
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand survey results in identification of PNP pincer complexes of iridium as long-lived and chemoselective catalysts for dehydrogenative borylation of terminal alkynes†
Chun-I
Lee
a,
Jessica C.
DeMott
a,
Christopher J.
Pell
a,
Alyson
Christopher
b,
Jia
Zhou
c,
Nattamai
Bhuvanesh
a and
Oleg V.
Ozerov
*a
aDepartment of Chemistry, Texas A&M University, College Station, TX 77842, USA. E-mail: ozerov@chem.tamu.edu
bDepartment of Chemistry, Brandeis University, MS 015, 415 South Street, Waltham, MA 02454, USA
cDepartment of Chemistry, Harbin Institute of Technology, Harbin 150001, China
First published on 4th August 2015
Abstract
Following the report on the successful use of SiNN pincer complexes of iridium as catalysts for dehydrogenative borylation of terminal alkynes (DHBTA) to alkynylboronates, this work examined a wide variety of related pincer ligands in the supporting role in DHBTA. The ligand selection included both new and previously reported ligands and was developed to explore systematic changes to the SiNN framework (the 8-(2-diisopropylsilylphenyl)aminoquinoline). Surprisingly, only the diarylamido/bis(phosphine) PNP system showed any DHBTA reactivity. The specific PNP ligand (bearing two diisopropylphosphino side donors) used in the screen showed DHBTA activity inferior to SiNN. However, taking advantage of the ligand optimization opportunities presented by the PNP system via the changes in the substitution at phosphorus led to the discovery of a catalyst whose activity, longevity, and scope far exceeded that of the original SiNN archetype. Several Ir complexes were prepared in a model PNP system and evaluated as potential intermediates in the catalytic cycle. Among them, the (PNP)Ir diboryl complex and the borylvinylidene complex were shown to be less competent in catalysis and thus likely not part of the catalytic cycle.
Introduction
Selective conversion of C–H bonds into C–B bonds (Fig. 1) has attracted broad attention over the last two decades.1,2 The resulting organoboron compounds are relatively stable, non-toxic, and can be easily transformed into C–O or C–C bonds via oxidation3–6 or Suzuki–Miyaura coupling.7–9 Many examples of alkane,10–12 arene,13,14 and alkene15,16 dehydrogenative borylation, as well as benzylic17 and allylic18 C–H borylation have been reported. The dehydrogenative arene borylation has been proven especially fruitful with very impressive advances by Hartwig et al.19,20 and Smith and Maleczka et al.21,22 already finding applications.2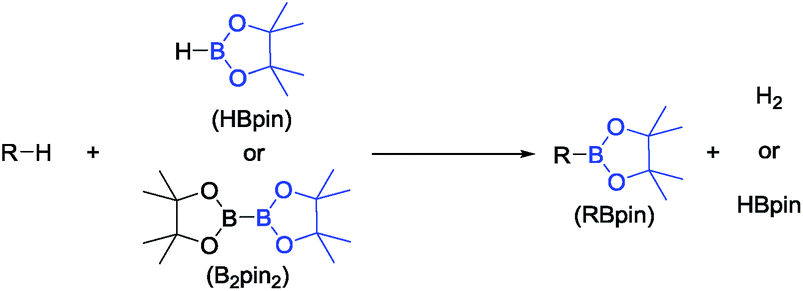 | ||
| Fig. 1 C–H borylation (dehydrogenative borylation). | ||
The development of C–H borylation methods did not include the C(sp)–H bonds of terminal alkynes. The products of dehydrogenative borylation of terminal alkynes (DHBTA), alkynylboronates, are versatile building blocks in synthetic chemistry. Their synthetic value derives not just from the direct use in C–Calkynyl coupling,23 but more so from pursuing the reactions of the triple bond. Cyclotrimerization,24 [3 + 2] cycloaddition,25 cyclopentenone synthesis,26 hydrozirconation,27 enyne metathesis,28 and others29–32 have been reported; these reactions yield more complex molecules that contain C–B bonds in positions that would be difficult to borylate by alternative means.
The classical synthesis of alkynylboronates was developed by Brown et al.: deprotonation of alkyne by n-BuLi, followed by reaction with a boric ester and quench with anhydrous acid.33 An Ag-catalyzed variation was reported in 2014 by Hu et al.34 Ingleson et al. also recently demonstrated that certain borenium cations can react with terminal alkynes to give alkynylboronates.35 Just as with borylation of sp2 and sp3 C–H bonds, catalysis of direct coupling of a C–H bond with a B–H bond (Fig. 1 and R = alkynyl for DHBTA) carries significant advantages (if the catalysis is efficient enough): better atom economy, as well as milder conditions allowing greater functional group compatibility. In comparison to C(sp2)–H and C(sp3)–H bonds, for the relatively acidic C(sp)–H bonds (pKa ∼ 25) of terminal alkynes, the C–H activation itself is generally not a difficult task and C–H bond selectivity would not typically be an issue. On the other hand, in contrast to the non-olefinic C(sp2)–H and C(sp3)–H substrates, a combination of a triple C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond, a B–H bond and a metal catalyst is very likely to lead to hydroboration.36,37 In addition, hydrogenation38 of the alkyne substrate or product with H2 (the by-product of DHBTA) may also be a concern.
C bond, a B–H bond and a metal catalyst is very likely to lead to hydroboration.36,37 In addition, hydrogenation38 of the alkyne substrate or product with H2 (the by-product of DHBTA) may also be a concern.
In 2013, we reported the first example of catalytic DHBTA performed by Ir complexes of a SiNN pincer39 ligand (1-Ir-COE and 1-Ir-Bpin2, Fig. 2).40 The reaction was strictly chemoselective and could be performed under very mild conditions (ambient temperature, ca. 10 turnovers per min) with a variety of alkyl-, aryl- and silyl- terminal alkynes in high yield. However, the catalyst longevity was limited to ca. 100 turnovers. Very recently, Tsuchimoto et al. described DHBTA catalysis by Zn(OTf)2/pyridine using 1,8-nathpthalenediamidoborane.41 The Tsuchimoto process displayed a wide scope similar to (SiNN)Ir, but operated much slower (ca. 1 turnover per hour) even at 100 °C. Our group also reported that (POCOP)Pd complexes are modest DHBTA catalysts for some substrates.42
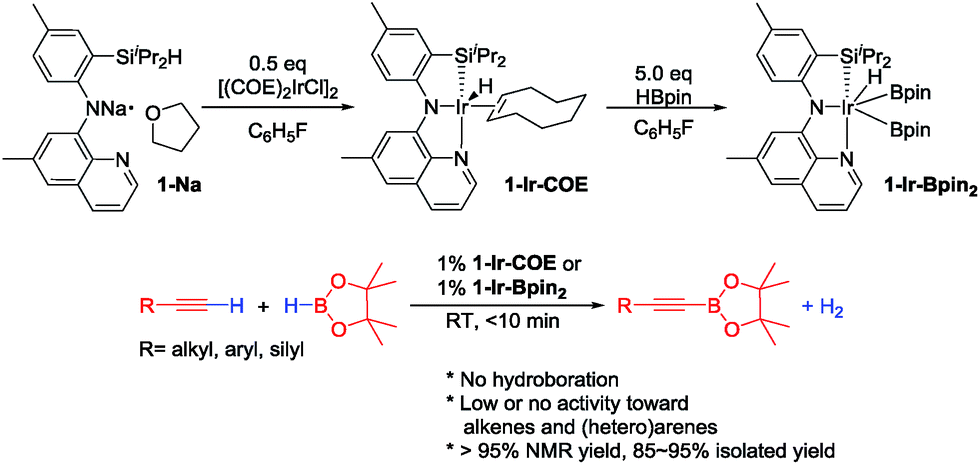 | ||
| Fig. 2 Synthesis of SiNN Ir complexes (top) and DHBTA catalyzed by SiNN Ir complexes (bottom). | ||
The discovery of the prowess of the SiNN ligand in DHBTA was rather serendipitous, and we sought to explore the associated ligand space in a more systematic fashion. Here we report the exploration of a series of Ir complexes of related ligands as potential catalysts in DHBTA that has led to the discovery of a new highly active, much more long-lived catalyst with a broader scope, as well as to the insight into the role of possible intermediates in DHBTA.
Results and discussion
Synthesis and screening of ligands for DHBTA
In light of the success of 1-Ir-COE in DHBTA, we decided to examine a series of ligands that systematically explored variations of the SiNN ligand features (Fig. 3). From 2-H to 7-H, we preserved the central amido donor and the quinoline fragment of SiNN but removed the silane side arm (2-H) or replaced it with hemilabile donors (3-H to 7-H). For 8-H and 9-H, the silane segment and the central amido donor were maintained while the quinoline moiety was eliminated (8-H) or substituted with a phosphine donor (9-H). We also included the PNP ligand (10-H) and the PCP/POCOP ligands (11-H to 14-H) because these are commonly used pincer ligands with a rich history of C–H activation chemistry with Ir.43–46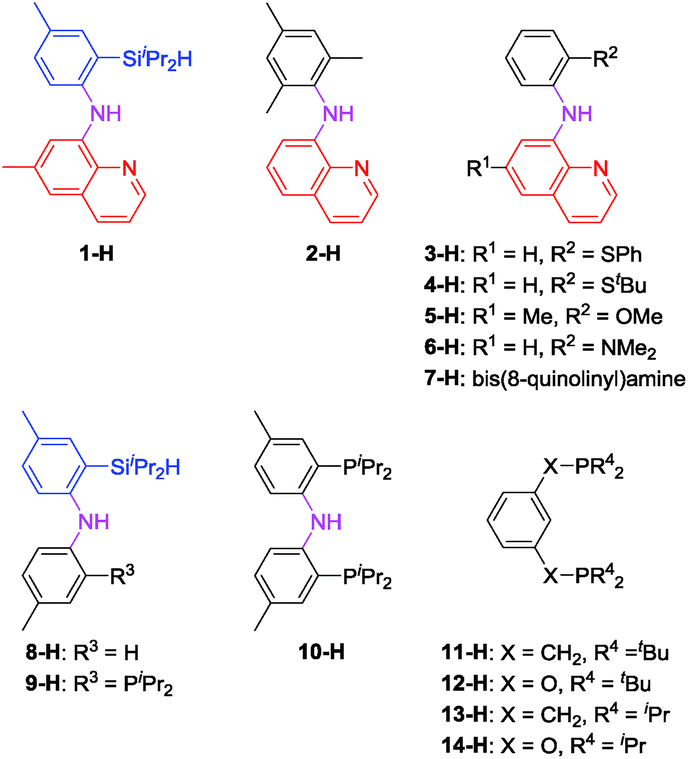 | ||
| Fig. 3 Ligands selected for DHBTA screening. | ||
The syntheses of ligands used in the screening of DHBTA are shown in Scheme 1. Quinoline derivatives (i.e.2-H,473-H, 4-H, 5-H, 6-H,487-H48) were readily synthesized via Buchwald–Hartwig coupling of 8-bromoquinoline with various anilines or 8-aminoquinoline with various bromoarenes. 8-H was prepared via the intermediate S1. The synthesis of S1 relied on the same selective dilithiation of bis(2-bromo-4-methylphenyl)amine we previously used in the synthesis of S2,49,50 followed by quenching with water. Pure S1 was isolated in 86% yield by column chromatography. Treatment of S1 with n-BuLi, followed by addition of iPr2SiHCl and workup gave 8-H in 73% yield. The new SiNP ligand 9-H was prepared from S2 through a similar protocol.
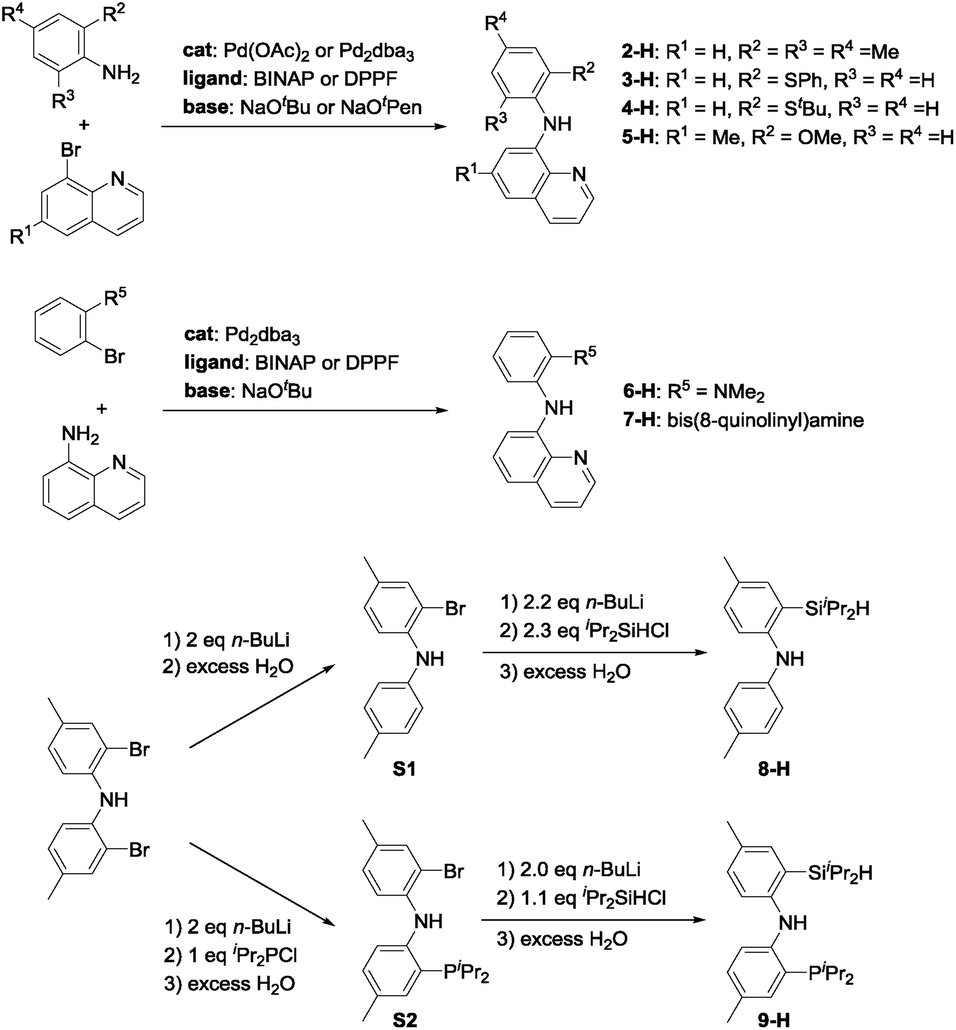 | ||
| Scheme 1 Synthesis of ligands used in screening for DHBTA. | ||
In our original DHBTA report,40 we demonstrated that generation of the 1-Ir-COE precatalyst in situ from 1-Na and [(COE)2IrCl]2 (Scheme 2) produced results equivalent to those obtained using isolated 1-Ir-COE. Therefore, we used a similar synthetic approach here for testing catalysis using the series of ligands with central amido donors (2-H to 10-H). They were deprotonated with 1 equiv. of NaN(SiMe3)2in situ, allowed to react with 0.5 equiv. of [(COE)2IrCl]2 in C6D6 and the resultant solutions were tested for catalytic DHBTA activity. In the case of PCP/POCOP ligands 11–14, we isolated dihydride complexes (11-Ir-H251 and 12-Ir-H252) or alkene complexes (13-Ir-C2H4 and 14-Ir-COE) to be used in DHBTA testing.
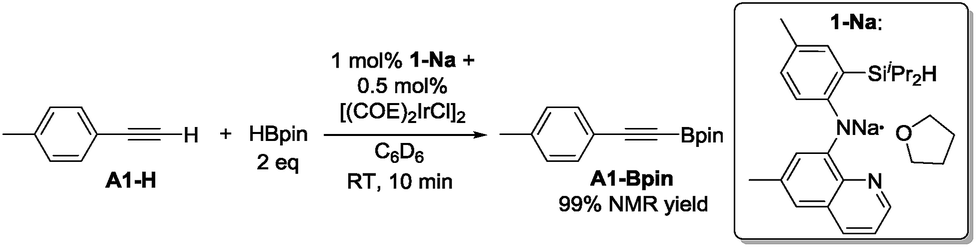 | ||
| Scheme 2 DHBTA catalyzed by 1-Ir-COE generated in situ from 1-Na. | ||
The precatalyst 13-Ir-C2H4 was obtained after modification of previously reported procedures for related compounds (Scheme 3, top).53–55 Thermolysis of 13-H with [(COE)2IrCl]2 at 80 °C overnight in toluene resulted in a dark red solution that contained ca. 85% of the desired product (31P NMR evidence). Column chromatography allowed for the collection of 99% pure 13-Ir-HCl in 22% yield. This portion of 13-Ir-HCl was then treated with a slight excess of NaOtBu in toluene, degassed, and then stirred under an atmosphere of ethylene for 30 min. After filtration and removal of volatiles under vacuum, analytically pure 13-Ir-C2H4 was obtained as a dark brown solid in 66% isolated yield (based on 13-Ir-HCl). 14-Ir-COE was synthesized by reacting the previously reported 14-Ir-HCl56,57 with a slight excess of NaOtBu and COE in C6D6 (Scheme 3, bottom).
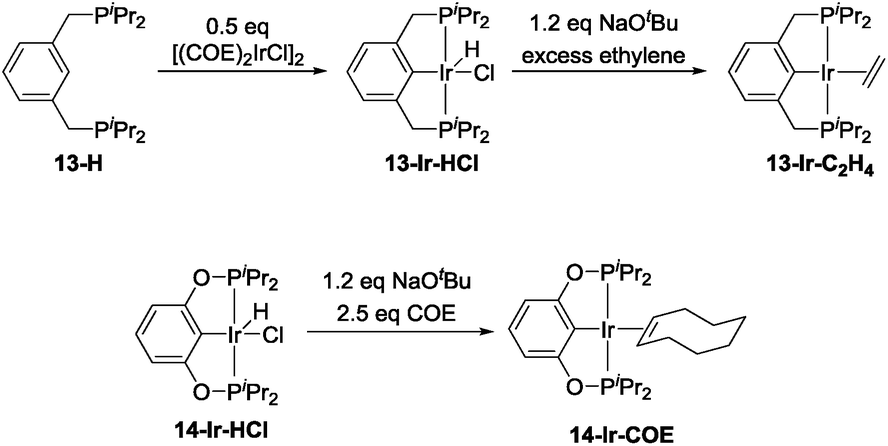 | ||
| Scheme 3 Synthesis of 13-Ir-C2H4 and 14-Ir-COE. | ||
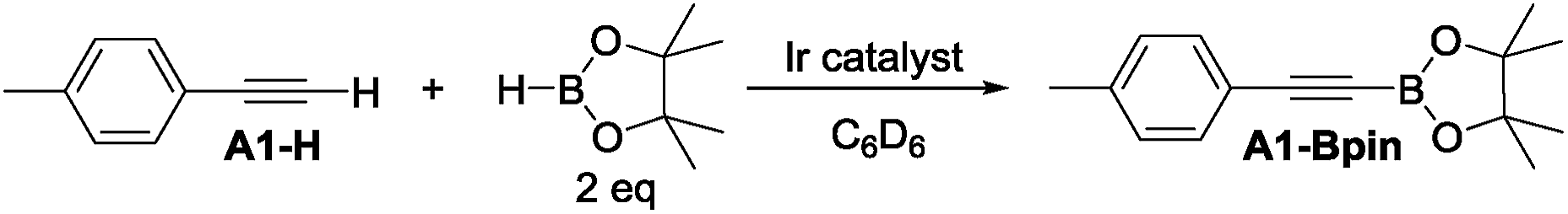
4-Ethynyltoluene (A1-H) was selected as the alkyne for testing. We mimicked the conditions that were successful for the SiNN ligand 1, with 1 mol% Ir loading and 2 equiv. of HBpin used at ambient temperature in C6D6 solvent. The results are summarized in Chart 1. Surprisingly, of all the ligands tested, only 10-H showed any DHBTA reactivity. In all other cases, no evidence for the DHBTA product A1-Bpin was visible by 1H NMR spectroscopy after 1 h. In general, sluggish and nonselective hydrogenation and hydroboration was observed for 2-H to 9-H, and for the iso-propyl PCP/POCOP iridium complexes (13-Ir-C2H4 and 14-Ir-COE). For tert-butyl PCP/POCOP iridium complexes (11-Ir-H2 and 12-Ir-H2), a mixture of trans-alkenylboronate (A1-1) and cis-alkenylboronate (A1-2) were observed as major products. The use of 10-H resulted in 76% A1-Bpin after 10 min and 90% (NMR evidence) after 1 h, with about 3% of 4-ethyltoluene (A1-3, from apparent hydrogenation of A1-H). We also tested one of the most active arene borylation catalyst systems ([(COD)Ir(OMe)]2 + 4,4′-di-tert-butyl-bipyridine)58 but no catalysis of any kind was observed after 1 h at RT.59 It is difficult to rationalize the results of the ligand screen other than to cautiously note that a central amido donor may be crucial and that selectivity for DHBTA is quite sensitive to the balance of steric and electronic factors.
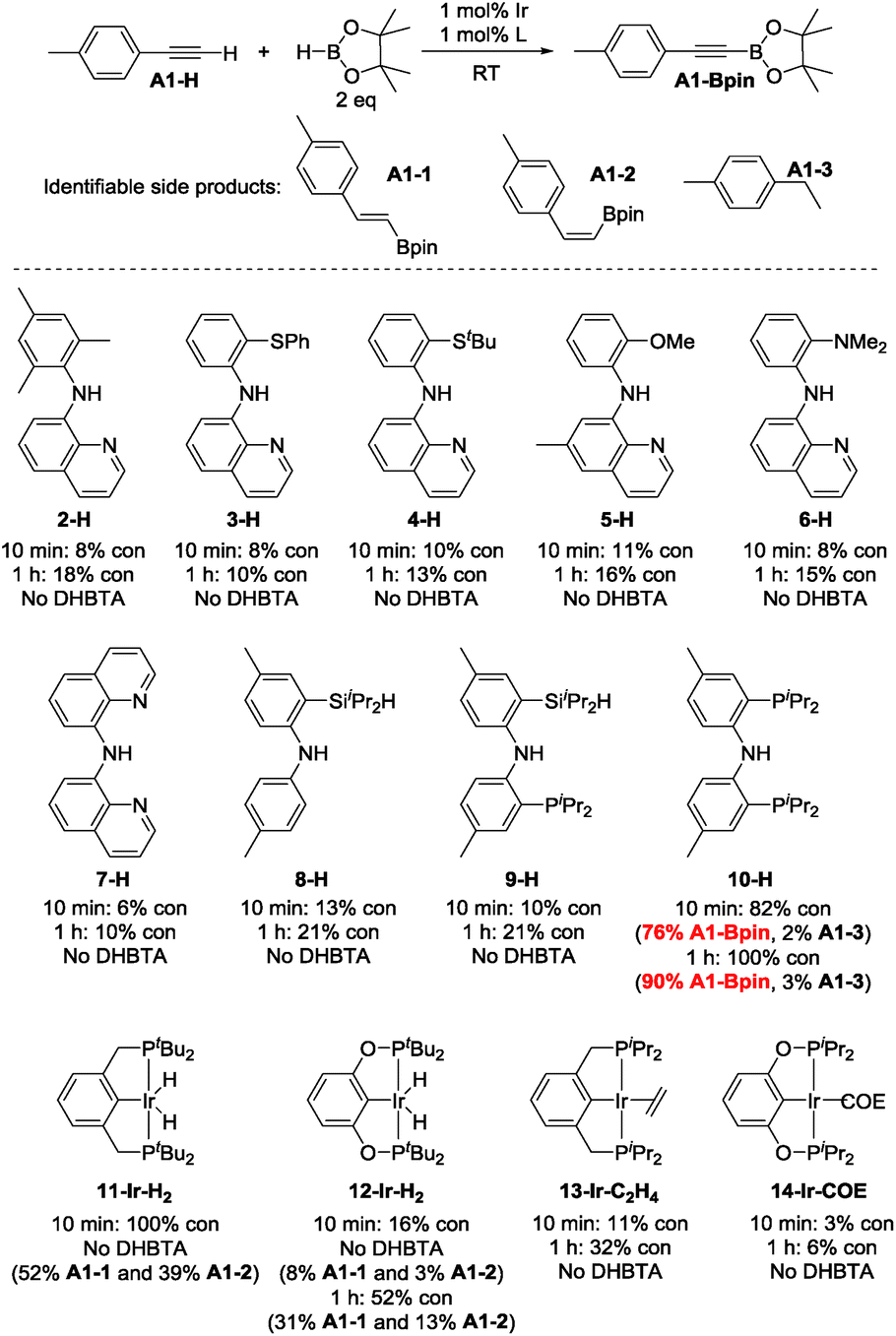 | ||
| Chart 1 Ligand screening in DHBTA. For 2-H to 10-H: in the following order, the ligand (0.0010 mmol), NaN(TMS)2 (0.0010 mmol), [(COE)2IrCl]2 (0.00050 mmol) and HBpin (0.20 mmol) were mixed in C6D6 in a J. Young tube. 4-Ethynyltoluene (0.10 mmol) was then added in 4 portions with 1 min intervals and the mixture was allowed to stand at ambient temperature for 10 min (see ESI† for details). For 11-H to 14-H: the iridium complex (0.0010 mmol) and HBpin (0.20 mmol) were mixed in C6D6 in a J. Young tube. 4-ethynyltoluene (0.10 mmol) was then added in 4 portions with 1 min intervals and the mixture was allowed to stand at ambient temperature for 10 min (see ESI† for details). The numbers for “% con” refer to the conversion of A1-H. | ||
With this lead in hand, we tested isolated 10-Ir-H259 as a catalyst in reactions with 4-ethynyltoluene (A1-H), trimethylsilylacetylene (A2-H), and 1-hexyne (A3-H) (Chart 2). The effectiveness of 10-Ir-H2 in the DHBTA of 4-ethynyltoluene (A1-H) was similar to the catalyst generated from 10-Hin situ. DHBTA of trimethylsilylacetylene (A2-H) was finished in 1 h and gave an excellent yield of A2-Bpin. The catalytic activity of 10-Ir-H2 towards A3-H was significantly lower than towards A1-H and A2-H and only 50% yield was achieved after 3 h. A small amount of the hydrogenation product A1-3 was observed in DHBTA of A1-H, but no hydrogenation products were detected in the reactions of A2-H and A3-H.
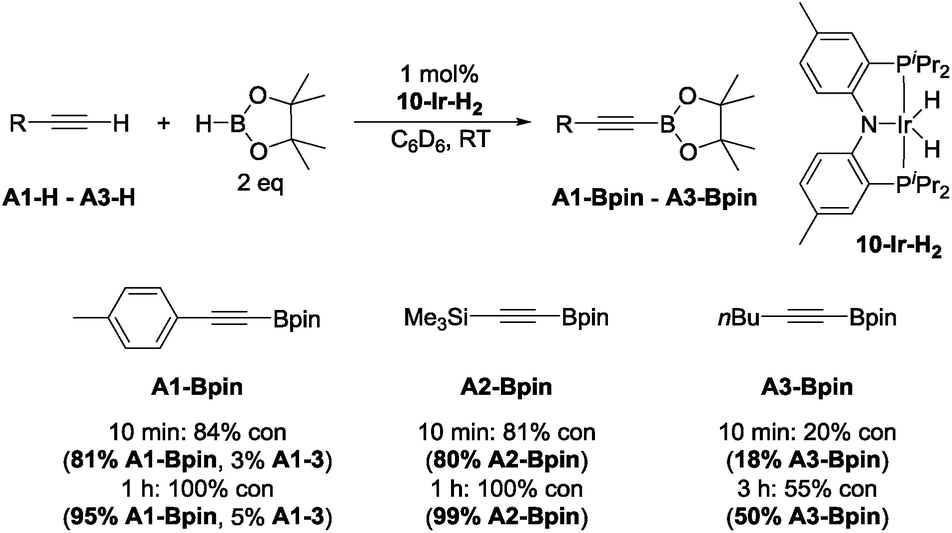 | ||
| Chart 2 DHBTA catalyzed by 10-Ir-H2. | ||
Testing of (PNP)Ir complexes with various phosphine substituents
The effectiveness of 10-Ir-H2 fell somewhat short of the SiNN-based catalysis, where >95% yield of A1/2/3-Bpin was obtained in <10 min and without any hydrogenation side products. Nonetheless, we were encouraged by the results because the PNP framework offers facile opportunities for optimization of the ligand via substituent variation. We selected previously reported PNP ligands 15-H,5016-H,60 and 17-H50,61 for further testing in DHBTA. The syntheses of the corresponding Ir-COE complexes are depicted in Scheme 4. 15-H is an oil that is difficult to purify; however, the Li derivative (15-Li) could be isolated in in 56% yield as a pure solid. 15-Li was then reacted with 0.5 equiv. of [(COE)2IrCl]2 to yield 15-Ir-COE. 16-Ir-COE62 and 17-Ir-COE were synthesized via one-pot reactions by deprotonation of the neutral ligands in situ and treatment with [(COE)2IrCl]2.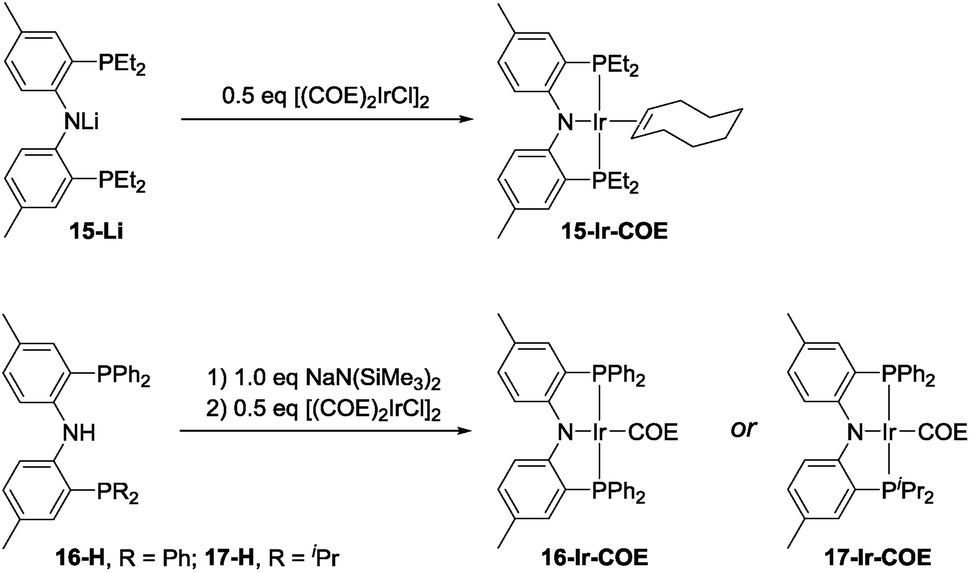 | ||
| Scheme 4 Synthesis of (PNP)Ir(COE) complexes. | ||
The newly synthesized and isolated (PNP)Ir(COE) complexes (15-Ir-COE, 16-Ir-COE, 17-Ir-COE), 10-Ir-H2, and the previously reported 1-Ir-COE were all tested in DHBTA by using A1-H as the substrate with 2 equiv. of HBpin at ambient temperature in C6D6 solvent. The results are summarized in Table 1. At 1 mol% catalyst loading, 15-Ir-COE, 17-Ir-COE, 10-Ir-H2, as well as 1-Ir-COE gave excellent yields of A1-Bpin at ambient temperature, whereas 16-Ir-COE did not (entry 4) and was eliminated from further consideration. At 0.25% Ir, 17-Ir-COE showed superior reactivity to 1-Ir-COE, 10-Ir-H2, and 15-Ir-COE by producing 92% A1-Bpin in 10 min. 1-Ir-COE gave 43% yield after 1 h (entry 6) and the yield did not increase with longer reaction times, suggesting faster catalyst decomposition for the SiNN-based catalyst. 17-Ir-COE was able to effect 100% conversion of A-1H and 85% yield of A1-Bpin (NMR evidence) even at 0.025% loading in only hours at ambient temperature. The reaction rate was higher at 60 °C (entry 12) without loss in yield. The 84% yield of A1-Bpin at 0.025 mol% catalyst loading (entry 12) corresponds, impressively, to 3400 turnovers. Under incomplete conversion with 0.01 mol% loading (entry 13), a turnover number of 6500 was achieved after 2 h at 60 °C. In terms of chemoselectivity, 1-Ir-COE is superior in DHBTA of A1-H as it gave A1-Bpin as the product exclusively; 2–10% of hydrogenation product A1-3 was observed in all reactions catalyzed by the (PNP)Ir complexes (Fig. 4). Because 15 and 17 gave faster catalysis than 10, it is possible that a less sterically encumbered ligand is advantageous. The lower reactivity of 16 may in turn reflect sensitivity to the electronic factors.
|
|
||||||
|---|---|---|---|---|---|---|
| # | Catalyst | [Ir] mol% | Time | Con (%) | Yield (%) | A1-3 (%) |
| a The iridium complex and HBpin (0.20 mmol) were mixed in C6D6 in a J. Young tube. A1-H (0.10 mmol) was then added in 4 portions with 1 min intervals and the mixture was allowed to stand at ambient temperature (see ESI for details). b 10 min: 81% yield. c 10 min: 37% yield. d 10 min: 34% yield. e 10 min: 45% yield. f 10 min: 13% yield. g Run at 60 °C. | ||||||
| 1 | 1-Ir-COE | 1 | 10 min | 100 | 99 | 0 |
| 2 | 10-Ir-H 2 | 1 | 1 h | 100 | 95b | 5 |
| 3 | 15-Ir-COE | 1 | 10 min | 100 | 97 | 2 |
| 4 | 16-Ir-COE | 1 | 10 min | 27 | 21 | 2 |
| 5 | 17-Ir-COE | 1 | 10 min | 100 | 97 | 2 |
| 6 | 1-Ir-COE | 0.25 | 1 h | 44 | 43c | 0 |
| 7 | 10-Ir-H 2 | 0.25 | 4 h | 100 | 90d | 6 |
| 8 | 15-Ir-COE | 0.25 | 2 h | 100 | 82e | 5 |
| 9 | 17-Ir-COE | 0.25 | 10 min | 100 | 92 | 2 |
| 10 | 17-Ir-COE | 0.05 | 2 h | 100 | 85 | 7 |
| 11 | 17-Ir-COE | 0.025 | 8 h | 100 | 85f | 9 |
| 12 | 17-Ir-COE | 0.025 | 1 hg | 100 | 84 | 10 |
| 13 | 17-Ir-COE | 0.01 | 2 hg | 77 | 65 | 5 |
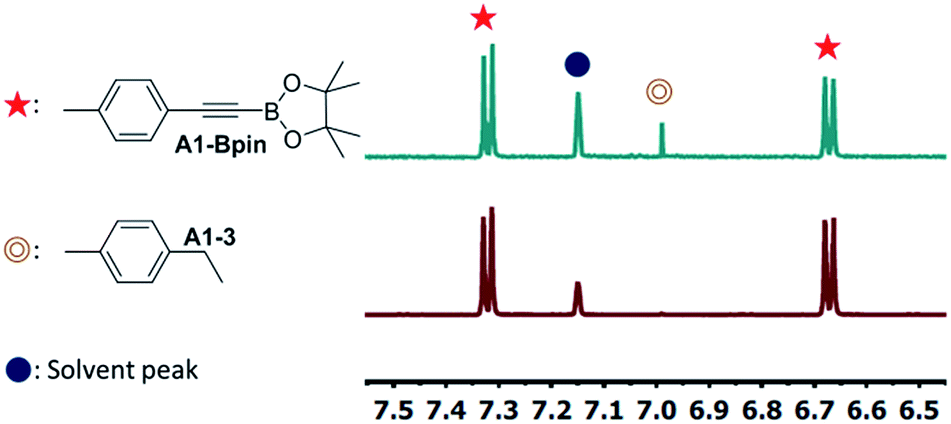 | ||
| Fig. 4 Partial 1H NMR spectra of DHBTA reaction mixtures catalyzed by (a) 1 mol% 15-Ir-COE (entry 3 in Table 1) and (b) 1 mol% 1-Ir-COE (entry 1 in Table 1). | ||
To further explore the catalytic reactivity of 17-Ir-COE, A1-H, A2-H, 5-chloro-1-pentyne (A4-H), as well as 3-methyl-3-trimethylsiloxy-1-butyne (A5-H), trimethylsilyl propargyl ether (A6-H), 4-dimethylamino-phenylacetylene (A7-H), N-tosylated allyl propargyl amine (A8-H), and dimethyl 2-allyl-2-(prop-2-yn-1-yl)malonate (A9-H) were chosen as representative substrates for aromatic, silyl, aliphatic terminal alkynes, propargyl derivatives and 1,6-enynes, respectively (Chart 3). For A1-H, 84% NMR yield was observed and accompanied with 10% hydrogenation product A1-3; similar results were obtained with A6-H and A7-H. 96–99% NMR yields were obtained for A2-, A4-, A5, and A9-Bpin with 0.025 to 0.1 mol% loading of 17-Ir-COE as the catalyst. Borylation of A2-H and A6-H was also performed with reduced amount of HBpin (1.1 eq.) and comparable yields/side product (A6-1 for A6-H) were obtained. No DHBTA products were observed for phenyl propargyl sulfide, 3-ethynylpyridine, 4-cyano-1-butyne, 3,3-diethoxy-1-propyne, and methyl propiolate with 0.1 mol% 17-Ir-COE. A1-Bpin, A5-Bpin and A8-Bpin could be easily purified by recrystallization and were isolated in good yields in preparative-scale reactions. In contrast, 1-Ir-COE requires 1% loading for high yields of A1-, A2-, A4-, and A5-Bpin, and is altogether ineffective for the synthesis of propargyl derivatives A6- and A8-Bpin (<10% yield at 1% catalyst loading). A mercury drop test63 was performed with 0.025 mol% 17-Ir-COE loading and A1-H as substrate. No significant yield changes were observed for either A1-Bpin or the major side-product A1-3 which suggested that the catalysis is homogeneous.
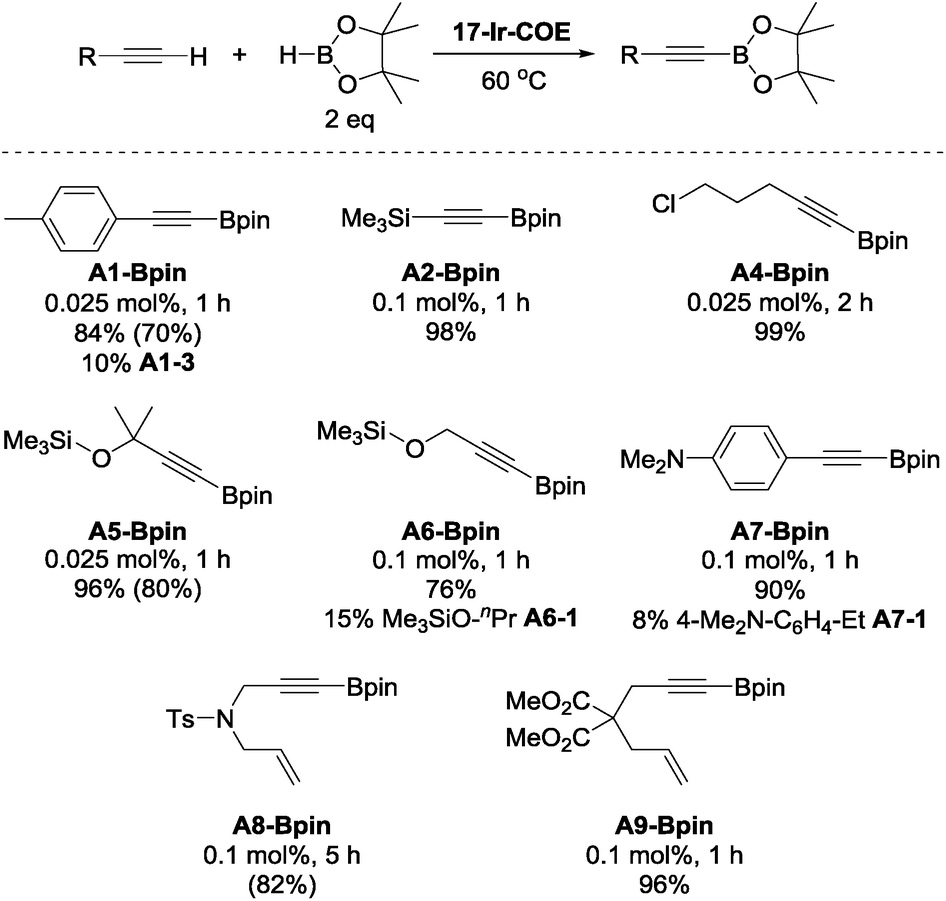 | ||
| Chart 3 DHBTA of representative terminal alkynes catalyzed by 17-Ir-COE. a17-Ir-COE and HBpin (0.20 mmol) were mixed in C6D6 in a J. Young tube. Alkyne (0.10 mmol) was then added in 4 portions with 1 min intervals at RT and the mixture was heated at 60 °C (see ESI† for details). b NMR yield. c Yields in parentheses are isolated yields in preparative-scale (10 mmol alkyne) reactions that used toluene or fluorobenzene as solvent instead of C6D6. | ||
Synthesis of plausible DHBTA intermediates
In order to gain new insight into the reaction mechanism, we set out to examine conceivable intermediates in DHBTA. Because of its NMR-friendly C2v-symmetric structure, and because a number of its iridium complexes are already known,44–46 we opted for ligand 10 for this study. We were particularly interested in determining the possible products arising from combining the 10-Ir fragment with HBpin, terminal alkynes, and alkynylboronates and their catalytic competence.In our report on the DHBTA activity of SiNN-based catalysts, we showed that the iridium diboryl complex 1-Ir-Bpin2 can be synthesized by reacting 1-Ir-COE with 5 equivalents of HBpin,40 and that isolated 1-Ir-Bpin2 exhibited the same catalytic activity as 1-Ir-COE. Treating 10-Ir-H2 with 5 equivalents of HBpin, however, led to a mixture of 10-Ir-H3Bpin and 10-Ir-HBpin in equilibrium with free H2 (top, Scheme 5). To access 10-Ir-Bpin2, we employed an alternative route of heating 10-Ir-HMes, a good synthon for 10-Ir,45 with 1 equiv. of B2pin2. This permitted isolation of 10-Ir-Bpin2 in 83% yield (bottom, Scheme 5).
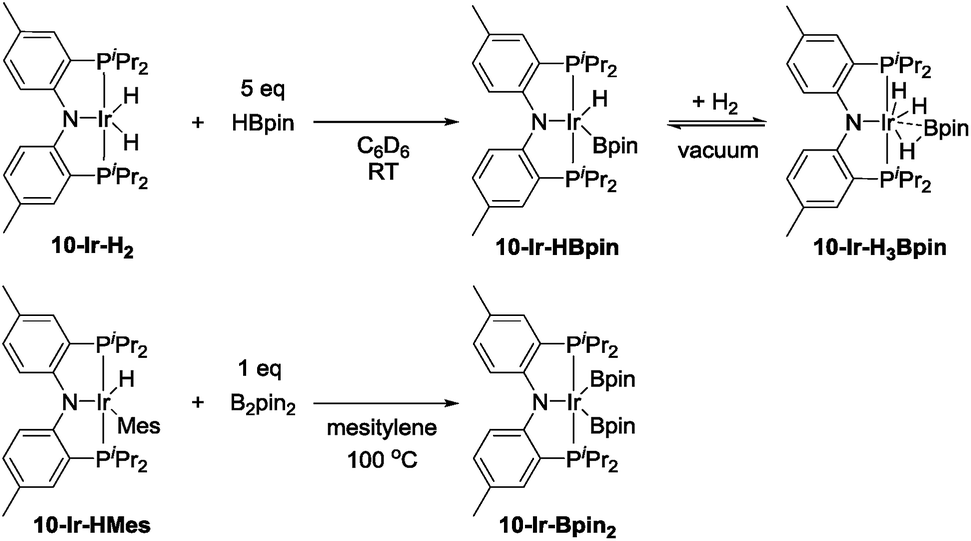 | ||
| Scheme 5 The synthesis of 10-Ir-HBpin and its equilibrium with 10-Ir-H3Bpin (top). The synthesis of 10-Ir-Bpin2 (bottom). | ||
10-Ir-HBpin exhibited an upfield signal at −19.8 ppm (t, JP-H = 8.4 Hz, 1H) in its 1H NMR spectrum, and the peak sharpened upon 11B decoupling (Fig. 5, left) which suggested that this proton interacted with a boron atom of the boryl. 10-Ir-H3Bpin displayed two broad upfield signals at −5.3 (1H, ω1/2 = 60 Hz) and −12.4 (2H, ω1/2 = 64 Hz) ppm in the 1H NMR spectrum at ambient temperature. The resonance at −5.3 ppm (ω1/2 = 35 Hz) sharpened upon 11B decoupling (Fig. 5, right), but the width of the peak at −12.4 ppm remained unchanged, indicating that only the proton associated with the resonance at −5.3 ppm displayed substantial coupling to the boron nucleus. The −12.4 ppm signal of 10-Ir-H3Bpin resolved into two distinct resonances (−9.43, −15.35 ppm, Fig. 6) upon cooling to 213 K. On the basis of the 1H{11B} and VT 1H NMR spectroscopic data, 10-Ir-H3Bpin is best described as an exo-σ-borane dihydride complex, similarly to 12-Ir-H3Bpin in the study by Goldberg and Heinekey.64
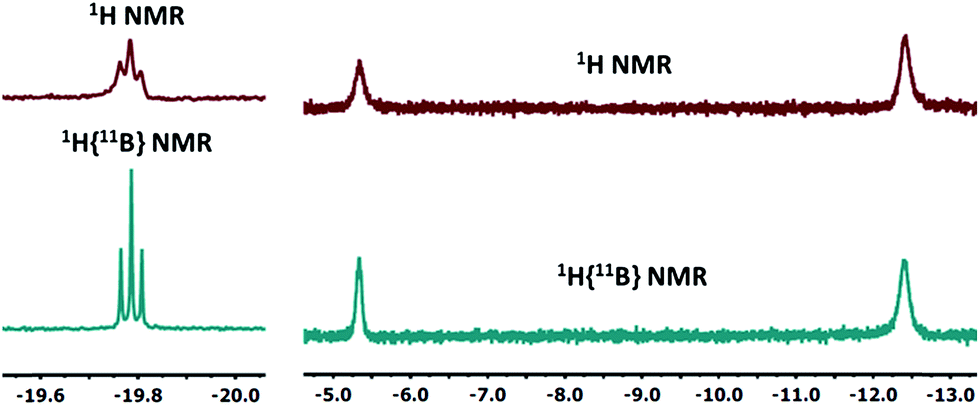 | ||
| Fig. 5 The upfield region of 1H and 1H{11B} NMR spectrum (400 MHz, C6D6) of 10-Ir-HBpin (left) and 10-Ir-H3Bpin (right). | ||
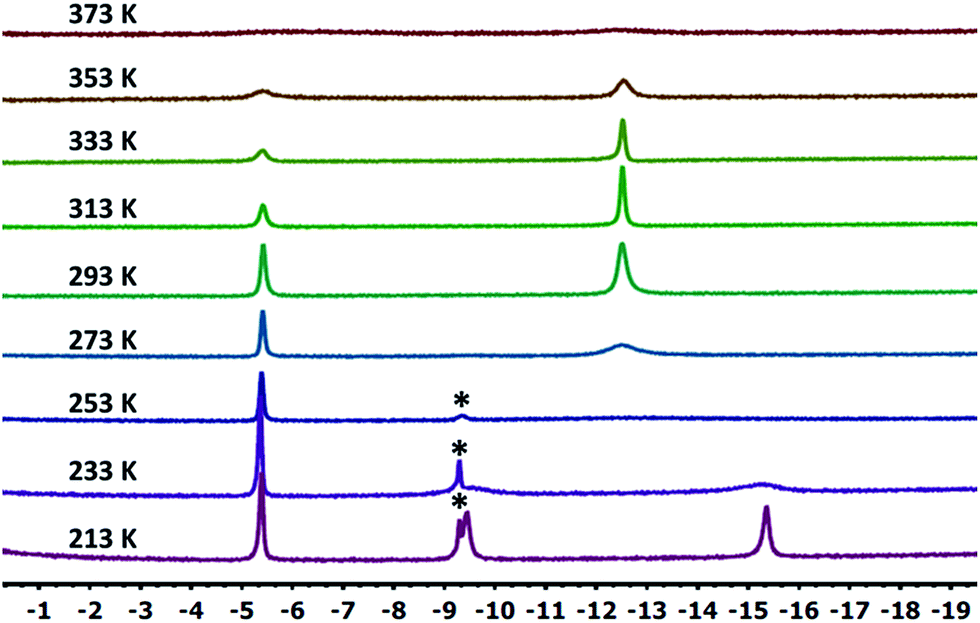 | ||
| Fig. 6 Partial (upfield region) 1H NMR spectrum (500 MHz, toluene-d8) of 10-Ir-H3Bpin as a function of temperature. Small amount of unidentified impurity (marked with asterisks) was shown near −9.2 ppm. | ||
Reactions of 10-Ir-H2 or 10-Ir-HMes with one equivalent or excess of A1-H under various conditions all led to mixtures of unidentified products that have resisted our attempts at isolation and separation. The phenomenon might be related to the fact that Rh analog 10-Rh-H2 has been shown to be an alkyne dimerization catalyst65 and PCP/POCOP iridium complexes reacted with alkynes to form a variety of allene or enyne complexes.66,67
For the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 combination of 10-Ir with A1-Bpin, we used DFT calculations (M06/SDD/6-311G(d,p) level of theory, see details in ESI†) to evaluate the relative thermodynamic stability of the three conceivable isomeric structures: the alkynylboronate π-complex 10-Ir-p-tol; the vinylidene complex 10-Ir-v-tol; and the alkynyl boryl complex 10-Ir-ynlBpin-tol (Fig. 7). 10-Ir-v-tol was calculated to be the lowest energy isomer, with 10-Ir-p-tol and 10-Ir-ynlBpin-tol lying 3.4 and 7.7 kcal mol−1 higher in energy, respectively.
:1 combination of 10-Ir with A1-Bpin, we used DFT calculations (M06/SDD/6-311G(d,p) level of theory, see details in ESI†) to evaluate the relative thermodynamic stability of the three conceivable isomeric structures: the alkynylboronate π-complex 10-Ir-p-tol; the vinylidene complex 10-Ir-v-tol; and the alkynyl boryl complex 10-Ir-ynlBpin-tol (Fig. 7). 10-Ir-v-tol was calculated to be the lowest energy isomer, with 10-Ir-p-tol and 10-Ir-ynlBpin-tol lying 3.4 and 7.7 kcal mol−1 higher in energy, respectively.
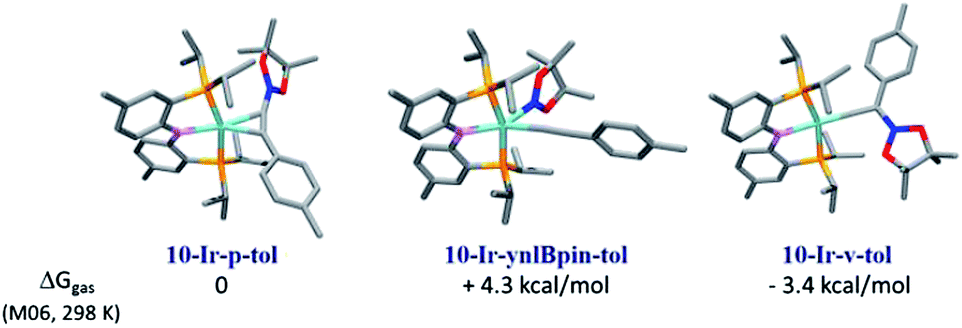 | ||
| Fig. 7 Relative free energies (DFT calculation) of three possible isomers of [10-Ir + A1-Bpin]. | ||
Mixing 10-Ir-HMes with one equivalent of A1-Bpin at ambient temperature overnight led to two products which appeared at 43.9 (5%) and 25.6 (11%) ppm respectively in the 31P{1H} NMR spectrum (Scheme 6). Further heating the mixture at 100 °C for 1 h cleanly converted all iridium complexes to a single product that resonated at 43.9 ppm in the 31P NMR spectrum, which was isolated and identified as 10-Ir-v-tol. The Me3Si-substituted vinylidene analog 10-Ir-v-TMS was also characterized by NMR spectroscopy in solution by using A2-Bpin as the reactant (Scheme 6). The vinylidene resonances were observed at 282.8 and 269.3 ppm in the 13C NMR spectrum as expected for 10-Ir-v-tol and 10-Ir-v-TMS, respectively.68 These results are consistent with the DFT prediction of 10-Ir-v-tol as the thermodynamically favored isomer.
 | ||
| Scheme 6 Synthesis of vinylidene complexes 10-Ir-v-tol and 10-Ir-v-TMS, and p-alkyne complexes 10-Ir-p-tol and 10-Ir-p-F3tol. | ||
Esteruelas and López69 were able to monitor the conversion of osmium boryl alkynyl complexes to vinylideneboronate esters, and we envisaged that other isomers of 10-Ir-v-tol might be obtained if a suitable (MePNPiPr)Ir precursor was reacted with A1-Bpin under milder conditions. We first attempted to mix 10-Ir-H2 with A1-Bpin with subsequent rapid removal of volatiles. The residue was redissolved in C6D6 and analyzed by 1H and 31P NMR spectroscopy. The major product was assigned as the alkynylboronate π-complex 10-Ir-p-tol (Scheme 6), and its resonance in the 31P NMR spectrum appeared at 25.6 ppm, which was identical to the observed intermediate in the synthesis of 10-Ir-v-tol. We also observed 9% of 10-Ir-HBpin formation indicating that Csp–B bond cleavage is facile; the amount of 10-Ir-HBpin increased over time. The assignment of 10-Ir-p-tol was supported by the considerable downfield shift (8.28 ppm) of the 1H NMR resonances of the ortho-hydrogens of the p-tolyl group in the coordinated A1-Bpin. Such downfield chemical shift is characteristic of internal aromatic alkyne π-complexes.67,70,71 However, the isomerization from 10-Ir-p-tol to 10-Ir-v-tol proceeded at an appreciable rate at ambient temperature (about 50% after 15 h) and precluded the isolation of pure 10-Ir-p-tol. We surmised that a more electron-poor alkyne should be thermodynamically less predisposed to form a vinylidene,68 and that the alkyne isomer may also be kinetically more long-lived. To this end, we replaced A1-Bpin with A10-Bpin as the reactant (Scheme 6) and mixed it with pre-cooled 10-Ir-H2 at −35 °C. We were able to isolate 10-Ir-p-F3tol as a pure red-orange solid in 74% yield. To the best of our knowledge, this is the first alkynylboronate π-complex that has been isolated and characterized. 31P NMR spectroscopic analysis showed a singlet at 26.1 ppm, which was similar to that of 10-Ir-p-tol. Consistent with our proposal, the conversion of 10-Ir-p-F3tol to the vinylidene complex 10-Ir-v-F3tol is significantly slower (about 5% after 15 h at ambient temperature) than the analogous transformation of 10-Ir-p-tol. Similarly to 10-Ir-p-tol, the aromatic proton signals of the 2,6-positions on A10-Bpin in 10-Ir-p-F3tol were shifted downfield to 8.22 ppm in the 1H NMR spectrum. In the 13C NMR spectrum, the carbon signal of alkynyl–C (![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) C–B) in 10-Ir-p-F3tol (δ 105.7) was slightly downfield of that in free A10-Bpin, as expected72 for a two-electron donor alkyne.
C–B) in 10-Ir-p-F3tol (δ 105.7) was slightly downfield of that in free A10-Bpin, as expected72 for a two-electron donor alkyne.
Select X-ray and computational structural studies
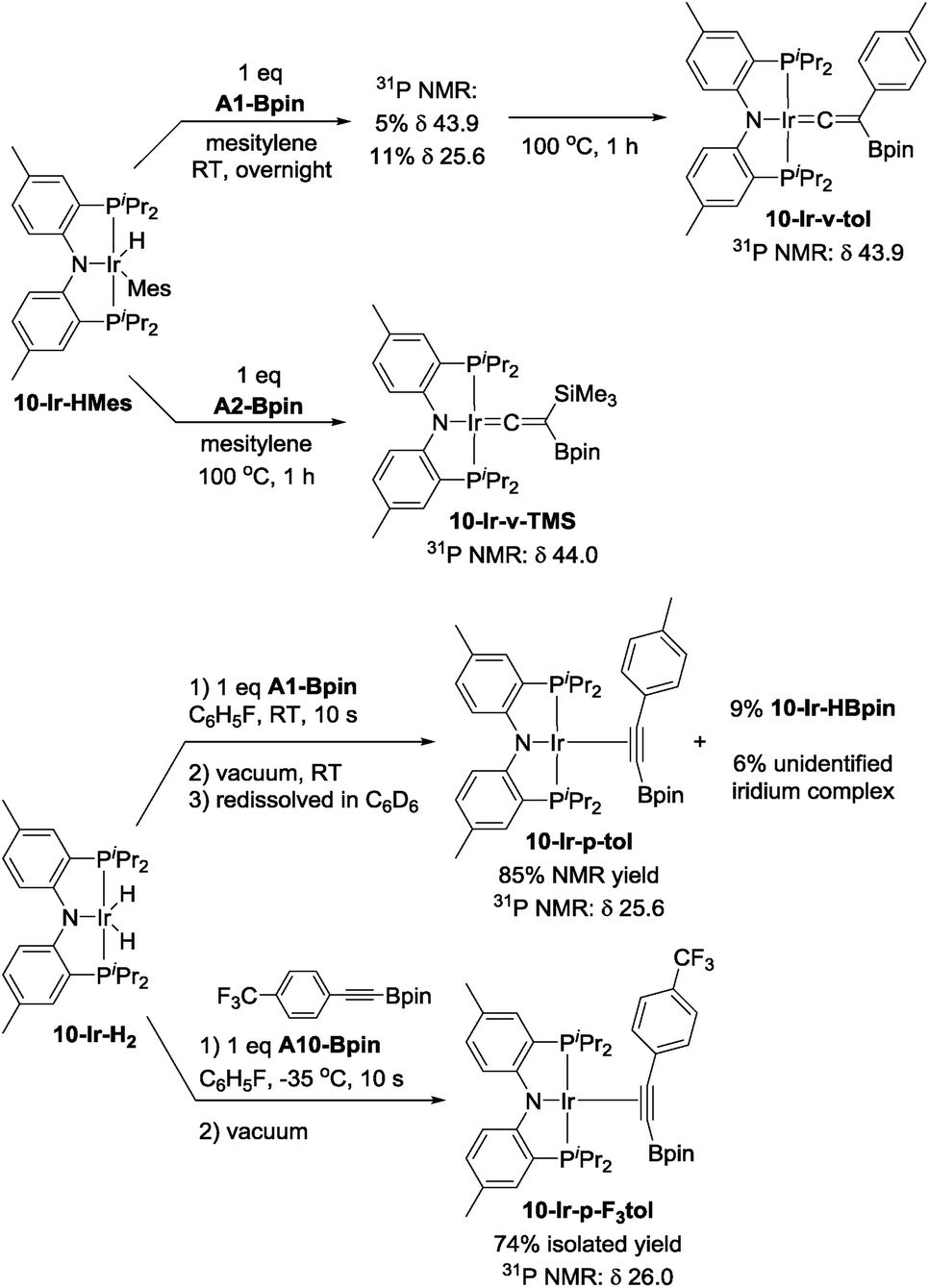
We were able to determine molecular structures of 10-Ir-HBpin, 10-Ir-Bpin2, 10-Ir-v-tol, and 10-Ir-p-F3tol in the solid state by X-ray diffractometry on corresponding single crystals. The structure of 10-Ir-HBpin (Fig. 8, top left) can be compared against the analogous POCOP complex 12-Ir-HBpin64 (Fig. 8, top right) reported by Heinekey et al.10-Ir-HBpin is Y-shaped five-coordinate if viewed as a hydride boryl complex. To reinforce the X-ray studies, especially with respect to the location of the Ir–H, density functional theory (DFT) analysis of 10-Ir-HBpin and 12-Ir-HBpin in the gas phase using the M06 functional was also performed. DFT calculations show 10-Ir-HBpin possesses shorter Ir–B and Ir–H bond distances and a B–H bond distance 0.2 Å longer than 12-Ir-HBpin which was judged to be a σ-borane complex, suggesting greater degree of B–H bond activation in 10-Ir-HBpin. The structure of 10-Ir-Bpin2 (Fig. 9, top left) can be described as Y-shaped five-coordinate where the Y is defined by N(amido) and the two boryls with an acute B–Ir–B angle (68.2°). The Y-shaped geometry is expected for a five-coordinate d6 complex73,74 when the equatorial plane contains a single good π-donor (N(amido)) and two strong σ-donors (two boryls). The two Ir-bound Bpin fragments display essentially the same metrics, and the associated Ir–B distances are similar to the analogous Ir–Bpin distances reported in the literature (2.02–2.07 Å).13,40 In general, all parameters of bond distances and bond angles in the NIrB2 plane are very close to the previously reported 1-Ir-Bpin2 (Fig. 9, top right).40 The B⋯B distance of 2.29 Å is too long for boron–boron interaction, thus 10-Ir-Bpin2 should be unambiguously viewed as an Ir(III) diboryl complex.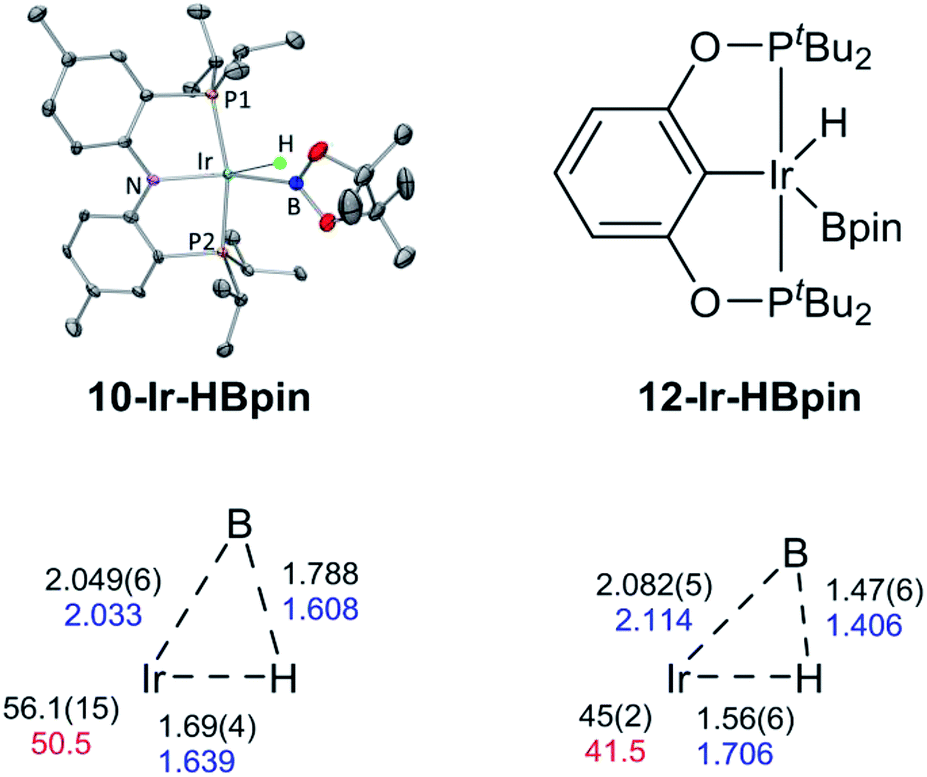 | ||
| Fig. 8 ORTEP drawing75 (50% probability ellipsoids) of 10-Ir-HBpin (top left) showing selected atom labeling, and depiction of 12-Ir-HBpin (top right). Hydrogen atoms are omitted for clarity in the ORTEP drawing, except for the hydride on the Ir atom. Metric parameters in the Ir/B/H triangles in compounds 10-Ir-HBpin (bottom left) and 12-Ir-HBpin64 (bottom right): DFT calculated distances (Å) in blue, B–Ir–H angles (°) in red, XRD-determined distances (Å) and B–Ir–H angles (°) in black. | ||
 | ||
| Fig. 9 ORTEP drawing75 (50% probability ellipsoids) of 10-Ir-Bpin2 (top left) showing selected atom labeling and depiction of 1-Ir-Bpin2 (top right). Hydrogen atoms are omitted for clarity in the ORTEP drawing. The selected bond distances (Å) and angles (deg) for 10-Ir-Bpin2 and 1-Ir-Bpin2 are summarized in the table at bottom. | ||
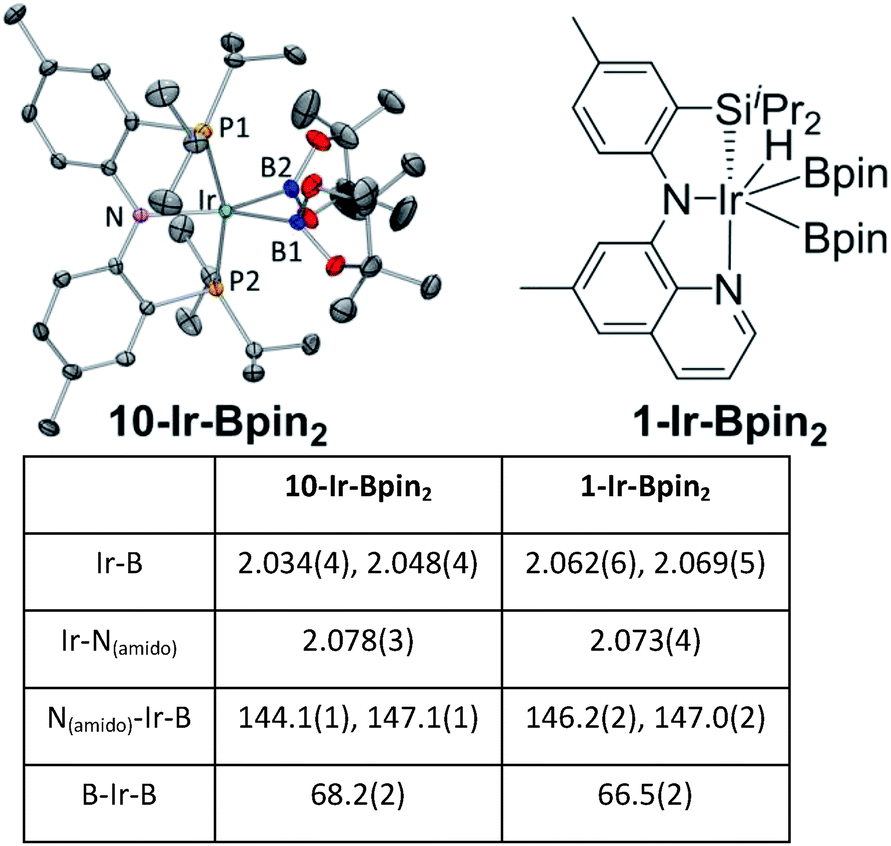
The coordination environment about Ir in the structures of 10-Ir-v-tol and 10-Ir-p-F3tol (Fig. 10) can be described as distorted square planar, with the greatest deviation corresponding to the P–Ir–P angles constrained by the pincer ligand. The C2–C1–Ir bond angle (178.2(3)°) in 10-Ir-v-tol is very close to 180° which is typical for a vinylidene complex.68 The Ir–C1 and C1–C2 bond lengths of 1.807(4) and 1.334(5) Å, respectively, are similar to the analogous distances in the [(Ph2PCH2SiMe2)2N]Ir![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2 vinylidene complex reported by Fryzuk.76 In the structure of 10-Ir-p-F3tol, A10-Bpin is bound to iridium in an η2 fashion (Ir–C1: 2.165(11) Å, Ir–C2: 2.101(12) Å) and the Ir–C distances are within the range of other square planar Ir(I) alkyne complexes.67,77,78 Both the elongation of CC bond (1.304 Å) and the bending of CC–Cipso (147.5(11)°) and CC–B (161.9(11)°) away from 180° indicate back-donation from the iridium center to the π* orbitals of CC bond.72,79
CCH2 vinylidene complex reported by Fryzuk.76 In the structure of 10-Ir-p-F3tol, A10-Bpin is bound to iridium in an η2 fashion (Ir–C1: 2.165(11) Å, Ir–C2: 2.101(12) Å) and the Ir–C distances are within the range of other square planar Ir(I) alkyne complexes.67,77,78 Both the elongation of CC bond (1.304 Å) and the bending of CC–Cipso (147.5(11)°) and CC–B (161.9(11)°) away from 180° indicate back-donation from the iridium center to the π* orbitals of CC bond.72,79
 | ||
| Fig. 10 ORTEP drawings75 (50% probability ellipsoids) of 10-Ir-v-tol (left) and 10-Ir-p-F3tol (right) showing selected atom labeling and hydrogen atoms are omitted for clarity. For 10-Ir-v-tol, one of two molecules in the asymmetric unit is shown, and a non-coordinated fluorobenzene molecule is omitted for 10-Ir-p-F3tol. Selected bond distances (Å) and angles (deg) for 10-Ir-v-tol: Ir–C1, 1.807(4); C1–C2, 1.334(5); P1–Ir–P2, 164.84(3); C2–C1–Ir, 178.2(3); C1–C2–C3, 120.1(3); C1–C2–B 113.1(3); C3–C2–B, 126.8(3). Selected bond distances (Å) and angles (deg) for 10-Ir-p-F3tol: Ir–C1, 2.165(11); Ir–C2, 2.101(12); C1–C2, 1.301(15); P2–Ir–P1, 163.81(10); C1–C2–C3, 147.5(11); B–C1–C2, 161.9(11). | ||
Stoichiometric reactions of (MePNPiPr)Ir complexes
To examine the possible roles that four new isolated (MePNPiPr)Ir complexes played in DHBTA, these compounds were examined in reactions with the three components in DHBTA: a terminal alkyne (substrate), HBpin (substrate), and H2 (by-product). 10-Ir-HBpin was reacted with three different terminal alkynes to study the boryl transfer ability: A1-H, A2-H and A10-H (Scheme 7, top). After 10 min at ambient temperature, approximately 50% yield of the corresponding alkynylboronate was observed in the 1H NMR spectrum for each of the three substrates. The amount of alkynylboronate did not increase with longer reaction times; however, multiple side reactions including hydrogenation occurred. By 31P NMR spectroscopic analysis, different degrees of unreacted 10-Ir-HBpin were observed along with multiple phosphorus-containing species formed, but they could not be assigned at this stage. Surprisingly, 10-Ir-Bpin2 was inert to all three major components in DHBTA: HBpin, terminal alkyne and H2 (Scheme 7, bottom) in stoichiometric reactions at ambient temperature. 31P NMR spectroscopic analysis showed 10-Ir-Bpin2 was the only observable phosphorus-containing compound in each reaction mixture. Even at 100 °C, 10-Ir-Bpin2 remained ostensibly intact in reactions with A1-H and HBpin. Only heating of 10-Ir-Bpin2 under 1 atm H2 at 100 °C overnight led to 41% 10-Ir-H3Bpin formation.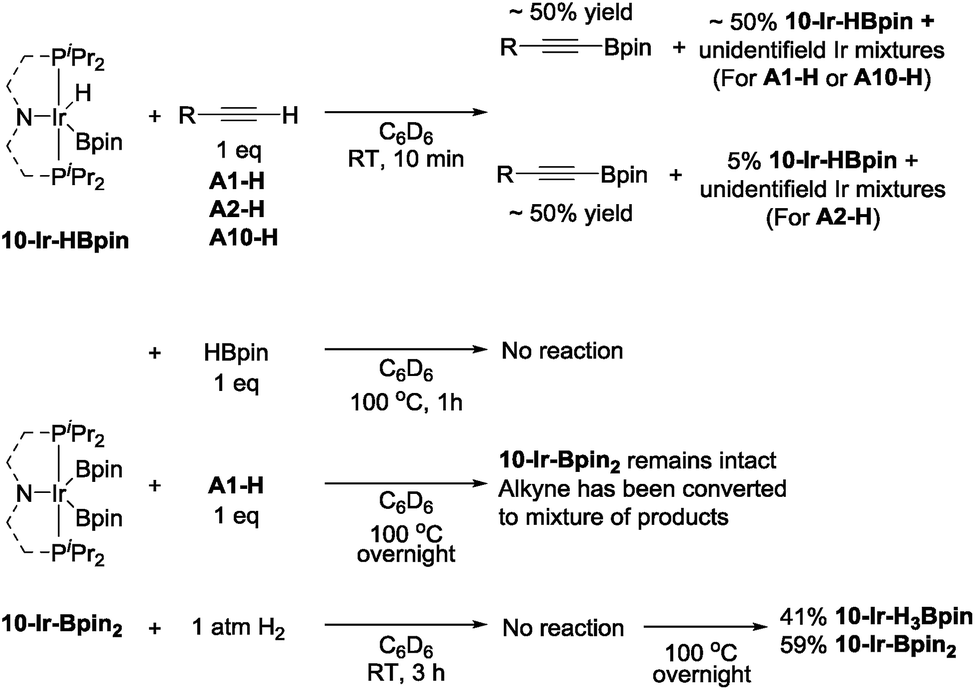 | ||
| Scheme 7 Boryl transfer from 10-Ir-HBpin to terminal alkynes (top) and reactivity of 10-Ir-Bpin2. | ||
10-Ir-v-tol was stable toward both HBpin and A1-H at ambient temperature (Scheme 8, top). On the other hand, treating 10-Ir-v-tol with H2 quickly resulted in 80% conversion and the formation of 60% gem-alkenylboronate (A1-4) and unidentified iridium compounds in 1 h at ambient temperature. After overnight, 10-Ir-H2 was the only observable species by 31P NMR spectroscopic analysis. Lack of observation of A1-4 in catalytic reaction mixtures suggested that 10-Ir-v-tol is not present in significant concentrations during catalysis. Treating 10-Ir-p-F3tol with 1 equivalent of HBpin at ambient temperature cleanly led to 83% 10-Ir-HBpin formation after 3 h (Scheme 8, bottom); meanwhile, equal amount of free A10-Bpin was observed in the 1H NMR spectrum. The reaction between 10-Ir-p-F3tol and A10-H was relatively sluggish with no noticeable change after 10 min, and only resulted in 8% conversion after 2 h at ambient temperature based on analysis by 31P NMR spectroscopy. Exposing 10-Ir-p-F3tol to 1 atm H2 quickly yielded 27% 10-Ir-H3Bpin and 24% unknown iridium species in 10 min, and the formation of 10-Ir-H3Bpin proved the Csp–B bond cleavage is facile. cis-Alkenylboronate (A10-1) was also observed.
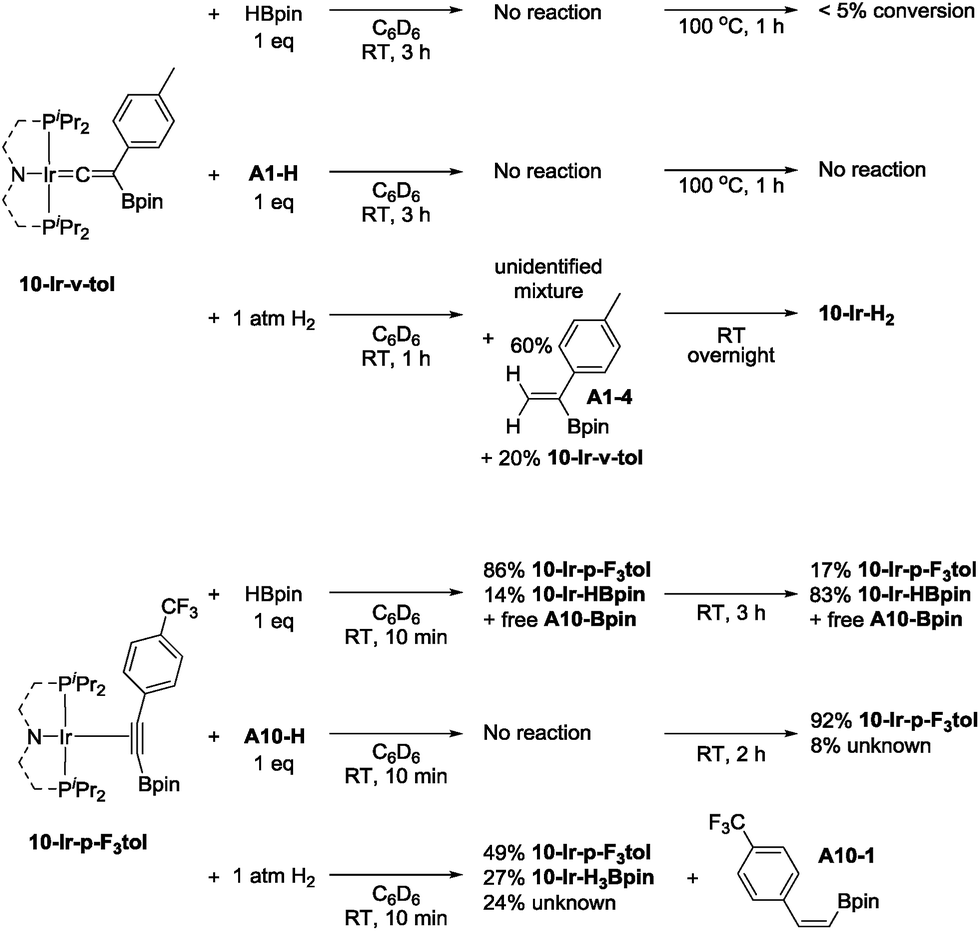 | ||
| Scheme 8 Reactivity of 10-Ir-v-tol (top) and 10-Ir-p-F3tol (bottom). | ||
Competence of isolated compounds in catalytic DHBTA
Chart 4 summarizes the results of catalytic DHBTA experiments that utilized various isolated (MePNPiPr)Ir compounds as pre-catalysts. The catalytic reactions were carried out in the fashion consistent with our other studies – the Ir compound was treated with 200 equiv. of HBpin, followed by 100 equiv. of the terminal alkyne (i.e., 1 mol% Ir). When 10-Ir-H2 was treated with excess HBpin, a yellow mixture of 10-Ir-HBpin and 10-Ir-H3Bpin immediately formed before the addition of alkyne. Not surprisingly, essentially identical yields of A1-Bpin and hydrogenation side-product A1-3 was observed when using 10-Ir-HBpin and 10-Ir-H2 as pre-catalysts. The use of 10-Ir-v-tol did lead to the formation of A1-Bpin, but in a significantly smaller yield than with 10-Ir-HBpin and 10-Ir-H2. In contrast, 10-Ir-Bpin2 showed no DHBTA at all after the first 10 min and only gave 37% A1-Bpin after 3 h. The inertness of 10-Ir-Bpin2 in the DHBTA correlated with its lack of reactivity in the stoichiometric reactions described above. In the two reactions with A10-H, 10-Ir-H2 and 10-Ir-p-F3tol led to the same yield of A10-Bpin and the hydrogenation side-product A10-1 (4-CF3-C6H4-C2H5) at the 10 min and the 1 h mark.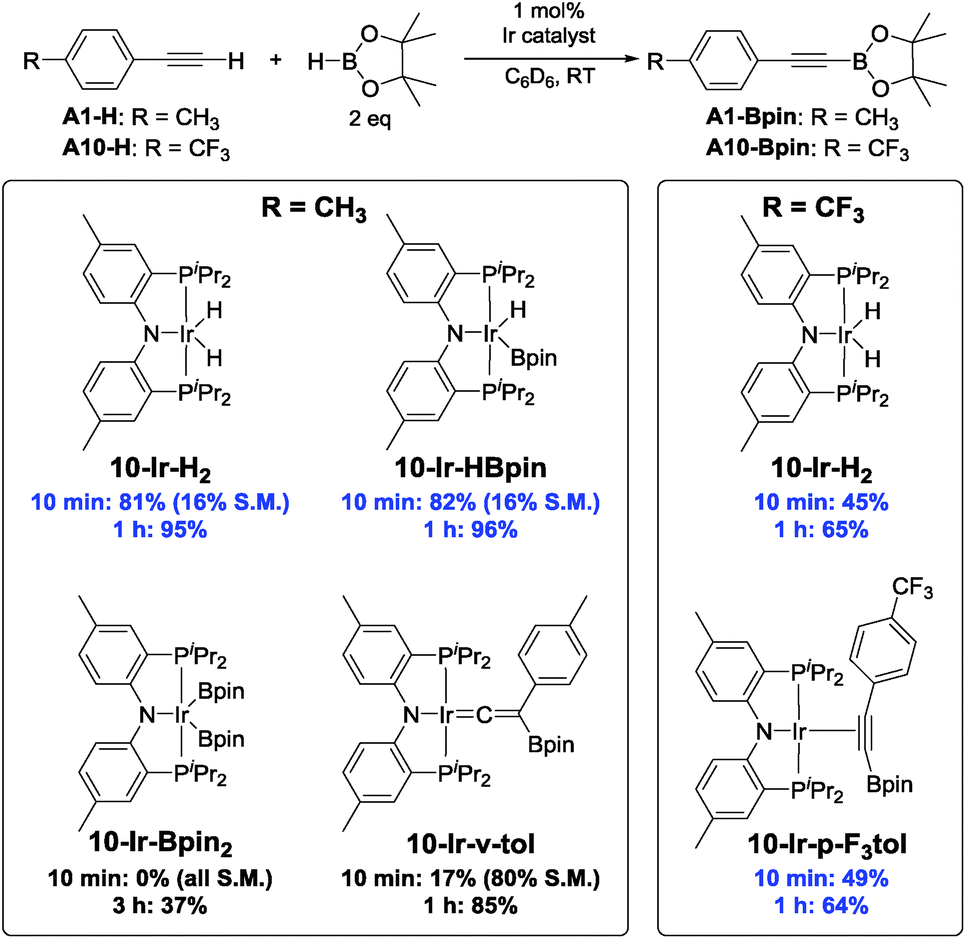 | ||
| Chart 4 Catalytic results for DHBTA using various 10-Ir complexes. | ||
On the basis of the stoichiometric and catalytic experiments the diboryl complexes analogous to 10-Ir-Bpin2 and the vinylidene complexes analogous to 10-Ir-v-tol can be firmly ruled out as intermediates in the DHBTA catalysis by (PNP)Ir complexes. Interestingly, this raises the question of whether the previously reported (SiNN)Ir catalysis actually requires 1-Ir-Bpin2 as an intermediate or if it is merely an off-cycle precursor that can access the catalytic cycle rapidly enough.
Conclusions
Building on a recent report of successful dehydrogenative borylation of terminal alkynes (DHBTA) with a pincer iridium catalyst,40 we examined a series of ligands structurally related to the successful SiNN ligand. Although most of the tested ligands failed to produce DHBTA products, we discovered that various PNP pincer ligands do result in active iridium DHBTA catalysts. Using the unsymmetric PNP-supported iridium complex 17-Ir-COE, useful yields were obtained with 0.025% loading of catalyst, corresponding to thousands of turnovers. Good to excellent yields were obtained under mild conditions for aryl-, silyl-, and alkyl-substituted terminal alkynes, even propargyl derivatives and 1,6-enynes. Unlike the strict chemoselectivity in the (SiNN)Ir system, <10% hydrogenation products were observed as the main side-products in all the (PNP)Ir systems with arylacetylene substrates. This has not precluded isolation of alkynylboronate products in 70–82% yields on preparative scale.Several iridium complexes of the symmetric PNP ligand 10 were synthesized and examined as potential intermediates in the catalytic cycle via testing in stoichiometric and catalytic reactions. The vinylidene (10-Ir-v-tol) and diboryl (10-Ir-Bpin2) complexes reacted too slowly with either terminal alkynes or HBpin under the conditions of catalysis, which ruled them out of the catalytic cycle of the 10-Ir system. The inactivity of 10-Ir-Bpin2 is in contrast to the analogous 1-Ir-Bpin240 which suggests that a diboryl intermediate is not essential for successful DHBTA. On the other hand, the hydride boryl complex (10-Ir-HBpin) and the alkynylboronate π-complex (10-Ir-p-F3tol) showed nearly identical performance to 10-Ir-H2 indicating that they either are intermediates in the catalytic cycle or are connected to such via a low-barrier pathway. Although the full mechanistic picture remains uncertain, the presently reported results strongly suggest that a pincer ligand containing an amido donor is key to an active DHBTA catalyst with iridium. An intriguing possibility is that this is related to the facile migration of boryl from the metal to the amido nitrogen we recently discovered for 1-Ir-Bpin2 and 1-Rh-Bpin2.80
Acknowledgements
We are grateful for the support of this research by the US National Science Foundation (grant CHE-1310007 to O. V. O.), the Welch Foundation (grant A-1717 to O. V. O.), and Fundamental Research Funds for the Central Universities of China (Grant No. AUGA5710013115 to J. Z.). We are also grateful to Ms. Linda Redd for editorial assistance.Notes and references
- J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- J. H. Brewster and E.-I. Negishi, Science, 1980, 207, 44 CAS.
- Y. Kondo, D. Garcia-Cuadrado, J. F. Hartwig, N. K. Boaen, N. L. Wagner and M. A. Hillmyer, J. Am. Chem. Soc., 2002, 124, 1164 CrossRef CAS PubMed.
- J. Shin, S. M. Jensen, J. Ju, S. Lee, Z. Xue, S. K. Noh and C. Bae, Macromolecules, 2007, 40, 8600 CrossRef CAS.
- R. E. Maleczka Jr, F. Shi, D. Holmes and M. R. Smith III, J. Am. Chem. Soc., 2003, 125, 7792 CrossRef PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed.
- E. M. Beck, R. Hatley and M. J. Gaunt, Angew. Chem., Int. Ed., 2008, 47, 3004 CrossRef CAS PubMed.
- A. D. Finke and J. S. Moore, Org. Lett., 2008, 10, 4851 CrossRef CAS PubMed.
- H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS.
- J. M. Murphy, J. D. Lawrence, K. Kawamura, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 13684 CrossRef CAS PubMed.
- H. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391 CrossRef CAS.
- T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS PubMed.
- J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka Jr and M. R. Smith III, Science, 2002, 295, 305 CrossRef CAS PubMed.
- J. M. Brown and G. C. Lloyd-Jones, J. Am. Chem. Soc., 1994, 116, 866 CrossRef CAS.
- R. B. Coapes, F. E. S. Souza, R. L. Thomas, J. J. Hall and T. B. Marder, Chem. Commun., 2003, 614 RSC.
- T. Ishiyama, K. Ishida, J. Takagi and N. Miyaura, Chem. Lett., 2001, 30, 1082 CrossRef.
- V. J. Olsson and K. J. Szabό, Angew. Chem., Int. Ed., 2007, 46, 6891 CrossRef CAS PubMed.
- Q. Li, C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 8755 CrossRef CAS PubMed.
- S. H. Cho and J. F. Hartwig, Chem. Sci., 2014, 5, 694 RSC.
- B. Ghaffari, S. M. Preshlock, D. L. Plattner, R. J. Staples, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr and M. R. Smith III, J. Am. Chem. Soc., 2014, 136, 14345 CrossRef CAS PubMed.
- S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr and M. R. Smith III, J. Am. Chem. Soc., 2013, 135, 7572 CrossRef CAS PubMed.
- D. Ogawa, J. Li, M. Suetsugu, J. Jiao, M. Iwasaki and Y. Nishihara, Tetrahedron Lett., 2013, 54, 518 CrossRef PubMed.
- V. Gandon, D. Leca, T. Aechtner, K. P. C. Vollhardt, M. Malacria and C. Aubert, Org. Lett., 2004, 6, 3405 CrossRef CAS PubMed.
- J. Huang, S. J. F. Macdonald and J. P. A. Harrity, Chem. Commun., 2009, 436 RSC.
- J. Barluenga, P. Barrio, L. Riesgo, L. A. López and M. Tomás, J. Am. Chem. Soc., 2007, 129, 14422 CrossRef CAS PubMed.
- L. Deloux, E. Skrzypczak-Jankun, B. V. Cheesman, M. Srebnik and M. Sabat, J. Am. Chem. Soc., 1994, 116, 10302 CrossRef CAS.
- J. Renaud, C.-D. Graf and L. Oberer, Angew. Chem., Int. Ed., 2000, 39, 3101 CrossRef CAS.
- M. M. Hussain, H. Li, N. Hussain, M. Ureña, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2009, 131, 6516 CrossRef CAS PubMed.
- A.-L. Auvinet and J. P. A. Harrity, Angew. Chem., Int. Ed., 2011, 50, 2769 CrossRef CAS PubMed.
- M. M. Hussain, J. H. Toribio, P. J. Carroll and P. J. Walsh, Angew. Chem., Int. Ed., 2011, 50, 6337 CrossRef CAS PubMed.
- Y. Nishihara, Y. Okada, J. Jiao, M. Suetsugu, M.-T. Lan, M. Kinoshita, M. Iwasaki and K. Takagi, Angew. Chem., Int. Ed., 2011, 50, 8660 CrossRef CAS PubMed.
- H. C. Brown, N. G. Bhat and M. Srebnik, Tetrahedron Lett., 1988, 29, 2631 CrossRef CAS.
- J.-R. Hu, L.-H. Liu, X. Hu and H.-D. Ye, Tetrahedron, 2014, 70, 5815 CrossRef CAS PubMed.
- J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon and M. J. Ingleson, Angew. Chem., Int. Ed., 2013, 52, 7518 CrossRef CAS PubMed.
- I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957 CrossRef CAS.
- B. M. Trost and Z. T. Ball, Synthesis, 2005, 6, 853 CrossRef.
- I. N. Michaelides and D. J. Dixon, Angew. Chem., Int. Ed., 2013, 52, 806 CrossRef CAS PubMed.
- For information on the chemistry of pincer ligands in general, see: (a) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (b) The Chemistry of Pincer Compounds, ed. D. Morales-Morales, and C. Jensen, Elsevier, Amsterdam, 2007 Search PubMed; (c) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS PubMed.
- C.-I. Lee, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2013, 135, 3560 CrossRef CAS PubMed.
- T. Tsuchimoto, H. Utsugi, T. Sugiura and S. Horio, Adv. Synth. Catal., 2015, 357, 77 CrossRef CAS PubMed.
- C. J. Pell and O. V. Ozerov, Inorg. Chem. Front., 2015, 2, 720 RSC.
- (a) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS PubMed; (b) G. E. Dobereiner, J. Yuan, R. R. Schrock, A. S. Goldman and J. D. Hackenberg, J. Am. Chem. Soc., 2013, 135, 12572 CrossRef CAS PubMed.
- L. Fan, S. Parkin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 16772 CrossRef CAS PubMed.
- Y. Zhu, L. Fan, C.-H. Chen, S. R. Finnell, B. M. Foxman and O. V. Ozerov, Organometallics, 2007, 26, 6701 CrossRef CAS.
- (a) M. T. Whited, Y. Zhu, S. D. Timpa, C.-H. Chen, B. M. Foxman, O. V. Ozerov and R. H. Grubbs, Organometallics, 2009, 28, 4560 CrossRef CAS; (b) M. T. Whited and R. H. Grubbs, Acc. Chem. Res., 2009, 42, 1607 CrossRef CAS PubMed.
- D. Liu, Y. Luo, W. Gao and D. Cui, Organometallics, 2010, 29, 1916 CrossRef CAS.
- J. C. Peters, S. B. Harkins, S. D. Brown and M. W. Day, Inorg. Chem., 2001, 40, 5083 CrossRef CAS PubMed.
- D. E. Herbert and O. V. Ozerov, Organometallics, 2011, 30, 6641 CrossRef CAS.
- J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2015, 54, 2916 CrossRef CAS PubMed.
- M. Gupta, C. Hagen, W. C. Kaska, R. E. Cramer and C. M. Jensen, J. Am. Chem. Soc., 1997, 119, 840 CrossRef CAS.
- I. Göttker-Schnetmann, P. S. White and M. Brookhart, Organometallics, 2004, 23, 1766 CrossRef.
- F. Liu, E. B. Pak, B. Singh, C. M. Jensen and A. S. Goldman, J. Am. Chem. Soc., 1999, 121, 4086 CrossRef CAS.
- A. C. Sykes, P. White and M. Brookhart, Organometallics, 2006, 25, 1664 CrossRef PubMed.
- R. Ahuja, M. Brookhart, A. S. Goldman, Z. Huang, and A. R. Macarthur, WO 2007008847 A2, 18 Jan 2007.
- D. Morales-Morales, R. Redón, C. Yung and C. M. Jensen, Inorg. Chim. Acta, 2004, 357, 2953 CrossRef CAS PubMed.
- L. P. Press, B. J. McCulloch and O. V. Ozerov, manuscript in preparation.
- T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS.
- See ESI† for details.
- J. D. Masuda, K. C. Jantunen, O. V. Ozerov, K. J. T. Noonan, D. P. Gates, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 2408 CrossRef CAS PubMed.
- R. B. Lansing, K. I. Goldberg and R. A. Kemp, Dalton Trans., 2011, 8950 RSC.
- E. Calimano and T. D. Tilley, Dalton Trans., 2010, 9250 RSC.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed.
- T. J. Hebden, M. C. Denney, V. Pons, P. M. B. Piccoli, T. F. Koetzle, A. J. Schultz, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, J. Am. Chem. Soc., 2008, 130, 10812 CrossRef CAS PubMed.
- W. Weng, C. Guo, R. Celenligil-Cetin, B. M. Foxman and O. V. Ozerov, Chem. Commun., 2006, 197 RSC.
- R. Ghosh, X. Zhang, P. Achord, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2007, 129, 853 CrossRef CAS PubMed.
- N. Phadke and M. Findlater, Organometallics, 2013, 33, 16 CrossRef.
- M. I. Bruce, Chem. Rev., 1991, 91, 197 CrossRef CAS.
- M. A. Esteruelas, A. M. López, M. Mora and E. Oñate, Organometallics, 2012, 31, 2965 CrossRef CAS.
- D. A. Ortmann, B. Weberndörfer, K. Ilg, M. Laubender and H. Werner, Organometallics, 2002, 21, 2369 CrossRef CAS.
- P. Steinert and H. Werner, Chem. Ber., 1997, 130, 1591 CrossRef CAS PubMed.
- J. L. Templeton, Adv. Organomet. Chem., 1989, 29, 1 CrossRef CAS.
- J. F. Riehl, Y. Jean, O. Eisenstein and M. Pelissier, Organometallics, 1992, 11, 729 CrossRef CAS.
- W. H. Lam, S. Shimada, A. S. Batsanov, Z. Lin, T. B. Marder, J. A. Cowan, J. A. K. Howard, S. A. Mason and G. J. McIntyre, Organometallics, 2003, 22, 4557 CrossRef CAS.
- ORTEP plots were created using Ortep-3 for Windows. L. Farugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef.
- M. D. Fryzuk, L. Huang, N. T. McManus, P. Paglia, S. J. Rettig and G. S. White, Organometallics, 1992, 11, 2979 CrossRef CAS.
- D. B. Grotjahn, J. M. Hoerter and J. L. Hubbard, J. Am. Chem. Soc., 2004, 126, 8866 CrossRef CAS PubMed.
- N. Grüger, H. Wadepohl and L. H. Gade, Eur. J. Inorg. Chem., 2013, 30, 5358 CrossRef PubMed.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, 2005 Search PubMed.
- C.-I. Lee, N. A. Hirscher, J. Zhou, N. Bhuvanesh and O. V. Ozerov, Organometallics, 2015, 34, 3099 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting information in the form of descriptions of experiments, characterization data, crystallographic information in the form of CIF files, and a video recording of hydrogen evolution from the mixture of 3-methyl-3-trimethylsiloxy-1-butyne, 17-Ir-COE and HBpin in toluene. CCDC 1014977–1014979 and 1015119. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02161h |
| This journal is © The Royal Society of Chemistry 2015 |