
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight driven mesoscale assembly of a coordination polymeric gelator into flowers and stars with distinct properties†
Rahul Dev
Mukhopadhyay
 ab,
Vakayil K.
Praveen
a,
Arpan
Hazra
c,
Tapas Kumar
Maji
c and
Ayyappanpillai
Ajayaghosh
*ab
ab,
Vakayil K.
Praveen
a,
Arpan
Hazra
c,
Tapas Kumar
Maji
c and
Ayyappanpillai
Ajayaghosh
*ab
aPhotosciences and Photonics Group, Chemical Sciences and Technology Division, India. E-mail: ajayaghosh@niist.res.in
bAcademy of Scientific and Innovative Research (AcSIR), CSIR-National Institute for Interdisciplinary Science and Technology (CSIR-NIIST), Trivandrum-695019, India
cChemistry and Physics of Materials Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bangalore 560064, India
First published on 12th August 2015
Abstract
Control over the self-assembly process of porous organic–inorganic hybrids often leads to unprecedented polymorphism and properties. Herein we demonstrate how light can be a powerful tool to intervene in the kinetically controlled mesoscale self-assembly of a coordination polymeric gelator. Ultraviolet light induced coordination modulation via photoisomerisation of an azobenzene based dicarboxylate linker followed by aggregation mediated crystal growth resulted in two distinct morphological forms (flowers and stars), which show subtle differences in their physical properties.
Introduction
Mesoscopic science is broadly defined as the space between nanoscopic (atoms, molecules and assemblies up to a few nanometers in size) and the macroscopic (hierarchical assemblies, few micrometers in size or more) domains. The evolution of mesoscopic materials is controlled by chemical and physical processes, wherein atoms and molecules interact through physical forces, guided by thermodynamic and kinetic phenomena. Molecular self-assembly is a key nucleation point of mesoscopic evolution of functional materials and occupies a unique space in the field of ‘nanoarchitectonics’,1 which is a novel conceptual idea for the construction of functional materials via control over reactions or assembly of molecules making use of different physical stimuli.1c In recent years, metal–organic frameworks (MOFs) or porous coordination polymers (PCPs),2,3 known for their highly crystalline porous nature and ubiquitous applications have been receiving attention in the field of nanoarchitectonics. While wide range applications are envisaged for MOFs and PCPs, reversible control on the porosity and gas adsorption properties of such materials remain a challenging task. Therefore, an innovative approach in tuning the properties of MOFs or PCPs is of paramount importance. Such considerations encourage chemists to explore the possibility of light sensitive metal–organic systems.4 In this context, azobenzene, a widely explored photoswitch,5 has been utilised recently by a number of research groups to design photoresponsive crystalline porous materials.6 However, photoisomerisation of directly linked azobenzene moieties in a rigid coordination framework is often found to be difficult.4b Therefore, an alternate approach is required to explore the utility of cis–trans photoisomerisation of azobenzene moieties in a metal–organic coordination framework.Unlike rigid crystalline MOFs or PCPs, amorphous organogels are an entirely different class of soft material possessing a flexible supramolecular network.7–10 Gels are known to provide an excellent medium to create a dynamic environment, ideal for crystal growth process via defect rectification during reversible self-assembly.11 A gel medium is also a clever option to create hierarchical mesoscopic structures having complex forms.12 While there are several reports on photoresponsive gels,5c,7,8e,13 reports on photoresponsive microporous coordination polymeric gels are rare.14 Moreover, a deeper understanding of the structural evolution of MOFs and PCPs at the mesoscopic scale under the influence of light is, as yet, unknown. This is rather surprising when considering the importance of mesoscopic structures in the field of materials science.15 Herein, we combine the chemistry of metal–organic materials and gels to create photoresponsive structures, to understand the effect of light on the kinetic pathways of mesoscopic structural evolution in synthetic molecular systems and study their properties. For this purpose, we designed a photoresponsive coordination polymeric gelator, which forms a gel having a flower-like morphology. Interestingly, pre-synthetic photoirradiation of the gelator resulted in star-shaped morphology with distinct rheological and gas adsorption properties.
Results and discussion
Synthesis and characterization of coordination polymeric gels CPG1 and CPG2
Our experience with supramolecular gels,8e encouraged us to exploit the Materials of Institute Lavoisier (MIL) family of coordination polymers,16 many of which are known for instantaneous gelation of the reaction mixture. Since the structural rigidity of MOFs and PCPs do not facilitate post-synthetic photoisomerisation of azobenzene on larger length scales,4b,6d we hypothesised that if a pre-synthetic isomerisation is carried out, the cis form of azobenzene should in principle get locked in the rigid matrix of the hybrid material thereby leading towards self-assembled forms with different properties. In a typical experiment a gently warmed mixture of the azobenzene-4,4′-dicarboxylic acid (ADA) and triethylamine in dimethyl sulfoxide (DMSO) was added to an equimolar quantity of Fe(NO3)3·9H2O in same volume of DMSO, leading to immediate gelation (Fig. 1). Gelation was observed in polar solvents such as DMSO and dimethyl formamide (DMF) at a critical gelator concentration (CGC) of 20 mg mL−1 (7.4 × 10−2 M) with respect to ADA and was found to be highly sensitive towards mechanical disturbance.8a However, the gel was not thermoreversible in nature by virtue of its coordination network.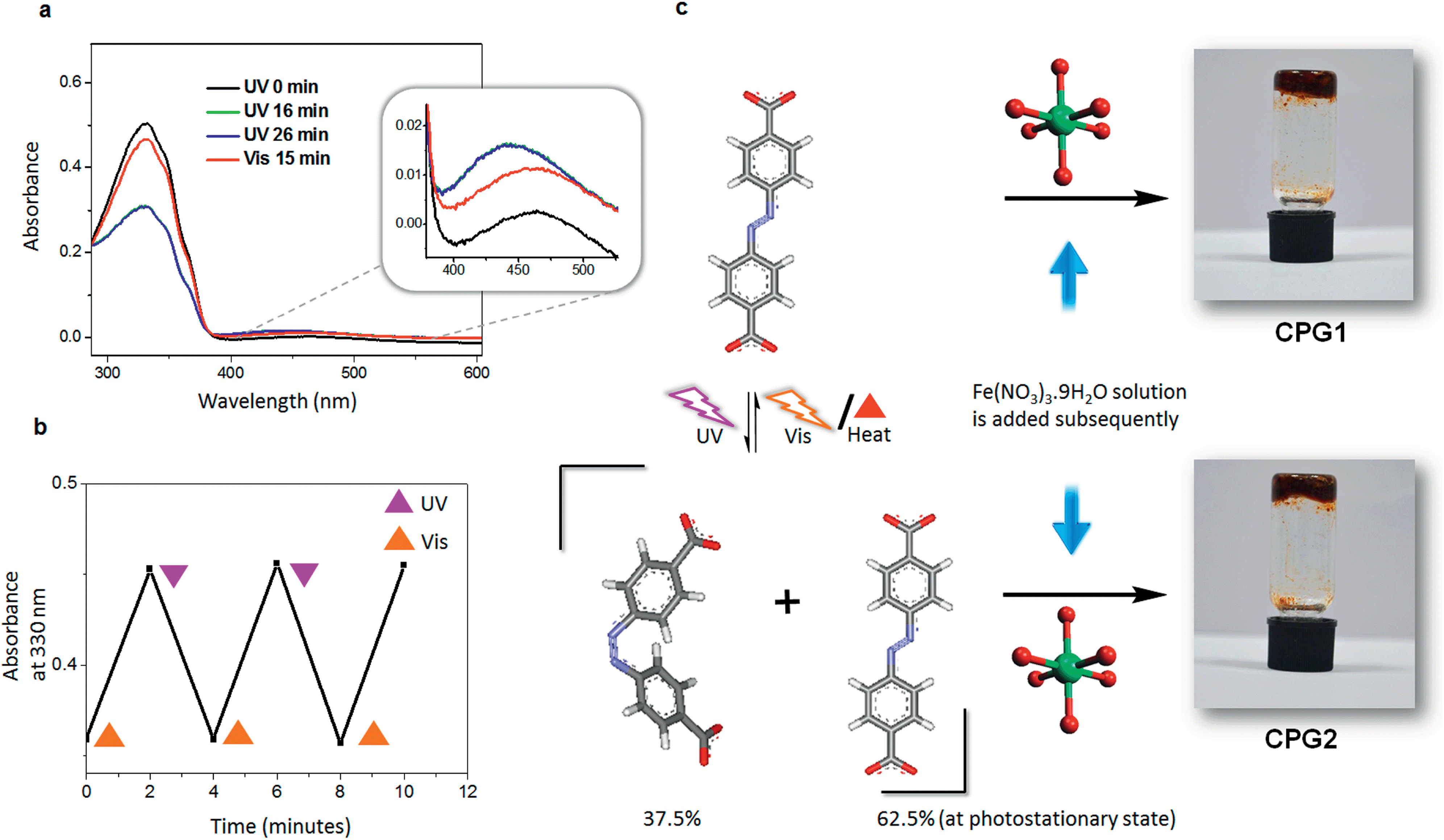 | ||
| Fig. 1 (a) Absorption spectra of the ADA solution in DMSO (1.85 × 10−5 M) before and after irradiation with ultraviolet (UV) and visible (Vis) light. Inset shows zoomed region of 379–527 nm corresponding to the n–π* transition band of cis azobenzene. (b) Reversibility of photoisomerisation, shown for three cycles. (c) Schematic representation of synthesis of CPG1 and CPG2 (corresponding gel images are shown). Trans and cis isomers of ADA (carbon – grey; nitrogen – blue; oxygen – red; hydrogen – white) and probable structure of inorganic octahedra (iron – green; oxygen – red) are also shown. | ||
The involvement of metal–ligand coordination responsible for such gelation was confirmed from Fourier-transform infrared (FT-IR) spectral studies.17 The intense IR band at 1688 cm−1 corresponding to asymmetric carbonyl stretching frequency of the free acid ADA diminishes with considerable broadening in the case of coordination polymeric gel (CPG1), with the appearance of a new band at 1413 cm−1, corresponding to the symmetric stretching frequency.17b Deconvolution of the broad band at ∼1700 cm−1 suggests that the separation between the asymmetric and symmetric carbonyl stretching frequency increases in CPG1 when compared to ADA, as evidenced from the higher wavenumber peak contributions at 1700, 1717 and 1731 cm−1. This observation primarily suggests the presence of unidentate coordination in CPG1 although the presence of bidentate binding modes cannot be completely ruled out as indicated by the peak contribution at 1671 cm−1 (Fig. S1†).17a
Scanning electron microscopy (SEM) analysis of the aforesaid gel revealed the formation of macroscopic sheets (Fig. 2). Furthermore, these sheets have a tendency to stack one above the other to form various metastable superstructures (Fig. S2†). Detailed morphological analysis of the gel sample by SEM showed the formation of the metal–organic gel through an evolutionary morphogenesis as reported previously in the case of certain infinite coordination polymers.18 A time-dependent analysis of the gel formation mechanism indicates that, at the initial stages of development, globular seeds are formed (Fig. 2b) which agglomerate to form linear as well as branched aggregates that grow into two-dimensional (2-D) sheets. However, the surface of larger sheets initially have a rough topography, which got smoothed by a gradual process of annealing (Fig. 2c). Folding of the smaller sheets lead to a cabbage like structure (Fig. 2d) followed by the sideways stacking of the sheets, which ultimately lead to metal–organic flowers (Fig. 2e). These micrometer sized flowers had nanoscale features in which the sheets were found to be about 25 nm thick (Fig. 2f). Transmission electron microscopy (TEM) analysis of CPG1 also confirmed a sheet like morphology. Lattice planes were clearly visible in the high-resolution transmission electron microscopy (HR-TEM) images, suggesting a highly ordered structure (Fig. 4a, inset).
 | ||
| Fig. 2 SEM analysis of CPG1. (a) Different morphological forms of CPG1 captured in one frame. Platelets (white box), rough sheets (green box), cabbage (blue box), large smooth sheets (yellow box) and flower (red box) like structures. (b–f) Time dependent SEM analysis of CPG1. (b) Nanoscale CPG1 platelets formed immediately, inset shows platelets undergoing fusion. (c and d) Images obtained after 5–6 h. (c) Gradual smoothening of rough sheets; smoothened portions marked with arrows. (d) Formation of cabbage like structure. (e) Matured metal–organic flowers formed after 1 day. (f) Zoomed view of the box marked in (e), showing nanoscale sheets (∼25 nm). | ||
In order to check the photoresponsive nature, we irradiated the gel sample with UV light. However, no noticeable change in morphology or properties was observed, and the gel remained intact even after hours of UV irradiation. Therefore, we thought of exploring the effect of the photoisomerisation of ADA before the preparation of the gel. The absorption spectrum of ADA in DMSO (1.85 × 10−5 M), showed two bands with maxima at 330 and 440 nm, corresponding to π–π* and n–π* transitions, respectively (Fig. 1a). Photoisomerisation studies revealed that the molecule reaches a photostationary state within 16 min, where ∼37.5% of the molecules are present in the cis form (Fig. 1a). The photoisomerisation of the azobenzene core was clearly reversible with respect to UV and visible light (λbandpass = 350 and 420 nm, LOT-Oriel 200 W high-pressure Hg Lamp) and was also reproducible over several repeated cycles (Fig. 1b). Thus, when a solution of ADA and triethylamine in DMSO (20 mg mL−1) was irradiated with UV light (λbandpass = 350 nm) for about 4 h (to confirm that maximum conversion has taken place) and then a solution of Fe(NO3)3·9H2O in DMSO was added to it, the solution mixture slowly turned into a gelatinous material, which was kept under UV light for about an hour and then analysed. The morphology of the irradiated metal–organic gel (CPG2) was quite different (Fig. 3) from its non-irradiated counterpart, CPG1 (Fig. 2). The morphology of CPG2 comprised of six armed metal–organic stars. Akin to the growth process of CPG1, CPG2 also showed a gradually evolving process starting from small leaf-like seeds (Fig. 3b), which grew with time to form thick leaves and tentacles (Fig. 3c) and ultimately evolved into star-like superstructures that possessed well-defined facets as that of a crystal (Fig. 3d). The transformation of tentacles to stars may be via a 2-D star-like architecture as found in several places in the SEM images (Fig. S2†). The presence of nano leaves and tentacles were confirmed from TEM analysis (Fig. S3†).
 | ||
| Fig. 3 SEM analysis of CPG2. (a) Different morphological forms of CPG2 captured in one frame. Leaves (white box), thick leaves (yellow box), tentacles (green box), stars in the formation (blue box), and crystalline stars (red box). (b–d) Time dependent SEM analysis of CPG2. (b) Nanoscale leaves of CPG2 after immediate gelation. (c) Aggregation of nano leaves after 1 h. (d) Crystalline metal–organic stars obtained after 1 day. | ||
Analysis of growth process of CPG1 and CPG2
Although in time-dependent experiments we could mostly observe a particular morphology after a certain time interval, more than one morphological form were also found (Fig. 2a and 3a) at a few places in both the samples, which suggest that the growth process was not only time dependent but also depends on the local concentration of the initial components. Most of the intermediate structures formed might be controlled by kinetic processes rather than thermodynamic control and are therefore limited by diffusion (Fig. S4†).19 At lower concentration of particles, the process of aggregation is controlled by diffusion leading to the formation of a patterned structure with random branching, whereas higher concentration leads to fast aggregation process leading to the formation of more compact structures. This process happens due to the large surface to volume ratio and high collision frequency of the particles. The occurrence of several unreacted sites on the surface of the seeds due to fast nucleation process, results in further polymerisation through coordination linkages and thereby fusion of the particles. With the gradual decrease in the concentration of the ligands and the metal ions, the nucleation of new particles is reduced and annealing of existing nuclei prevails much like the Ostwald ripening process.5b Unlike a single step thermodynamically controlled pathway, a kinetically controlled crystal growth process usually takes place via a sequence of steps involving amorphous intermediates. Such transformations can be easily tuned by various chemical stimuli (additives), which influence the energy of activation of nucleation (n), growth (g) and phase transformation (t) processes.20 Photoirradiation of trans-ADA molecules with UV light leads to the formation of energetically unstable cis-ADA isomer, which in combination with trans-ADA molecules can significantly influence the nucleation–growth–transformation processes. Therefore, a new kinetic pathway is followed, which is shown in a modified energy profile diagram as proposed previously by Mann and co-workers (Fig. S5†).20a In order to confirm our hypothesis, we have carried out the synthesis of nanoscale CPG1 and CPG2 under thermodynamic conditions at a very low concentration where we were able to arrive at only nano platelets and leaves (Fig. S6†).According to the Bravais, Friedel, Donnay and Harker (BFDH) law, the morphology of a crystal is mainly determined by the slowest growing facet of the crystal.21 The 2-D nucleus on a particular crystal facet determines the overall crystal morphology depending on the probabilities of growth or dissolution at a particular site. A crystal in principle should grow in a particular direction unless the energetics is changed along a particular facet. Now, taking a close look at the formation mechanism (Fig. S7†) for both CPG1 and CPG2, one can assume that the transformation of platelets (for CPG1) to leaves (for CPG2) essentially occurs by a similar process of anisotropic crystal growth. At the photostationary state ∼37.5% of cis-ADA can trigger this anisotropic growth mechanism by varying the crystal growth rates along different facets. It is known that the cis isomer of azobenzene can form energetically less favourable self-assembled structures.22 In case of a coordination bond driven self-assembly, the non-planar nature of cis-ADA can further hamper the chain building process as the geometry of the inorganic cluster brings cis-ADA closer to the coordination polymeric chain, where it remains in a sterically unfavourable position to undergo further reaction. Thus, cis-ADA can essentially act as a capping agent, which stops the crystal growth along a particular facet. The presence of these capping agents is difficult to verify even experimentally, and therefore, the explanation of such process mainly relies on speculation, in the absence of a completely understood crystal growth mechanism.23 It is hypothesised that the rate of crystal growth may range from moderate to maximum along different facets (Fig. 5). This anisotropy arises only after smaller fragments (oligomers) of the coordination polymeric chain, containing trans-ADA (∼62.5%) nucleate to form platelet like structures at the threshold of nanoscale. Transformation of platelets to leaves essentially takes place at the mesoscale followed by aggregation of preformed crystalline nano-leaves leading to the formation of microscopic superstructures. While trying to understand the growth process in these materials, it came to our notice that the morphology of colloidal hematite, a naturally occurring mineral, is also affected by similar aggregation mediated crystal growth process, showcasing several polymorphs ranging from platelets, ellipsoidal particles to stars. One can have a chemical control over the growth of different facets to facilitate such anisotropic crystal growth.24 The same control has been achieved in the present case via photonic manipulation. A number of reports towards engineering the self-assembly process of framework materials at the mesoscopic domain are known.25 It is important to note that in all these cases, a third component, for instance, a coordination modulator or a template was necessary to achieve such mesoscopic control over self-assembly. Whereas, in the present case, a certain fraction of the isomerised organic ligand is enough to bifurcate the bottom-up self-assembly process, while other cis-ADA molecules may get entrapped in the microporous structure. The cis-ADA molecules can gradually reconvert back to the thermodynamically more stable trans-ADA. The fact that cis-ADA reconverted to trans-ADA in CPG2 was confirmed from the FT-IR studies where no new peak at higher wavenumber region (∼720 cm−1) corresponding to cis-ADA was observed.6c The peak corresponding to the trans isomer (700 cm−1) in free ADA was shifted to 709 cm−1 in CPG1 and observed as a shoulder in CPG2 along with a prominent peak at 700 cm−1 corresponding to free trans-ADA. This was further confirmed from the deconvoluted FT-IR spectra of CPG2, where the peaks at 1688, 1424 and 1294 cm−1 corresponding to free ADA, which were absent in CPG1, were found to be present in CPG2 (Fig. S1†). The results also indicate the shifting of the asymmetric stretching frequency of ADA towards the lower wavenumber region, basically indicating that the bidentate coordination modes become more prominent in CPG2.
One of the problems of working with Fe(III) or Al(III) (hard acid) carboxylate (hard base) coordination polymers is the immediate formation of strong metal–ligand coordination, that does not allow defect rectification during the crystal growth process, leading towards precipitates or gels in most cases.26 The in situ formation of multiple structure directing inorganic building blocks under kinetic conditions often adds to this problem.16a,b,26b Therefore, the actual structure determination in the gel state, containing a mixture of polymorphic structures, is difficult to assign, even via computational simulation. However, we can derive some idea from inverse fast Fourier transform (IFFT) reconstructed HR-TEM images, which showed the existence of coordination polymeric chains arranged in a parallel manner (Fig. 4b and c). This image matches with the recently reported crystal structure of PCN-243,26b a MOF formed from ADA and an Fe(III) based inorganic cluster, when viewed along the a-axis. The distance between the polymeric chains roughly corresponds to ∼0.3 nm. Further, the presence of ADA (molecular length ∼1.12 nm) in the polymeric chains can be understood from the slightly kinked structure of the azobenzene core. Similarly, the rhombic pores visualised in Fig. 4d can be compared with the same crystal structure, if viewed along the b-axis. Elemental analysis, energy dispersive X-ray (EDX) analysis and thermogravimetric analysis (TGA) of the two forms showed similar composition, solvent weight loss and thermal decomposition pattern (Fig. S9 and S10†). The powder X-ray diffraction (XRD) pattern of the two forms CPG1 and CPG2 also did not show considerable difference (Fig. 6a) pointing that both the morphological form possess similar metal-linker connectivity. The simulated XRD pattern obtained from PCN-243 matches well with the observed pattern in CPG1 and CPG2 although the possibility of other amorphous phases cannot be completely ruled out, as evident from the presence of a broad halo (2θ 12–30°) in the XRD pattern of both the materials. However, the XRD peaks corresponding to hematite (α-Fe2O3) phase with rhombohedral structure (a, b = 0.5 nm, c = 1.38 nm, JCPDS card no. 24-0072) is completely absent in either CPG1 or CPG2 (Fig. 6a). Also the FT-IR peaks corresponding to the lattice vibrations in α-Fe2O3 (∼500–600 cm−1) is absent in either case.24b These data confirm that the observed unusual splitting of the growth process in our material was not affected by pseudomorphic growth replication process over hematite or presence of hematite phase as an impurity (Fig. S1†).25a From the above investigations, we conclude that the strong hard acid–hard base interactions results in fast unidentate coordination, and thereby gelation. With time, most of the amorphous intermediates are converted to more crystalline forms, which show bidentate coordination modes. Selected area electron diffraction (SAED) analysis of both CPG1 and CPG2 revealed multiple diffraction patterns ranging from amorphous, polycrystalline to single crystalline behavior (Fig. S11†). It is also observed that the presence of cis-ADA in the formation of CPG2 basically slows down the crystallisation process within the gel matrix resulting in more crystalline products. As a result, the presence of amorphous to highly crystalline species in the xerogel matrix makes the system highly heterogeneous for correlation of structure with properties.
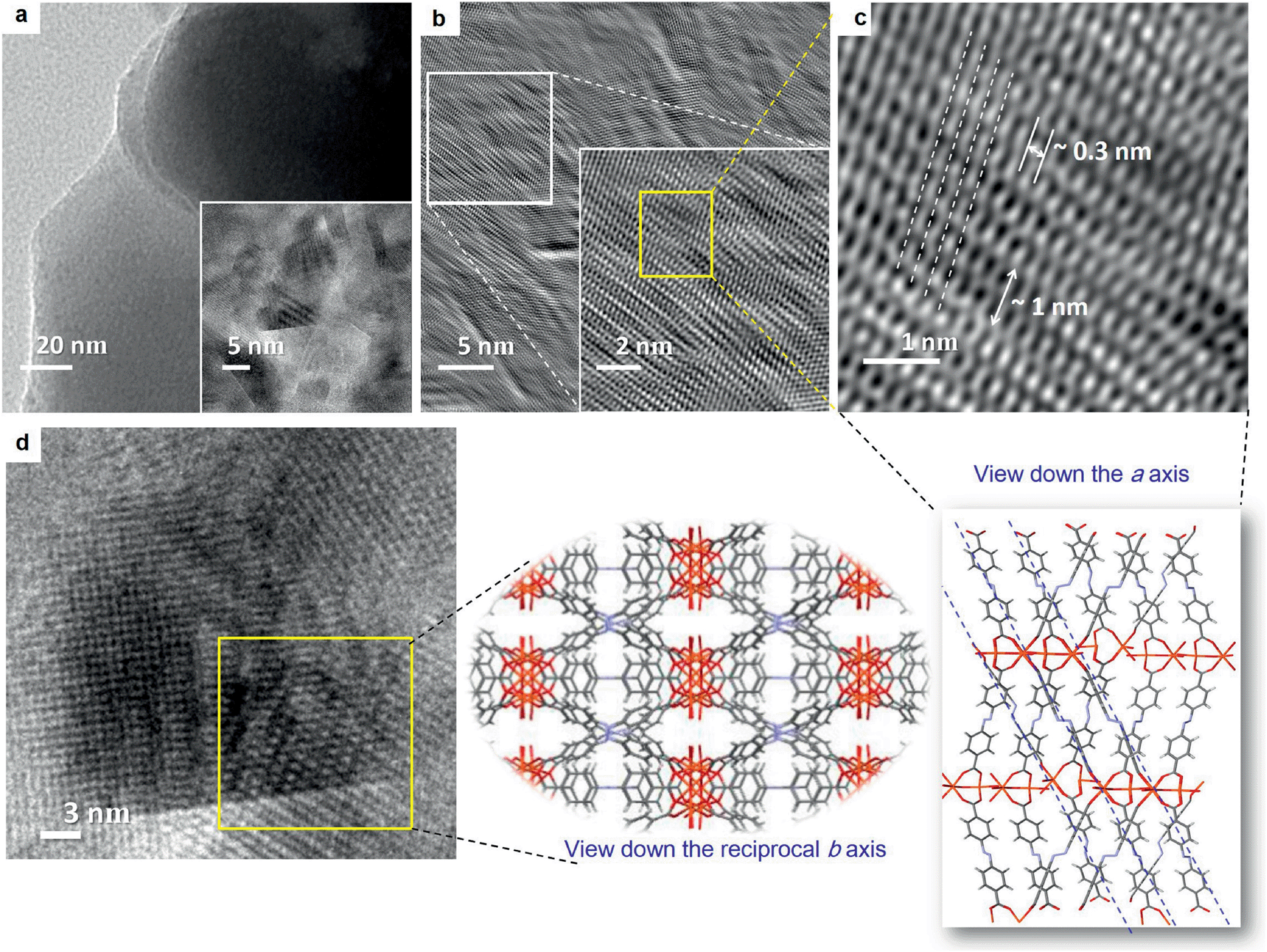 | ||
| Fig. 4 (a) TEM image of CPG1 nanosheets. HRTEM image of a nanosheet is shown in the inset. (b) IFFT reconstruction of the HR-TEM image shown as inset in (a). Zoomed view of the marked box region is shown as inset. (c and d) Molecular packing of the coordination polymeric chains observed from the reconstructed HR-TEM image. Crystal structure viewed down axis ‘a’ and reciprocal cell axis ‘b’ of PCN 243,26b are shown as a guide to the eye. | ||
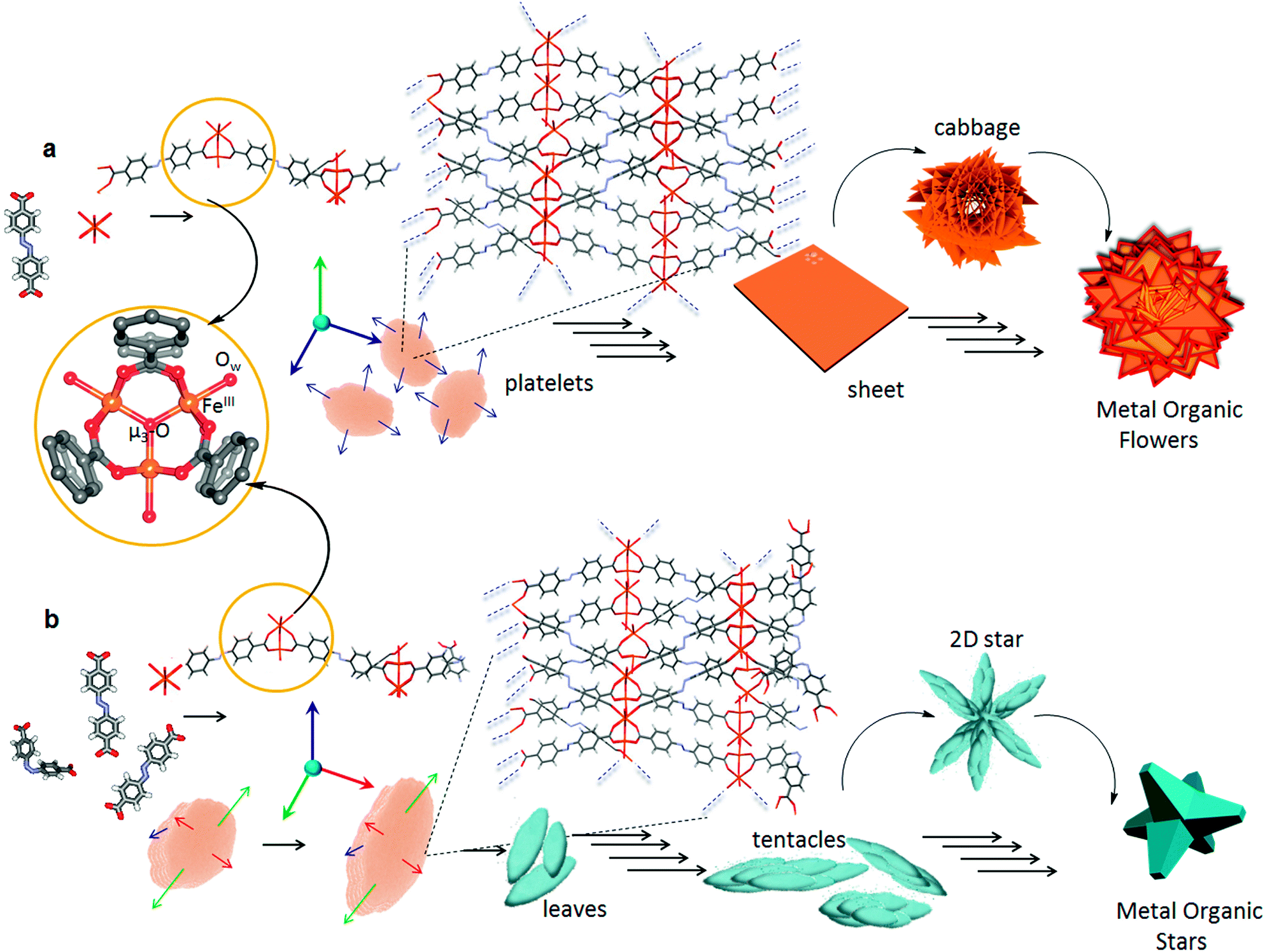 | ||
| Fig. 5 Proposed formation pathways of (a) flowers in CPG1 and (b) stars in CPG2. Detailed view of the secondary building unit (SBU) containing the [Fe3(μ3-O)(COO)6] cluster with coordinated solvent molecules (Ow). Each FeIII center is located in a highly distorted octahedral environment. Fusion of platelets in CPG1 leads to the formation of 2-D sheets and finally into flowers. The anisotropic growth along different facets in CPG2 leads to the formation of stars. Blue, green and red arrows represent maximum, moderate and restricted rates of crystal growth. Blue broken lines indicate directions of polymeric extension. Not all short contacts are shown so as to retain visual clarity. A different crystallographic view of PCN 243,26b can be found in Fig. S8.† | ||
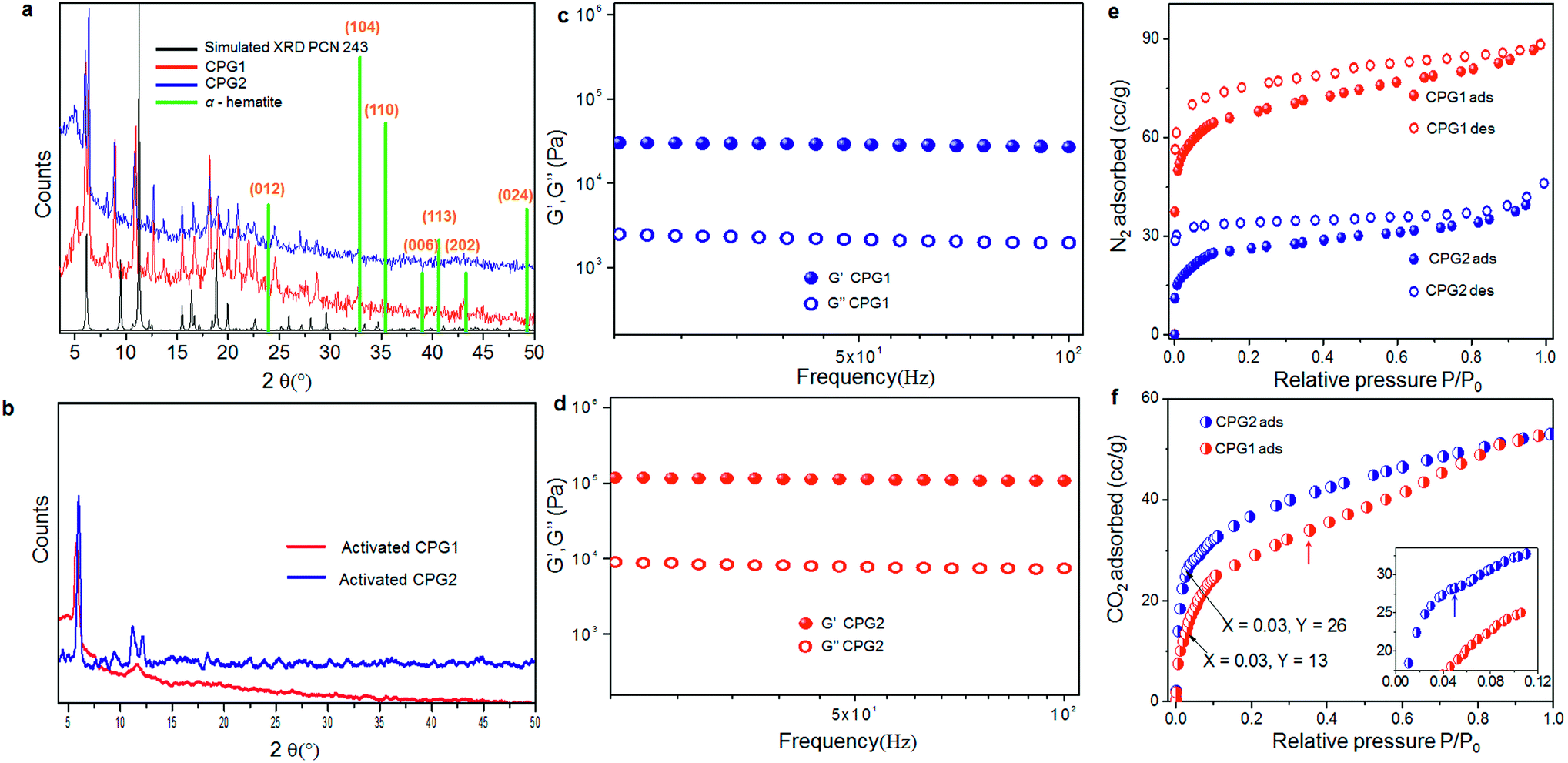 | ||
| Fig. 6 (a) XRD pattern of CPG1 and CPG2. Simulated XRD pattern of PCN 243 and stick pattern of rhombohedral hematite (α-Fe2O3, JCPDS card no. 24-0072) phase also shown for comparison. (b) XRD pattern of CPG1 and CPG2 activated at 150 °C. Rheological behaviour of (c) CPG1 and (d) CPG2. (e) Comparative N2 adsorption (ads) and desorption (des) isotherms of CPG1 and CPG2 at 77 K. (f) Comparative CO2 adsorption isotherms of CPG1 and CPG2 at 195 K. Inset shows the zoomed low-pressure region. Red and blue arrows indicate stepwise uptake of CO2 for CPG1 and CPG2, respectively. | ||
Further, in order to confirm that the mesoscopic change in the morphology is only due to the photoresponsive moiety and not due to formation of different inorganic cluster formation under UV light irradiation, a similar system derived from a non-photoisomerisable organic ligand, trimesic acid was subjected to UV light irradiation (Fig. S12†). In this case, we could not observe any change in the morphology, revealing that the inorganic cluster was not affected by any external physical stimulus such as light, unless it comes in contact with a photoresponsive organic material.
Comparative rheological behaviour and porosity analysis
Gel-like behaviour of CPG1 and CPG2 was confirmed from rheological studies where for both the materials, the value of elastic storage modulus (G′) was found to be invariant with the applied frequency and it also exceeded the value of elastic loss modulus (G′′) by an order of magnitude. These studies also lead to the conclusion that in comparison to CPG1, CPG2 has a greater storage modulus, and is more viscoelastic in nature (Fig. 6c and d). The XRD pattern of both the gels activated prior to gas adsorption studies when compared with that of the as-synthesised form indicate a lesser degree of transformation from the pristine crystalline form for activated CPG2, while activation of CPG1 transformed it almost into an amorphous material (Fig. 6b). Thus, application of heat or stress leads to less change in structure in the case of CPG2, corroborating greater rigidity and ability to retain its parent structure as compared to CPG1. This rigidity may be attributed to the compact packing of the molecular motifs in CPG2 as compared to CPG1.In order to have an idea about the porosity of the two materials, N2 gas adsorption measurements were performed on both CPG1 and CPG2. The adsorption isotherm obtained in both cases showed a typical type I behaviour according to International Union of Pure and Applied Chemistry (IUPAC) classification, confirming microporosity of the samples.6g,27 Significant hysteresis was observed in the process of desorption for both samples. The micropore dimensions calculated from the pore size distribution plot (10.2 Å) (Fig. S13†) matches well with the length of ADA. The maximum amount of N2 adsorbed at saturation pressure in the case of CPG1 was found to be 88 mL g−1 and it possessed a Brunauer–Emmett–Teller (BET) surface area ∼223 m2 g−1. In the case of CPG2, maximum uptake was found to be 46 mL g−1 of the sample and the BET surface area was substantially decreased to ∼90 m2 g−1 (Fig. 6e). The difference in gas adsorption properties has been compared with previous literature reports, where N2 and CO2 adsorption has been measured as a function of morphology or photoresponsive behaviour, and results are summarised in Tables S1 and S2,† which indicate that the porosity difference obtained in our material is quite significant. Apart from this, the surface area is higher when compared to previously reported xerogels and aerogels,28 although aerogels obtained by supercritical CO2 treatment are known to exhibit better porosity in most cases.16e In the absence of a particular crystalline phase, the drop-off in N2 gas uptake in CPG2 may be ascribed to the loss of surface area due to downsizing of the coordination network combined with considerable amount of pore blocking due to the presence of encapsulated ADA molecules, as evidenced from the FT-IR studies. As a result, CPG2 possess lower available free volume due to considerable pore blocking. The presence of encapsulated ADA molecules, leading towards a more close packed structure, supports the improvement in rheological behaviour. However, the CO2 adsorption isotherm (Fig. 6f) showed that at low partial pressure (0.03), CPG2 adsorbs almost double the amount of CO2 than CPG1. This comparatively high uptake of CO2 at lower pressure can be assigned to the ability of CPG2 to undergo a spatial adjustment to expose the lone pairs of electrons over nitrogen atoms towards the pores,29 which can then act as potential sites for CO2 adsorption (Fig. 6f, inset).30 CPG1 on the other hand shows a broad second step in the adsorption isotherm at higher partial pressure (0.46) to finally match the total amount of adsorbed CO2 found in the case of CPG2. This can be ascribed to the guest-induced second-order structural transformations taking place in CPG1.25b At higher pressures, CPG2 did not show any such change, indicating its densely packed rigid structure.
Conclusions
Results of this study point toward the conclusion that, irrespective of the presence of a photoresponsive group in CPG1 or CPG2, light does not have a post-synthetic influence on its bulk structure (Fig. S14†). However, pre-synthetic photoisomerisation of the precursor of the coordination polymer leads to morphologically different macroscopic structures. Time-dependent morphological investigations suggest that light-induced structural evolution nucleates at the molecular level, transforms at the mesoscopic level via controlled facetted crystal growth process, leading to different macroscopic morphological forms of CPG2 in the present case. Photoinduced coordination modulation can therefore be applied as a promising tool for ‘nanoarchitectonics’, to control the kinetic self-assembly of photoresponsive coordination polymers. It is further observed that the aforesaid synthetic strategy can have a profound influence on the material properties of porous metal–organic materials. Such porous gels like CPG2 with better rheological properties along with comparatively lower intake of N2 and better CO2 capture properties can easily replace the pore blocking binder materials used in generating shaped bodies from MOFs for industrial applications.31 Also the generation of iron oxide nanoparticles from such gel templates would be of interest for further investigation.32Acknowledgements
We thank the Council of Scientific and Industrial Research (CSIR), Government of India for financial support. A.A. is grateful to Department of Atomic Energy for a DAE-SRC Outstanding Researcher award. R.D.M. is thankful to CSIR, Government of India for a research fellowship. Dr J. D. Sudha, Mr C. K. Chandrakanth and Mr Kiran Mohan are acknowledged for rheology, SEM and TEM experiments, respectively.Notes and references
- (a) M. Aono, Sci. Technol. Adv. Mater., 2011, 12, 040301 CrossRef; (b) K. Ariga, Q. Ji, J. P. Hill, Y. Bando and M. Aono, NPG Asia Mater., 2012, 4, e17 CrossRef PubMed; (c) K. Ariga, Q. Ji, W. Nakanishi, J. P. Hill and M. Aono, Mater. Horiz., 2015, 2, 406 RSC; (d) K. Ariga, Y. Yamauchi, G. Rydzek, Q. Ji, Y. Yonamine, K. C.-W. Wu and J. P. Hill, Chem. Lett., 2014, 43, 36 CrossRef CAS.
- (a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS; (b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- (a) M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725 CrossRef CAS PubMed; (b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
- (a) N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H.-F. Hsu and M. Yoshizawa, J. Am. Chem. Soc., 2013, 135, 12976 CrossRef CAS PubMed; (b) R. D. Mukhopadhyay, V. K. Praveen and A. Ajayaghosh, Mater. Horiz., 2014, 1, 572 RSC.
- (a) A. Gopal, M. Hifsudheen, S. Furumi, M. Takeuchi and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 10505 CrossRef CAS PubMed; (b) S. Mahesh, A. Gopal, R. Thirumalai and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 7227 CrossRef CAS PubMed; (c) R. Rajaganesh, A. Gopal, T. M. Das and A. Ajayaghosh, Org. Lett., 2012, 14, 748 CrossRef CAS PubMed; (d) G. Abellán, E. Coronado, C. Martí-Gastaldo, A. Ribera, J. L. Jordá and H. García, Adv. Mater., 2014, 26, 4156 CrossRef PubMed.
- (a) A. Modrow, D. Zargarani, R. Herges and N. Stock, Dalton Trans., 2011, 40, 4217 RSC; (b) J. Park, D. Yuan, K. T. Pham, J. R. Li, A. Yakovenko and H. C. Zhou, J. Am. Chem. Soc., 2012, 134, 99 CrossRef CAS; (c) N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 4501 CrossRef CAS PubMed; (d) R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695 CrossRef CAS PubMed; (e) J. W. Brown, B. L. Henderson, M. D. Kiesz, A. C. Whalley, W. Morris, S. Grunder, H. Deng, H. Furukawa, J. I. Zink, J. F. Stoddart and O. M. Yaghi, Chem. Sci., 2013, 4, 2858 RSC; (f) L. Heinke, M. Cakici, M. Dommaschk, S. Grosjean, R. Herges, S. Brase and C. Woll, ACS Nano, 2014, 8, 1463 CrossRef CAS PubMed; (g) J. Park, L. B. Sun, Y. P. Chen, Z. Perry and H. C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 5842 CrossRef CAS PubMed.
- (a) S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520 RSC; (b) X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38 RSC.
- (a) H. A. Barnes, J. Non-Newtonian Fluid Mech., 1997, 70, 1 CrossRef CAS; (b) P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699 RSC; (c) M.-O. M. Piepenbrock, G. O. Llyod, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed; (d) J. Zhang and C.-Y. Su, Coord. Chem. Rev., 2013, 257, 1373 CrossRef CAS; (e) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed.
- (a) T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171 CrossRef CAS PubMed; (b) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- (a) J. Li, I. Cvrtila, M. Colomb-Delsuc, E. Otten and S. Otto, Chem.–Eur. J., 2014, 20, 15709 CrossRef CAS PubMed; (b) S. Saha, G. Das, J. Thote and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 14845 CrossRef CAS PubMed; (c) H. B. Aiyappa, S. Saha, P. Wadge, R. Banerjee and S. Kurungot, Chem. Sci., 2015, 6, 603 RSC.
- (a) O. M. Yaghi, G. Li and H. Li, Chem. Mater., 1997, 9, 1074 CrossRef CAS; (b) J. A. Foster, M.-O. M. Piepenbrock, G. O. Llyod, N. Clarke, J. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037 CrossRef CAS PubMed.
- K. J. C. van Bommel, A. Friggeri and S. Shinkai, Angew. Chem., Int. Ed., 2003, 42, 980 CrossRef CAS PubMed.
- H. Yamaguchi, Y. Kobayashi, R. Kobayashi, Y. Takashima, A. Hashidzume and A. Harada, Nat. Commun., 2012, 3, 603 CrossRef PubMed.
- S. C. Wei, M. Pan, K. Li, S. Wang, J. Zhang and C. Y. Su, Adv. Mater., 2014, 26, 2072 CrossRef CAS PubMed.
- (a) R. F. Service, Science, 2012, 335, 1167 CrossRef CAS PubMed; (b) M. Nakatsuji, Biomater. Sci., 2013, 1, 9 RSC.
- (a) C. Serre, F. Millange, S. Surble and G. Ferey, Angew. Chem., Int. Ed., 2004, 43, 6285 CrossRef PubMed; (b) T. R. Whitfield, X. Wang, L. Liu and A. J. Jacobson, Solid State Sci., 2005, 7, 1096 CrossRef CAS PubMed; (c) J. Zhang and C.-Y. Su, Coord. Chem. Rev., 2013, 257, 1373 CrossRef CAS PubMed; (d) Q. Wei and S. L. James, Chem. Commun., 2005, 1555 RSC; (e) M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056 RSC.
- (a) G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS; (b) T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427 CrossRef CAS PubMed.
- (a) M. Oh and C. A. Mirkin, Nature, 2005, 438, 651 CrossRef CAS PubMed; (b) S. Jung and M. Oh, Angew. Chem., Int. Ed., 2008, 47, 2049 CrossRef CAS PubMed; (c) Y. M. Jeon, G. S. Armatas, D. Kim, M. G. Kanatzidis and C. A. Mirkin, Small, 2009, 5, 46 CrossRef CAS PubMed; (d) A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218 RSC.
- (a) T. Witten and L. Sander, Phys. Rev. Lett., 1981, 47, 1400 CrossRef CAS; (b) J. Ge, J. Lei and R. N. Zare, Nat. Nanotechnol., 2012, 7, 428 CrossRef CAS PubMed; (c) J. Zeng and Y. Xia, Nat. Nanotechnol., 2012, 7, 415 CrossRef CAS PubMed.
- (a) H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350 CrossRef PubMed; (b) J.-L. Li and X.-Y. Liu, Adv. Funct. Mater., 2010, 20, 3196 CrossRef CAS PubMed.
- (a) J. D. H. Donnay and D. Harker, Am. Mineral., 1937, 22, 446 CAS; (b) A. Umemura, S. Diring, S. Furukawa, H. Uehara, T. Tsuruoka and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 15506 CrossRef CAS PubMed.
- F. Rakotondradany, M. A. Whitehead, A. M. Lebuis and H. F. Sleiman, Chem.–Eur. J., 2003, 9, 4771 CrossRef CAS PubMed.
- (a) W. Cho, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2008, 130, 16943 CrossRef CAS PubMed; (b) T. Tsuruoka, S. Furukawa, Y. Takashima, K. Yoshida, S. Isoda and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4739 CrossRef CAS PubMed.
- (a) E. Matijević and P. Scheiner, J. Colloid Interface Sci., 1978, 63, 509 CrossRef; (b) M. Ocaña, M. P. Morales and C. J. Serna, J. Colloid Interface Sci., 1995, 171, 85 CrossRef.
- (a) J. Reboul, S. Furukawa, N. Horike, M. Tsotsalas, K. Hirai, H. Uehara, M. Kondo, N. Louvain, O. Sakata and S. Kitagawa, Nat. Mater., 2012, 11, 717 CrossRef CAS PubMed; (b) Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata and S. Kitagawa, Science, 2013, 339, 193 CrossRef CAS PubMed.
- (a) L. Li, S. Xiang, S. Cao, J. Zhang, G. Ouyang, L. Chen and C.-Y. Su, Nat. Commun., 2013, 4, 1774 CrossRef PubMed; (b) D. Feng, K. Wang, Z. Wei, Y. P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T. F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H. C. Zhou, Nat. Commun., 2014, 5, 5723 CrossRef CAS PubMed.
- (a) H. Sato, R. Matsuda, K. Sugimoto, M. Takata and S. Kitagawa, Nat. Mater., 2010, 9, 661 CrossRef CAS PubMed; (b) W. Morris, C. J. Doonan and O. M. Yaghi, Inorg. Chem., 2011, 50, 6853 CrossRef CAS PubMed.
- (a) Y.-O. Liu, L. He, J. Zhang, X. Wang and C.-Y. Su, Chem. Mater., 2009, 21, 557 CrossRef CAS; (b) J. Zhang, X. Wang, L. He, L. Chen, C.-Y. Su and S. L. James, New J. Chem., 2009, 33, 1070 RSC; (c) S. Samai and K. Biradha, Chem. Mater., 2012, 24, 1165 CrossRef CAS.
- Y. Takashima, V. M. Martinez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef.
- C. M. Nagaraja, R. Haldar, T. K. Maji and C. N. R. Rao, Cryst. Growth Des., 2012, 12, 975 CAS.
- (a) A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284 RSC; (b) S. Mitchell, N. L. Michels, K. Kunze and J. Perez-Ramirez, Nat. Chem., 2012, 4, 825 CrossRef CAS PubMed.
- (a) S. Alam, C. Anand, K. Ariga, T. Mori and A. Vinu, Angew. Chem., Int. Ed., 2009, 48, 7358 CrossRef CAS PubMed; (b) W. Cho, S. Park and M. Oh, Chem. Commun., 2011, 47, 4138 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Text, figures, and tables depicting different synthetic procedures, detailed experimental procedures, characterisation data including FT-IR, microscopic analyses, TGA, EDAX and SAED analyses, pore size distribution. See DOI: 10.1039/c5sc02233a |
| This journal is © The Royal Society of Chemistry 2015 |