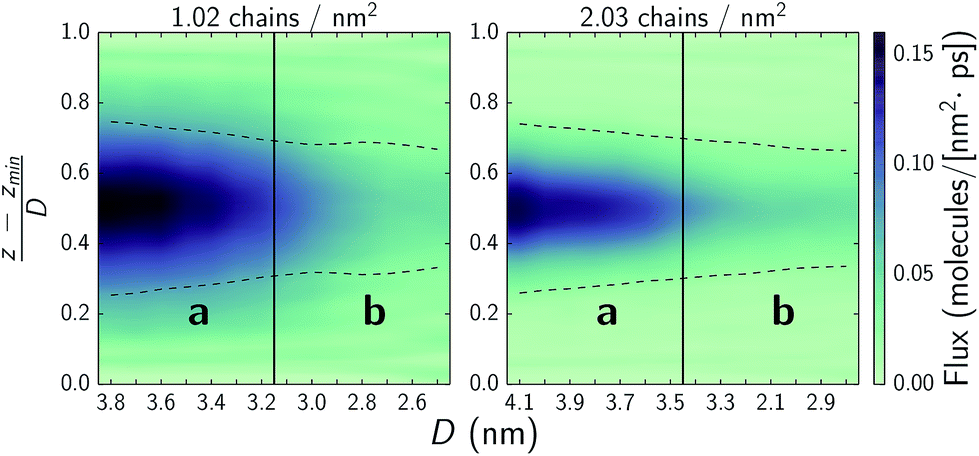 | ||
| Fig. 8 Water flux across xy-planes as a function of normal force. The y-axis denotes the normalized position in the pore from the bottom substrate at 0 to the top substrate at 1. Solid vertical lines denote approximate transitions between regimes a and b as discussed in text and highlighted in Fig. 3. The dashed lines denote the monolayer film thickness, wherein 90% of the monolayer atoms remain over the course of a simulation and which the choline headgroups frequently protrude from. | ||
4 Conclusions
Overall, these results qualitatively support the hypothesized hydration lubrication mechanism where tight binding of water molecules to charged groups allows materials to support high normal loads while simultaneously allowing water molecules to rapidly relax between bound states thus providing a fluid shear response.17 The results show that substrates sparsely functionalized with zwitterionic MPC monolayers in water provide strong resistance to normal loads by allowing water molecules to swell into the monolayers and allowing strongly repulsive monomer–monomer interactions to resist compression. Simultaneously, when shearing at normal loads where water mobility remains high, shear stresses fluctuate about zero and yield friction coefficients as low as 0.014. Our results show two regimes with distinct shear responses. In the high shear stress regime, monomer–monomer interactions dominate the shear response which thus resembles the response of previously studied dense alkylsilane monolayers without water.38–42 The low shear regime, where friction coefficients compare to those observed in experimental brush systems,4,7–9 is characterized by a thin layer of water separating monomers in opposing substrates. Estimates of friction coefficients in these regimes approximately agree with other simulation and experimental studies of systems with respectively comparable environments. In the low shear regime, hydration shells surrounding opposing headgroups efficiently slide past one another by exchanging water molecules via the hydration lubrication mechanism. While experimental studies have thus far examined large polymer brushes composed of the monomers studied here, our simulations present a picture of the local environment surrounding individual monomers and how phenomena occurring on this scale result in favorable frictional properties. The examination of zwitterionic monolayers further demonstrates the importance of water mobility and how sparse zwitterionic monolayers behave differently than dense monolayer systems, both in their shearing mechanisms and their ability to sustain lower coefficients of friction at higher normal loads.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant no. OCI-1047828. We also acknowledge the National Energy Research Supercomputing Center, which is supported by the Office of Science of the U.S. Department of Energy under Contract no. DE-AC02-05CH11231.References
- C. Clarke, R. Contini and R. M. Kenedi, J. Lubr. Technol., 1975, 97, 358 CrossRef.
- B. J. Roberts, A. Unsworth and N. Mian, Ann. Rheum. Dis., 1982, 41, 217–224 CrossRef CAS.
- J. Klein, Proc. Inst. Mech. Eng., Part J, 2006, 220, 691–710 CrossRef CAS.
- M. Kobayashi, Y. Terayama, N. Hosaka, M. Kaido, A. Suzuki, N. Yamada, N. Torikai, K. Ishihara and A. Takahara, Soft Matter, 2007, 3, 740 RSC.
- M. Chen, W. H. Briscoe, S. P. Armes and J. Klein, Science, 2009, 323, 1698–1701 CrossRef CAS PubMed.
- M. Chen, W. H. Briscoe, S. P. Armes, H. Cohen and J. Klein, Eur. Polym. J., 2011, 47, 511–523 CrossRef CAS PubMed.
- Z. Zhang, A. J. Morse, S. P. Armes, A. L. Lewis, M. Geoghegan and G. J. Leggett, Langmuir, 2011, 27, 2514–2521 CrossRef CAS PubMed.
- M. Kyomoto, T. Moro, K. Saiga, M. Hashimoto, H. Ito, H. Kawaguchi, Y. Takatori and K. Ishihara, Biomaterials, 2012, 33, 4451–4459 CrossRef CAS PubMed.
- X. Banquy, J. Burdyńska, D. W. Lee, K. Matyjaszewski and J. Israelachvili, J. Am. Chem. Soc., 2014, 136, 6199–6202 CrossRef CAS PubMed.
- J. Klein, E. Kumacheva, D. Mahalu, D. Perahia and L. J. Fetters, Nature, 1994, 370, 634–636 CrossRef CAS.
- U. Raviv, S. Giasson, N. Kampf, J.-F. Gohy, R. Jérôme and J. Klein, Nature, 2003, 425, 163–165 CrossRef CAS PubMed.
- N. Kampf, U. Raviv and J. Klein, Macromolecules, 2004, 37, 1134–1142 CrossRef CAS.
- M. Kyomoto, T. Moro, Y. Takatori, H. Kawaguchi and K. Ishihara, Clin. Orthop. Relat. Res., 2011, 469, 2327–2336 CrossRef PubMed.
- U. Raviv and J. Klein, Science, 2002, 297, 1540–1543 CrossRef CAS PubMed.
- W. H. Briscoe, S. Titmuss, F. Tiberg, R. K. Thomas, D. J. McGillivray and J. Klein, Nature, 2006, 444, 191–194 CrossRef CAS PubMed.
- A. Gaisinskaya, L. Ma, G. Silbert, R. Sorkin, O. Tairy, R. Goldberg, N. Kampf and J. Klein, Faraday Discuss., 2012, 156, 217 RSC.
- J. Klein, Friction, 2013, 1, 1–23 CrossRef PubMed.
- L. Ma, A. Gaisinskaya-kipnis, N. Kampf and J. Klein, Nat. Commun., 2015, 6, 1–6 Search PubMed.
- S. T. Cui, P. T. Cummings and H. D. Cochran, J. Chem. Phys., 2001, 114, 7189 CrossRef CAS PubMed.
- T. Onodera, Y. Morita, A. Suzuki, M. Koyama, H. Tsuboi, N. Hatakeyama, A. Endou, H. Takaba, M. Kubo, F. Dassenoy, C. Minfray, L. Joly-Pottuz, J.-M. Martin and A. Miyamoto, J. Phys. Chem. B, 2009, 113, 16526–16536 CrossRef CAS PubMed.
- T. Onodera, Y. Morita, R. Nagumo, R. Miura, A. Suzuki, H. Tsuboi, N. Hatakeyama, A. Endou, H. Takaba, F. Dassenoy, C. Minfray, L. Joly-Pottuz, M. Kubo, J.-M. Martin and A. Miyamoto, J. Phys. Chem. B, 2010, 114, 15832–15838 CrossRef CAS PubMed.
- J.-M. Y. Carrillo, D. Russano and A. V. Dobrynin, Langmuir, 2011, 27, 14599–14608 CrossRef CAS PubMed.
- D.-C. Yue, T.-B. Ma, Y.-Z. Hu, J. Yeon, A. C. T. van Duin, H. Wang and J. Luo, J. Phys. Chem. C, 2013, 117, 25604–25614 CAS.
- C. D. Lorenz, M. Chandross, J. M. D. Lane and G. S. Grest, Modell. Simul. Mater. Sci. Eng., 2010, 18, 034005 CrossRef.
- J. Zheng, Y. He, S. Chen, L. Li, M. T. Bernards and S. Jiang, J. Chem. Phys., 2006, 125, 174714 CrossRef PubMed.
- Y. He, S. Chen, J. C. Hower, M. T. Bernards and S. Jiang, J. Chem. Phys., 2007, 127, 084708 CrossRef PubMed.
- Y. He, Q. Shao, S. Chen and S. Jiang, J. Phys. Chem. C, 2011, 115, 15525–15531 CAS.
- M. Chandross, G. S. Grest and M. J. Stevens, Langmuir, 2002, 18, 8392–8399 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS , http://lammps.sandia.gov.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- C. Lorenz, E. Webb, M. Stevens, M. Chandross and G. Grest, Tribol. Lett., 2005, 19, 93–98 CrossRef CAS.
- M. Tuckerman, B. J. Berne and G. J. Martyna, J. Chem. Phys., 1992, 97, 1990 CrossRef CAS PubMed.
- I.-C. Yeh and M. L. Berkowitz, J. Chem. Phys., 1999, 111, 3155 CrossRef CAS PubMed.
- W. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef.
- S. Nose, J. Chem. Phys., 1984, 81, 511 CrossRef CAS PubMed.
- T. Reilly and A. M. Williams, Science and Soccer, Psychology Press, 2003, p. 111 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- J. B. Lewis, S. G. Vilt, J. L. Rivera, G. K. Jennings and C. McCabe, Langmuir, 2012, 28, 14218–14226 CrossRef CAS PubMed.
- B. D. Booth, S. G. Vilt, J. B. Lewis, J. L. Rivera, E. A. Buehler, C. McCabe and G. K. Jennings, Langmuir, 2011, 27, 5909–5917 CrossRef CAS PubMed.
- J. L. Rivera, G. K. Jennings and C. McCabe, J. Chem. Phys., 2012, 136, 244701 CrossRef PubMed.
- M. Chandross, E. Webb, M. Stevens, G. Grest and S. Garofalini, Phys. Rev. Lett., 2004, 93, 166103 CrossRef CAS.
- M. Chandross, C. D. Lorenz, G. S. Grest, M. J. Stevens and E. B. W. Iii, JOM, 2005, 57, 55–61 CrossRef CAS.
- R. Qiao and N. R. Aluru, J. Chem. Phys., 2003, 118, 4692 CrossRef CAS PubMed.
- Y. Leng and P. T. Cummings, J. Chem. Phys., 2006, 125, 104701 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of system charges and chain arrangements as well as film thickness results. See DOI: 10.1039/c4sm02883j |
| This journal is © The Royal Society of Chemistry 2015 |