
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controllability of dynamic double helices: quantitative analysis of the inversion of a screw-sense preference upon complexation†
Ryo
Katoono
*,
Shunsuke
Kawai
,
Kenshu
Fujiwara
and
Takanori
Suzuki
Department of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan. E-mail: katoono@sci.hokudai.ac.jp; Fax: +81 11 706 2714; Tel: +81 11 706 3396
First published on 7th August 2015
Abstract
We describe a quantitative analysis of the complexation-induced inversion of a screw-sense preference based on a conformationally dynamic double-helix structure in a macrocycle. The macrocycle is composed of two twisting units (terephthalamide), which are spaced by two strands (1,3-bis(phenylethynyl)benzene), and is designed to generate a double-helix structure through twisting about a C2 axis in a conrotatory manner. The attachment of chiral auxiliaries to the twisting units induces a helical preference for a particular sense of (M)- or (P)-helicity through the intramolecular transmission of chirality to dynamic double helices. The twisting unit can also act as a binding site for capturing a guest molecule, and, in a complexed state, the preferred screw sense of the dynamic double-helix structure is reversed to exhibit the contrary preference. We quantitatively monitored the complexation-induced inversion of the screw-sense preference using 1H NMR spectroscopy, which enabled us to observe independently two species with (M)- or (P)-helicity in both the absence and presence of a guest molecule. Inversion of the screw-sense preference was induced upon complexation with an achiral guest as well as a chiral guest.
Introduction
Artificial double-helix structures1 have attracted a wide range of interest, including how they can be constructed as well as how they can be controlled to prefer a particular chiral sense. Supramolecular assemblies, based on hydrogen bonding, metal coordination or π–π stacking, of two oligomeric2 or polymeric3 strands with or without a chiral source have been extensively investigated. Alternative approaches to the construction of a double-helix structure have been based on the sequential connection of doubly helical fragments4 or twisting of a macrocycle.5–7 We were particularly interested in such a double helix with a well-defined molecular structure, which might be advantageous with respect to quantitative considerations. Chiroptical properties based on a double-helix structure have often been studied with stereochemically stable chiral macrocycles,6 in which only a particular sense of double helices was completely dominant and persistent. However, the control of dynamic chiroptical properties based on a double-helix structure with labile helical chirality7 has not been well studied. In this context, we studied a macrocycle with the purpose of developing such a rare dynamic system.Here we report the successful control of a screw-sense preference based on a conformationally dynamic double-helix structure in a macrocycle, which includes not only simple biasing of the helical preference to a particular sense (Scheme 1a), but also inversion of the preferred screw sense to exhibit the contrary preference upon complexation with a guest molecule (Scheme 1b). In this article, we describe the complexation-induced inversion of a screw-sense preference of dynamic double helices through quantitative 1H NMR spectroscopy in addition to CD spectroscopy.
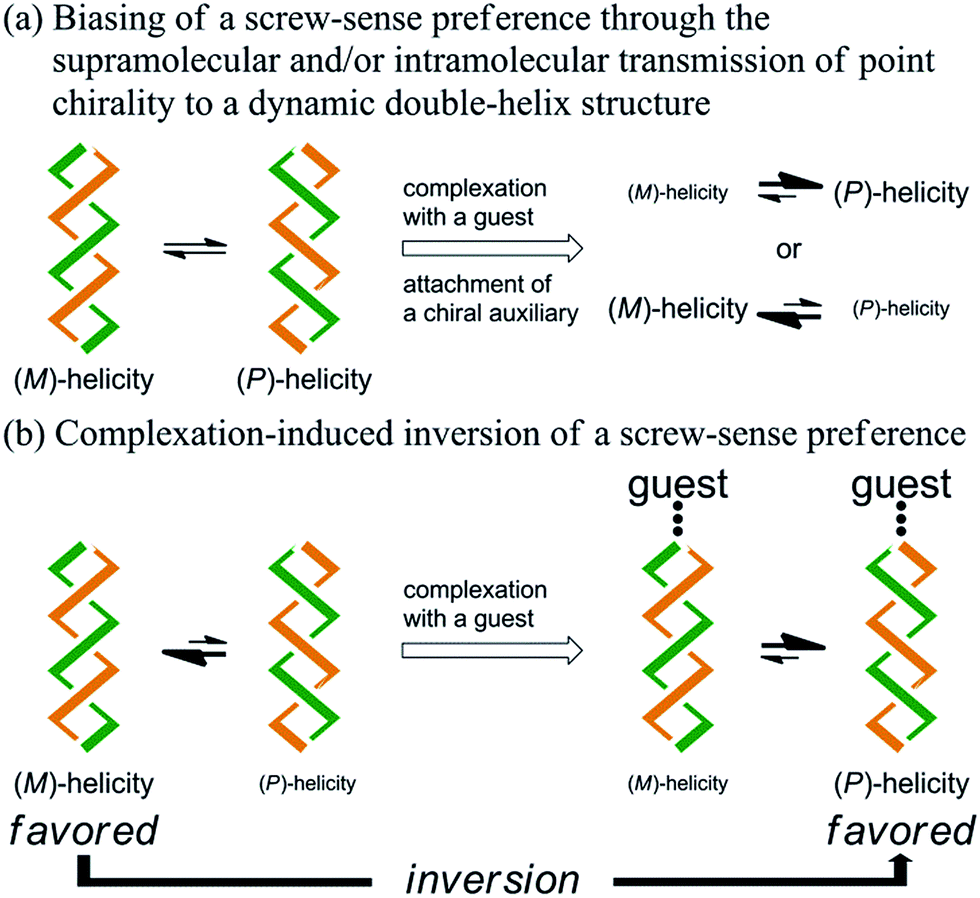 | ||
| Scheme 1 | ||
Results and discussion
Molecular design and preparation of macrocycles 1–3
We designed a series of macrocycles 1–3, in which a double-helix structure is generated through conrotatory twisting about a C2 axis toward (M)- or (P)-helicity (Fig. 1), as a new category of dynamic helical molecules based on terephthalamides.8,9 These macrocycles are composed of two twisting units (terephthalamide) that are spaced by two strands (1,3-bis(phenylethynyl)benzene), and are alternatively arranged to form a macrocyclic framework. Conrotatory twisting of the two amide groups in each twisting unit leads to two helical forms in the macrocycle, which can dynamically interconvert to each other through rotation about the CC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–Ccentral bonds. The direction of the twisting can be controlled to prefer a particular sense through the transmission of chirality in a supramolecular10 and/or intramolecular11 manner upon complexation with a chiral ditopic12 guest at the two amide carbonyls (supramolecular) or the attachment of a chiral auxiliary to each amide nitrogen in the twisting units (intramolecular). We inserted a spacer between two strands so that they would be fixed and entangled to form a double-helix structure. We assumed that the motility of the double helices in the macrocyclic framework would be modulated by the spacing groups, for which we used ethynylene (1), ethylene (2), and butadiynylene (3) bonds. The substituent X on each amide nitrogen is an n-butyl group with no asymmetric element (a) or an (R)-C*HMe(cHex) group as a chiral auxiliary (b) in each macrocycle.
O–Ccentral bonds. The direction of the twisting can be controlled to prefer a particular sense through the transmission of chirality in a supramolecular10 and/or intramolecular11 manner upon complexation with a chiral ditopic12 guest at the two amide carbonyls (supramolecular) or the attachment of a chiral auxiliary to each amide nitrogen in the twisting units (intramolecular). We inserted a spacer between two strands so that they would be fixed and entangled to form a double-helix structure. We assumed that the motility of the double helices in the macrocyclic framework would be modulated by the spacing groups, for which we used ethynylene (1), ethylene (2), and butadiynylene (3) bonds. The substituent X on each amide nitrogen is an n-butyl group with no asymmetric element (a) or an (R)-C*HMe(cHex) group as a chiral auxiliary (b) in each macrocycle.
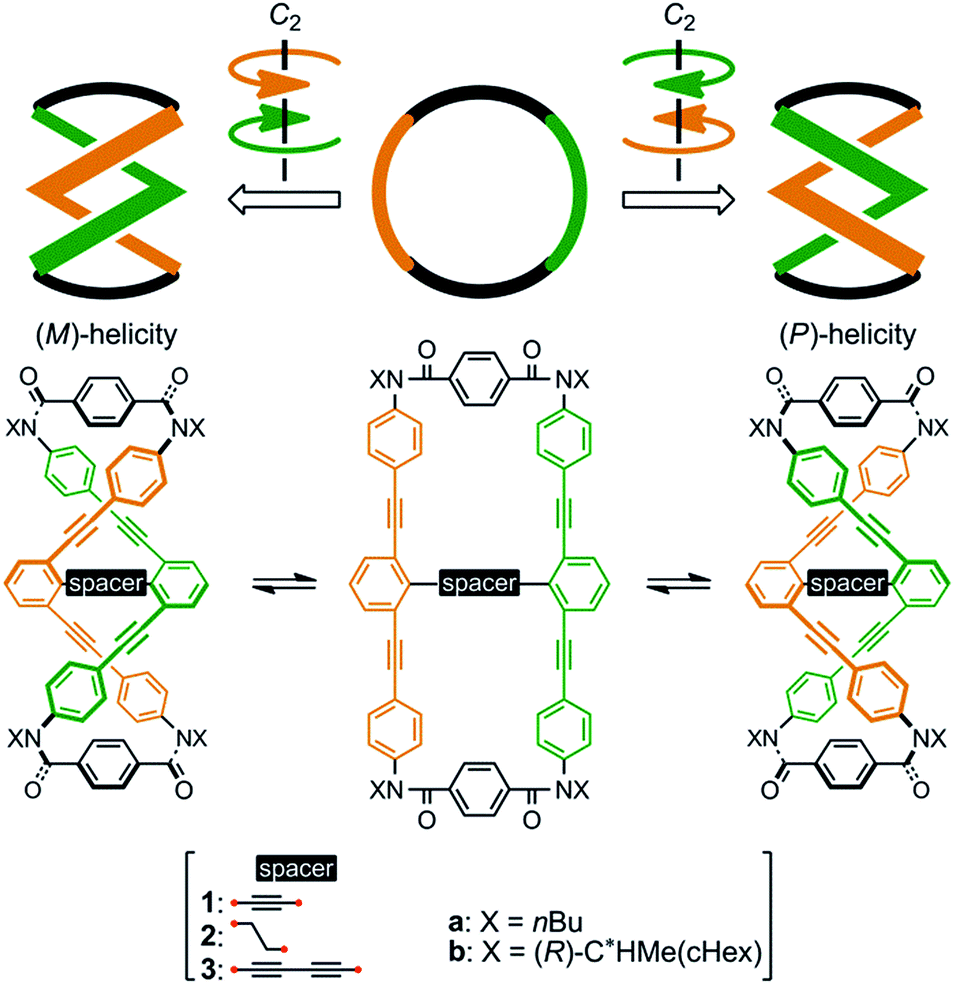 | ||
| Fig. 1 Chemical structures of macrocycles 1–3, and dynamic interconversion between two double-helix structures with (M)- or (P)-helicity. | ||
We prepared macrocycles 1–3 through a four-fold condensation reaction of two equivalents of terephthaloyl chloride with the corresponding tetraanilines 11–13, which were derived from the corresponding tetraynes 8–10 through a Sonogashira coupling reaction with N-trifluoroacetylated iodoaniline 14a or (R)-14b,9b followed by deprotection of the trifluoroacetyl groups. The preparation of tetraynes 8–10 is summarized in Scheme S1.† We used diammonium salts 4,9c (R,R)-5 and (S,S)-59a as ditopic guests to form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex with these macrocycles (Scheme 2).
:2 complex with these macrocycles (Scheme 2).
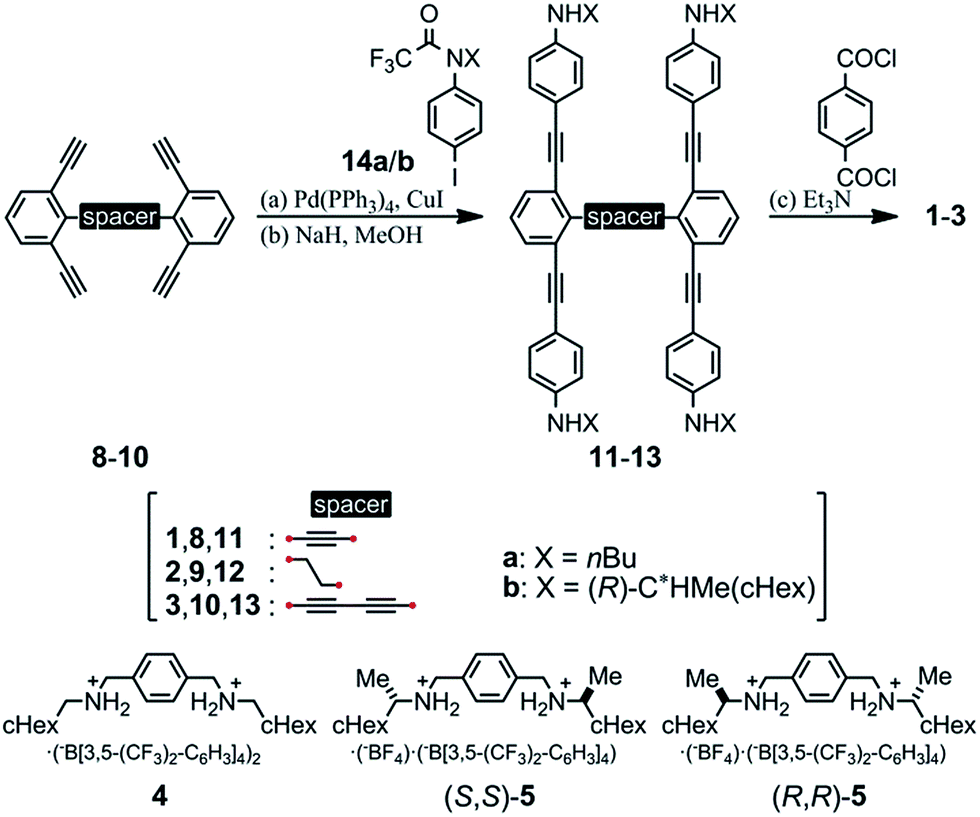 | ||
| Scheme 2 Preparation of macrocycles 1–3. Yields: (a) 75%, (b) 54% for 11a; (a) 76%, (b) 81% for 11b; (a) 39%, (b) 96% for 12a; (a) 82%, (b) 71% for 12b; (a) 75%, (b) 98% for 13a; (a) 87%, (b) 92% for 13b; (c) 82% for 1a; 18% for 1b; 63% for 2a; 39% for 2b; 25% for 3a; 38% for 3b. Chemical structures of ditopic guests 4, (R,R)-5 and (S,S)-5. | ||
Molecular structures of macrocycles 1–3
We obtained a single crystal of 1a that was appropriate for X-ray analysis, which revealed that the two amide groups of each terephthalamide were twisted in a conrotatory manner and led to global twisting of the molecule to give a double-helix structure in the macrocycle (Fig. 2a). In a crystal, two helical forms with (M)- or (P)-helicity appeared as a racemate. We found a similar conformation as the most energy-minimized structure in a conformational search for model 1a′ [NMe] (Fig. 2b), and expected that it would be the dominant conformation even in solution.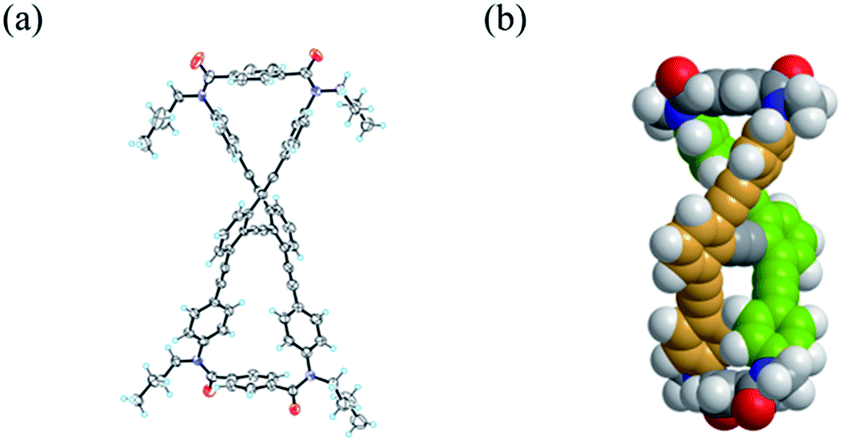 | ||
| Fig. 2 (a) X-ray structure of (M)-1a (P-1, Z = 2) in rac-1a·(benzene)1.5 crystal. Only one enantiomer is depicted. The solvent molecule was omitted for clarity. (b) Energy-minimized structure for 1a′ [NMe], obtained by a conformational search using the MacroModel software (v9.9 OPLS_2005, Monte Carlo Multiple Minimum method, non-solvated, 50000 steps). Only one enantiomer is depicted. | ||
We investigated the dynamic structures of macrocycles in solution using 1H NMR spectroscopy at various temperatures (Fig. 3a). At room temperature, the spectrum of 1a exhibited an anisochronous pair of signals for the methylene protons, which are closest to the amide nitrogen, and this indicated that they are in diastereotopic environments (HF and HF′) in the molecule and that therefore the molecule is chiral on the NMR timescale at this temperature. The two signals coalesced at an elevated temperature (358 K in dimethylsulfoxide-d6). We assumed the averaging of two diastereotopic protons on the NMR-timescale to be the result of dynamic interconversion (enantiomerization)13 between two equivalent forms with (M)- or (P)-helicity in light of the above-mentioned conformational insights.14 Notably, there were almost no significant changes in the chemical shift during the VT measurements, although broadening and splitting were observed for some aromatic protons due to slow local rotations about the phenylene rings at lower temperatures. These results showed that the macrocyclic framework could mostly convert to the equivalent form.
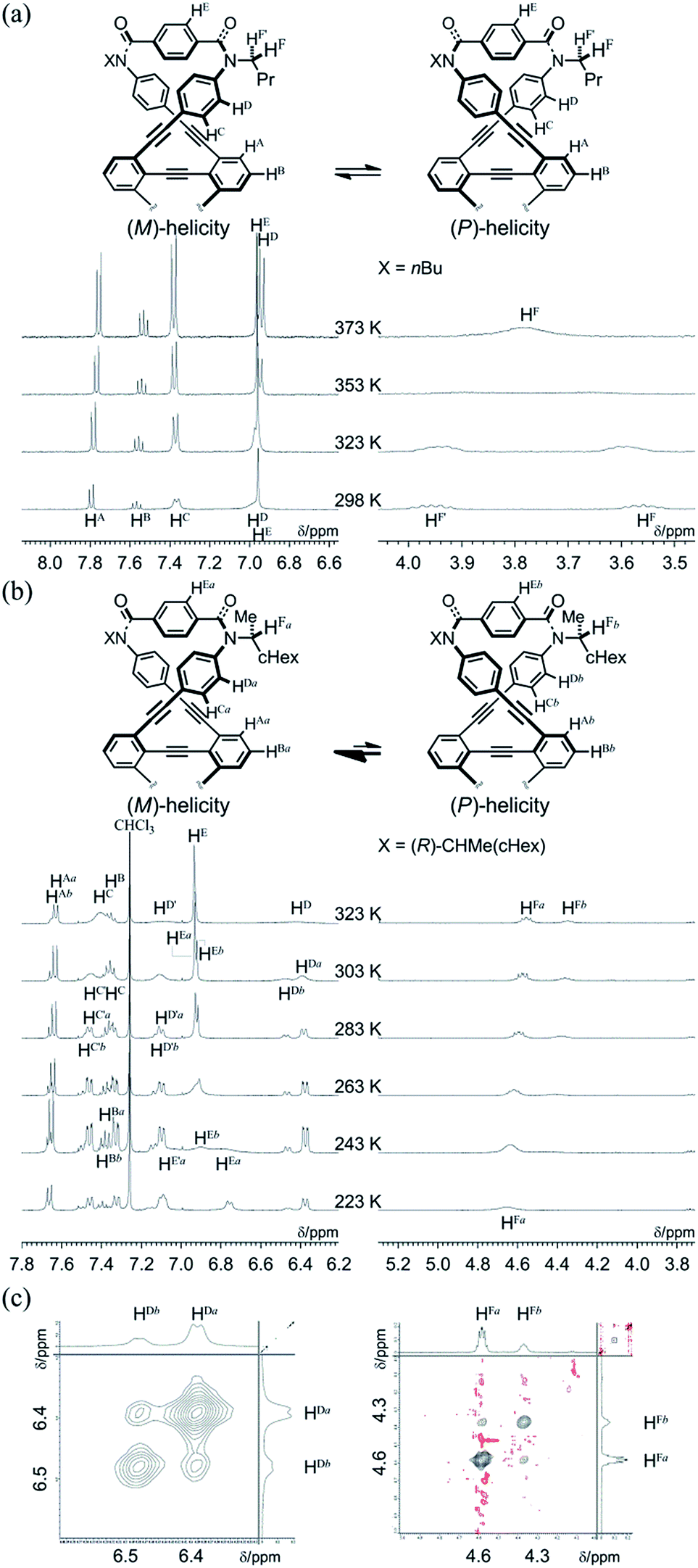 | ||
| Fig. 3 (a) Partial VT-1H NMR spectra (400 MHz) of 1a, measured in dimethylsulfoxide-d6 at 293–383 K. (b) Partial VT-1H NMR spectra (400 MHz) of (R,R,R,R)-1b, measured in chloroform-d at 223–323 K. Two protons (H and H′, or Ha and Hb) in a pair of split resonances are tentatively assigned. (c) Partial 2D-ROESY/EXSY spectra (600 MHz) of (R,R,R,R)-1b, measured in chloroform-d at 292 K (mixing time: 250 ms). | ||
The 1H NMR spectra of (R,R,R,R)-1b, measured in chloroform-d at 223–323 K, exhibited two non-equivalent sets of averaged resonances that were both assigned to C2 symmetry (Fig. 3b). The populations of two non-equivalent species changed with temperature, which indicated dynamic interconversion (stereoisomerization)11f–j,15 between two diastereomeric forms with (M)- or (P)-helicity in solution. This consideration was confirmed from the 2D-ROESY/EXSY spectrum of (R,R,R,R)-1b,16 measured at 292 K, in which we found cross-peaks that could be attributed to chemical exchange, not to NOE, for phenylene protons (HDa and HDb) and methine protons (HFa and HFb) (Fig. 3c).15,17 We considered the non-equivalent populations of the two diastereomeric species to be the result of a successful intramolecular transmission of point chirality (R) to dynamic double helices with (M)- or (P)-helicity in the macrocycle.18 The fact that we could observe two non-equivalent species independently at ambient temperature was important for quantitative monitoring of the complexation-induced inversion using 1H NMR spectroscopy (described later).
Macrocycle 2b in dichloromethane showed absorption at 299 (logε 5.11) and 317 (sh. 5.02) nm, which were similar in appearance to those in the spectra for single-stranded substructure 6 [298 (4.75) and 309 (4.75)] and diphenylacetylene derivative 79e [291 (4.47) and 307 (4.45)] (Fig. 4). The two strands in 2b are spaced by saturated carbons and are present independently regarding the electronic structure to show a similarity in appearance to 6 and 7. The molar absorptivity of 2b was approximately twice that of 6, which in turn was twice that of 7. The absorption maximum and end in the spectra of 1b and 3b with a conjugated spacer were bathochromically shifted [λmax 314 nm (logε 5.13) for 1b and 326 nm (5.08) for 3b, end at around 400 nm] (Fig. 4). Similar observations were noted with 1a [λmax 315 (logε 5.12)], 2a [315 (sh. 4.98) and 299 (5.08)], and 3a [325 (5.08)], which have an n-butyl group on each amide nitrogen, instead of (R)-C*HMe(cHex).
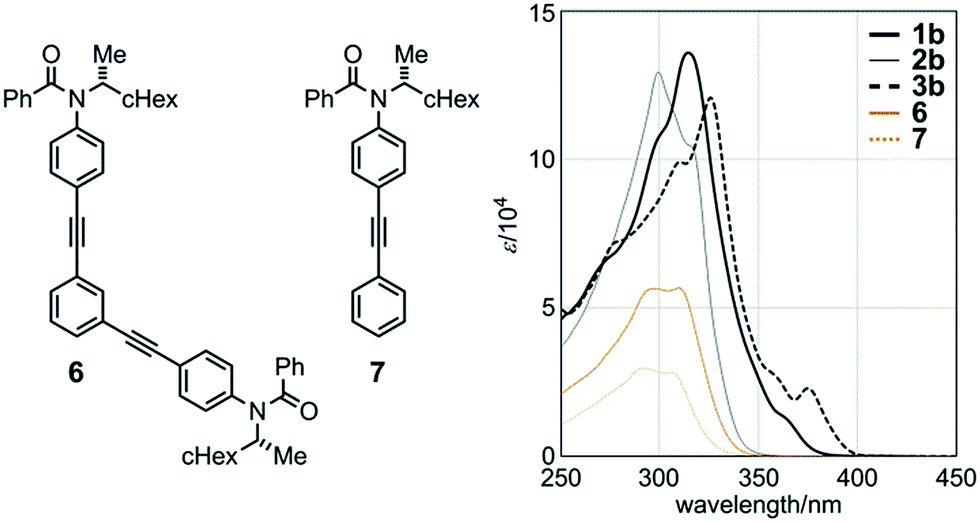 | ||
| Fig. 4 UV spectra of macrocycles 1b (bold solid line), 2b (thin solid line), and 3b (bold dashed line), single-stranded substructure 6 (orange solid line), and diphenylacetylene 7 (orange dashed line), measured in CH2Cl2 at room temperature. | ||
Complexation of macrocyclic hosts 1a–3a with chiral ditopic guests (R,R)-5 and (S,S)-5
We first examined the complexation of 1a with a chiral ditopic guest 519 using 1H NMR spectroscopy. The spectra of 1a in the presence of 5, measured in chloroform-d at room temperature, showed upfield shifts for both the phenylene protons HE in 1a and the phenylene protons Ha in 5 (Fig. S4†), which indicated that the guest was captured at the two amide carbonyls in the twisting unit through the formation of hydrogen bonds. We confirmed that the stoichiometry was 1:2 using Job plots based on continuous changes in the chemical shifts induced for both 1a and 5 (Fig. 5). There were marginal changes (Δδχ1a < 0.003) in the chemical shifts induced for other aromatic protons far from the binding site. This result showed that the host did not deform into any other structure during complexation. Similar upfield shifts with a slight difference in the chemical shift were induced when we used 2a or 3a as a host (Fig. S4†).
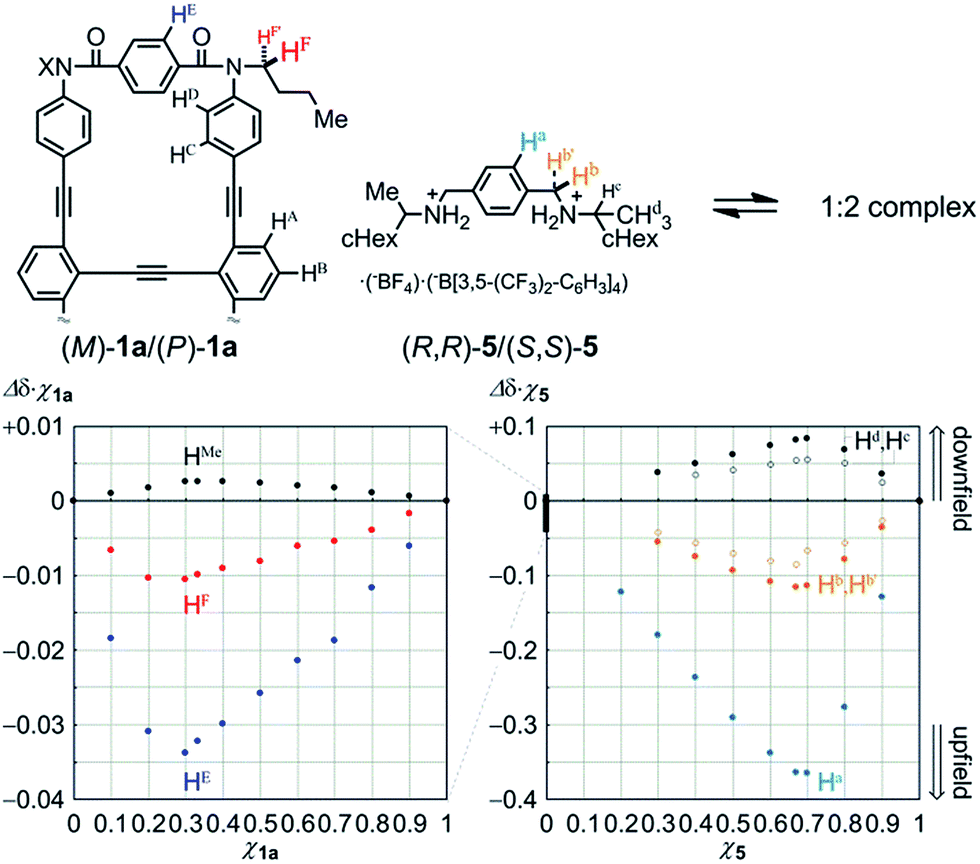 | ||
| Fig. 5 Job plots for the complexation of 1a with an equivalent mixture of (R,R)-5 and (S,S)-5, measured in CDCl3 at 303 K ([1a] + [5] = 2 mM), based on continuous changes in the chemical shifts induced for 1a (left) and 5 (right). | ||
Next, we monitored the 1:2 complexation of 1a with the chiral ditopic guests (R,R)-5 and (S,S)-5 in dichloromethane to investigate the supramolecular transmission of point chirality to dynamic double helices. In the absence of any guest, we assumed an equivalent mixture of two enantiomeric forms with (M)- or (P)-helicity, since there is no asymmetric element in 1a other than dynamic chirality. To a solution of 1a (9.5 × 10−5 M) we gradually added up to eight equivalents of (R,R)-5. We found that compositive Cotton effects were continuously induced throughout the absorption region of 1a (Fig. 6a, blue lines). When we used (S,S)-5 instead of (R,R)-5, a set of mirror-imaged Cotton effects emerged (Fig. 6a, red lines). We considered these complexation-induced Cotton effects to be the result of a successful supramolecular transmission of point chirality in the guest to dynamic chirality in the host. In a complexed state, the host preferred a particular sense in response to the chirality in each guest. During the addition of a guest, there were almost no significant changes in the UV spectrum of 1a (Fig. S5†), which indicated that the host mostly maintained the framework even in a complexed state. Similar observations were noted with 2a and 3a (Fig. 6b and c). Based on these titration experiments (see inset), we estimated binding constants (2 × 104 M−1 for each binding site), and the details are summarized in Table S1.†
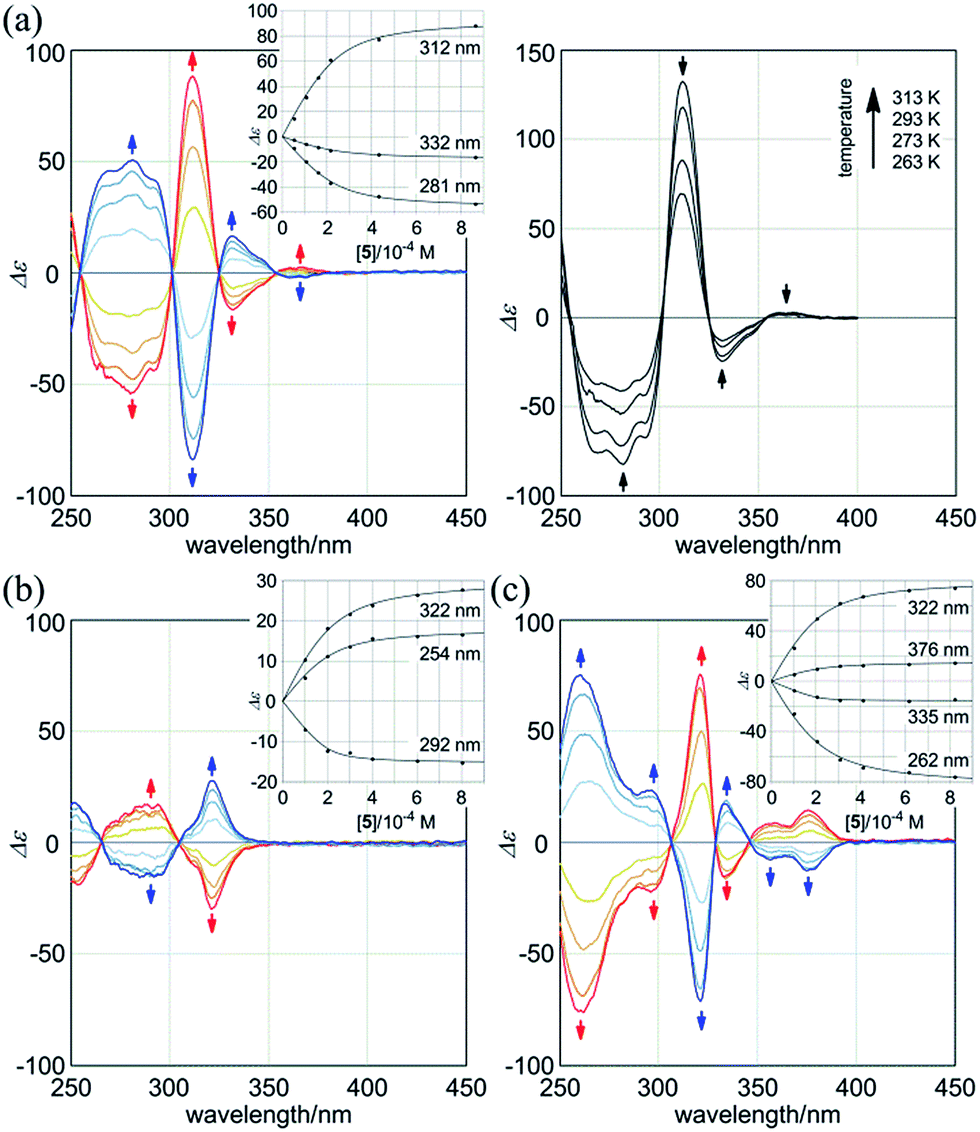 | ||
| Fig. 6 CD spectra of (a, left) 1a (8.9–9.5 × 10−5 M), (b) 2a (1.0 × 10−4 M), and (c) 3a (1.0 × 10−4 M) in the presence of (R,R)-5 (blue lines) or (S,S)-5 (red lines) (1, 2, 4, and 8 equiv.), measured at 293 K [inset: plots of induced molar CD versus concentration of the guest added]. All spectra were measured in CH2Cl2 at 293 K. (a, right) VT-CD spectra of 1a in the presence of (S,S)-5 (8 equiv.), measured at 263–313 K. | ||
We further investigated the dynamic interconversion between two non-equivalent forms in a complexed state through VT measurements at lower and elevated temperatures (273 and 313 K). During the measurements in the presence of eight equivalents of (S,S)-5, molar CDs were enhanced with a decrease in temperature and attenuated with an increase in temperature, while the appearance of the spectrum did not change (Fig. 6a, right). This result showed that there was a dynamic equilibrium between two non-equivalent complexes [(M)-1a·(S,S)-52 and (P)-1a·(S,S)-52], and a particular form was favored over the other. These observations can be explained by one or both of the following considerations. First, some supramolecular assemblies such as hydrogen-bonded complexes driven by a gain of enthalpy are often favored at lower temperatures. Second, a preferred conformer is more favored at lower temperatures in an equilibrium between two non-equivalent conformers, as demonstrated in the VT 1H NMR measurements for (R,R,R,R)-1b and (R,R,R,R)-2b.
Intramolecular transmission of point chirality to dynamic double helices in the macrocycle
Macrocycles (R,R,R,R)-1b, (R,R,R,R)-2b, and (R,R,R,R)-3b showed compositive Cotton effects in each absorption region (Fig. 7). The spectral shape was similar to that seen for a complex of 1a, 2a, or 3a with (S,S)-6 (Fig. 6, red lines). These Cotton effects changed isosbestically with temperature. We considered the thermal response to be the result of dynamic interconversion between two diastereomeric forms with (M)- or (P)-helicity, and a particular form was favored over the other through the intramolecular transmission of internal chirality to dynamic chirality. For a solution of (R,R,R,R)-1b, we noted helical excess11g–j,20 at each temperature, which was estimated from a plot of lnK versus 1/T (Fig. S2a†). Single-stranded substructure (R,R)-6 with the same internal chirality on each amide nitrogen showed Cotton effects that were totally different from those for macrocycles, but similar to those for (R)-7 (Fig. 7d). We considered that these Cotton effects shown by (R,R)-6 or (R)-7 were due to a local chiral conformation around the internal chirality with no relation to a framework.
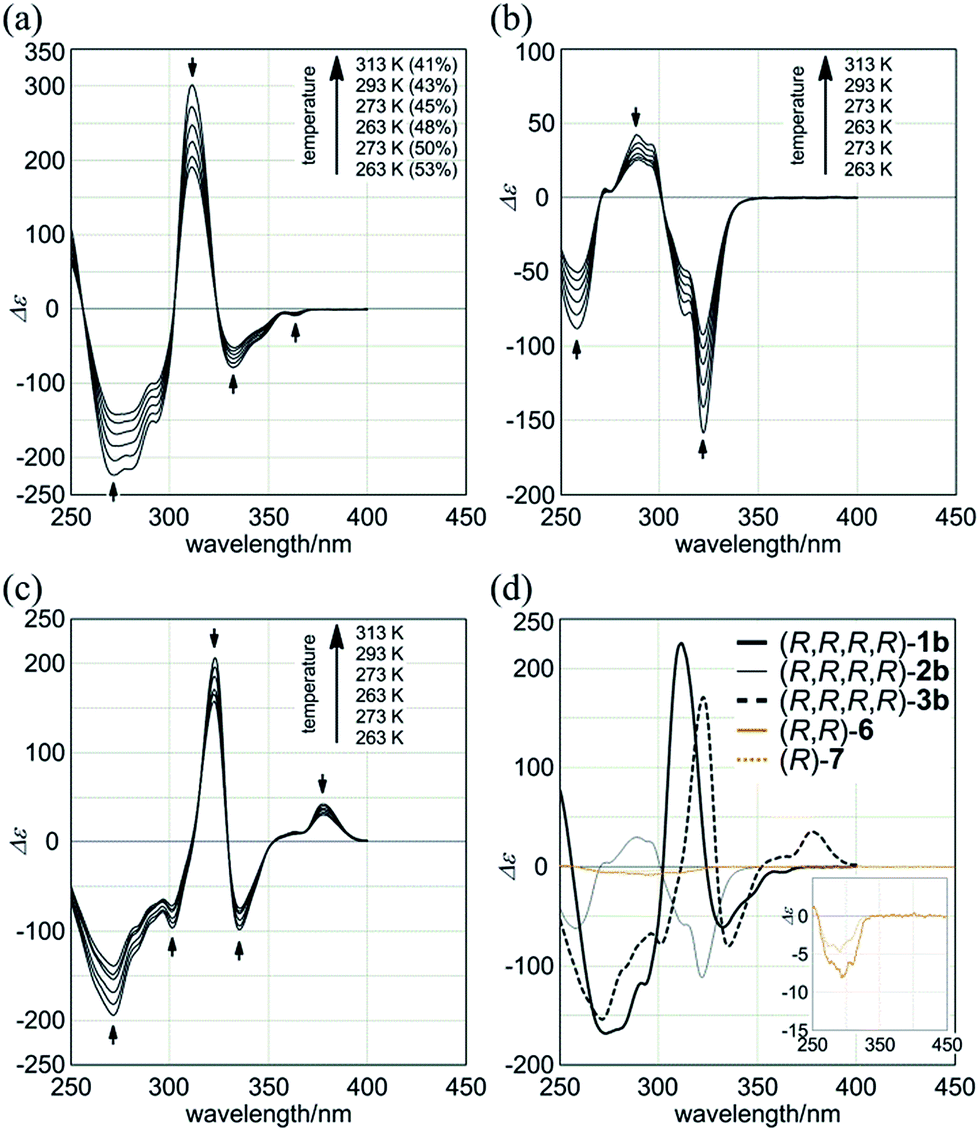 | ||
| Fig. 7 VT-CD spectra of (a) (R,R,R,R)-1b, (b) (R,R,R,R)-2b, and (c) (R,R,R,R)-3b, measured in CH2Cl2 at 263–313 K. (d) CD spectra of (R,R,R,R)-1b, (R,R,R,R)-2b, (R,R,R,R)-3b, single-stranded substructure (R,R)-6, and diphenylacetylene derivative (R)-7, measured in CH2Cl2 at 293 K. In (a), helical excess (%) is noted in parenthesis. | ||
Complexation-induced inversion of a screw-sense preference
Some dynamic helical molecules have been designed to undergo an inversion of helicity in response to a change in the environmental conditions21–24 or the addition of a guest molecule,25 and satisfy the following requirements: the dynamic molecule should have a helical preference for a particular sense in advance, and the preferred sense should be reversed to prefer the contrary sense in a different state. Here we demonstrate the complexation-induced inversion of a screw-sense preference based on dynamic double-helices in the macrocycles (R,R,R,R)-1b, (R,R,R,R)-2b, and (R,R,R,R)-3b, which were shown to have a preferred screw sense through the intramolecular transmission of chirality, as mentioned above.First, the preferred screw sense of the dynamic double-helices was reversed to prefer the contrary sense upon 1:2 complexation with an achiral guest 4 (Scheme 3). This was the case for (R,R,R,R)-1b and (R,R,R,R)-2b. In both the uncomplexed and complexed states, the screw-sense preference was controlled through the intramolecular transmission of point chirality, since the achiral guest has no preference for a particular sense in the direction of twisting. The chiral auxiliaries associated with the host led to two contrary preferences before and after complexation.
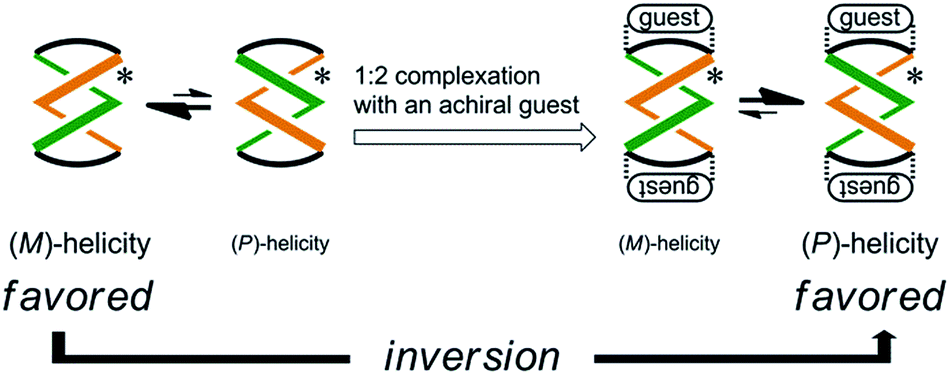 | ||
| Scheme 3 Inversion of a screw-sense preference upon complexation with an achiral guest. | ||
Next, the preferred sense was stereospecifically reversed upon 1:2 complexation with enantiomeric guests (R,R)-5 and (S,S)-5 (Scheme 4). This was the case for (R,R,R,R)-3b, which reversed the screw-sense preference only when it formed a complex with (R,R)-5. Alternatively, when we used the antipodal guest (S,S)-5, the host made the preference marginal even though a 1:2 complex was similarly formed. The details of these observations are described below.
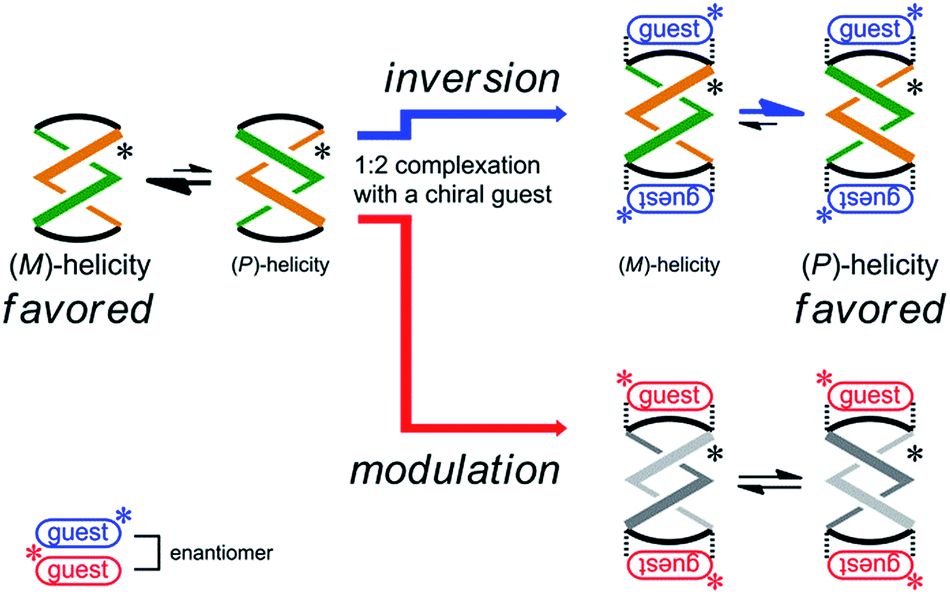 | ||
| Scheme 4 Stereospecific inversion of a screw-sense preference upon complexation with enantiomeric guests. | ||
When (R,R,R,R)-1b and 4 were mixed in dichloromethane, the CD spectrum of (R,R,R,R)-1b changed continuously to show pseudo mirror-imaged Cotton effects (Fig. 8a). In both the absence and presence of 4, the chiral auxiliaries associated with 1b were dominantly transferred to dynamic double helices to exhibit contrary preferences. The two-way intramolecular transmission of point chirality was confirmed upon complexation even with chiral guests (R,R)-5 and (S,S)-5 (Fig. S6†). The intensity of induced Cotton effects in a complexed state was enhanced compared to that of the original effects whenever any ditopic guest was used, and this showed that the intramolecular transmission was more effective in a complexed state.26 Similar complexation-induced inversions of the screw-sense preference were noted with (R,R,R,R)-2b (Fig. 8b and Fig. S6†).
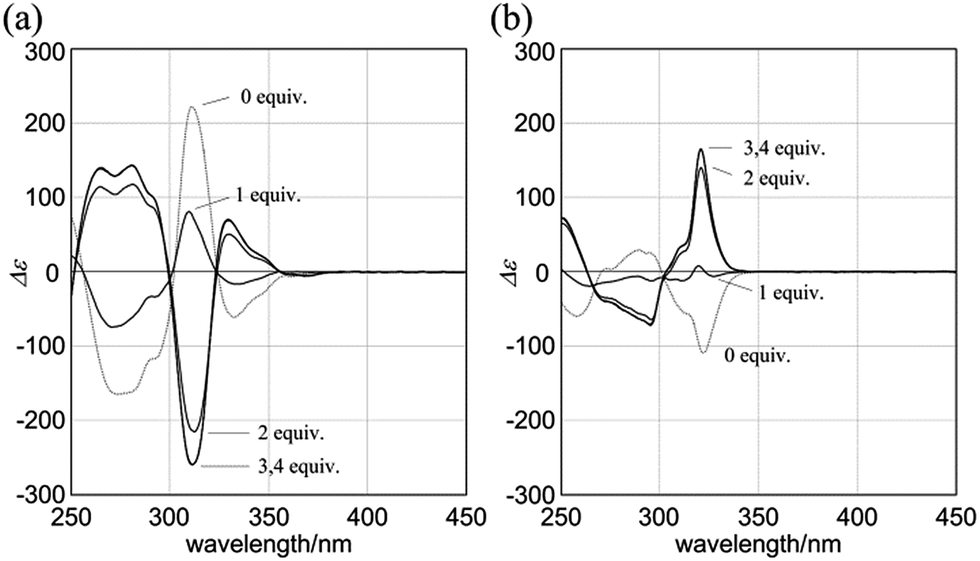 | ||
| Fig. 8 CD spectra of (a) (R,R,R,R)-1b (8.4 × 10−5 M) and (b) (R,R,R,R)-2b (1.0 × 10−4 M) in the presence of achiral ditopic guest 4 (solid lines) [0 (1 only, dashed line), 1, 2, 3 and 4 equiv.]. All spectra were measured in CH2Cl2 at 293 K. | ||
Next, we mixed (R,R,R,R)-3b and (R,R)-5 in dichloromethane. The CD spectrum of (R,R,R,R)-3b changed continuously to show pseudo mirror-imaged Cotton effects (Fig. 9a), although the intensity was reduced to some extent compared to the original effects, unlike in the above cases. The Cotton effects induced in a complexed state were similar to those seen in the spectrum of 3a in the presence of (R,R)-5. Alternatively, when we added the antipodal (S,S)-5 to a solution of (R,R,R,R)-3b, the original Cotton effects were continuously modulated to show marginal effects with a different appearance (Fig. 9b). Notably, the titration profiles (see inset) showed similar saturation points when up to six equivalents of each guest was added, and indicated that binding constants should be in a similar order (∼104 M−1, Table S2†), although the ultimate shapes in each spectrum were different due to the local diastereomeric situation around asymmetric points. The modulated Cotton effects in a complexed state were considered to be the result of competition in the transmission of each chirality in the host and guest. These results indicated that the external chirality could contribute to determining the preference in a complexed state where the internal chirality was mostly negated but still present,27 and thus we identified the stereospecific inversion of a screw-sense preference based on a dynamic double-helix structure.
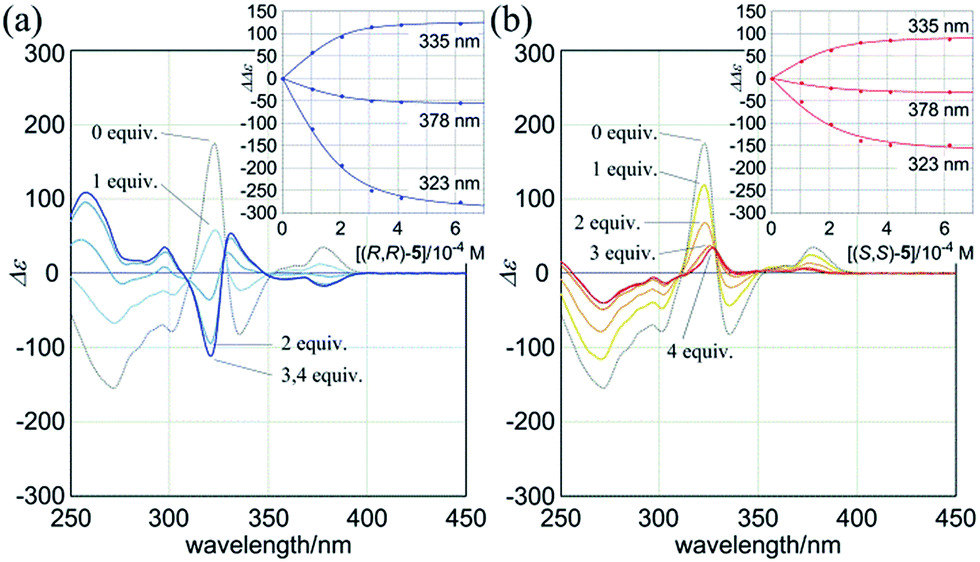 | ||
| Fig. 9 CD spectra of (R,R,R,R)-3b (1.0 × 10−4 M) in the presence of (a) (R,R)-5 [0 (3b only, black dashed line), and 1–4 equiv. (blue lines)], (b) (S,S)-5 [0 (3b only), and 1–4 equiv. (red lines)] [inset: plots of induced molar CD (ΔΔε = Δε3b·5 − Δε3b) versus concentration of the added guest (0–6 equiv.)]. All spectra were measured in CH2Cl2 at 293 K. | ||
Quantitative analysis of the complexation-induced inversion of a screw-sense preference based on 1H NMR spectroscopy
In most cases, an inversion of helicity has been demonstrated by the reversal of a sign of Cotton effects in CD spectroscopy.21–25 Alternatively, a quantitative analysis of the inversion of a screw-sense preference by the observation of two independent diastereomeric species that dynamically interconvert into each other is limited to only a few examples.28As mentioned above, only (R,R,R,R)-1b exhibited two non-equivalent species independently at room temperature by 1H NMR spectroscopy. In the spectrum, a particular set of averaged resonances was favored over the other. To a solution of (R,R,R,R)-1b (1 mM), (R,R)-5 was gradually added as a ditopic guest that could render a complexed species dissolved under the complexation conditions. The spectra in the presence of (R,R)-5 showed that the populations of two non-equivalent species (Ha and Hb) continuously changed to ultimately be reversed (Fig. 10). Each set was assumed to be averaged between uncomplexed and complexed species due to fast exchange on the NMR-timescale. We confirmed that the original ratio was restored by the addition of methanol, which showed that the induced changes were due to the formation of hydrogen bonds.
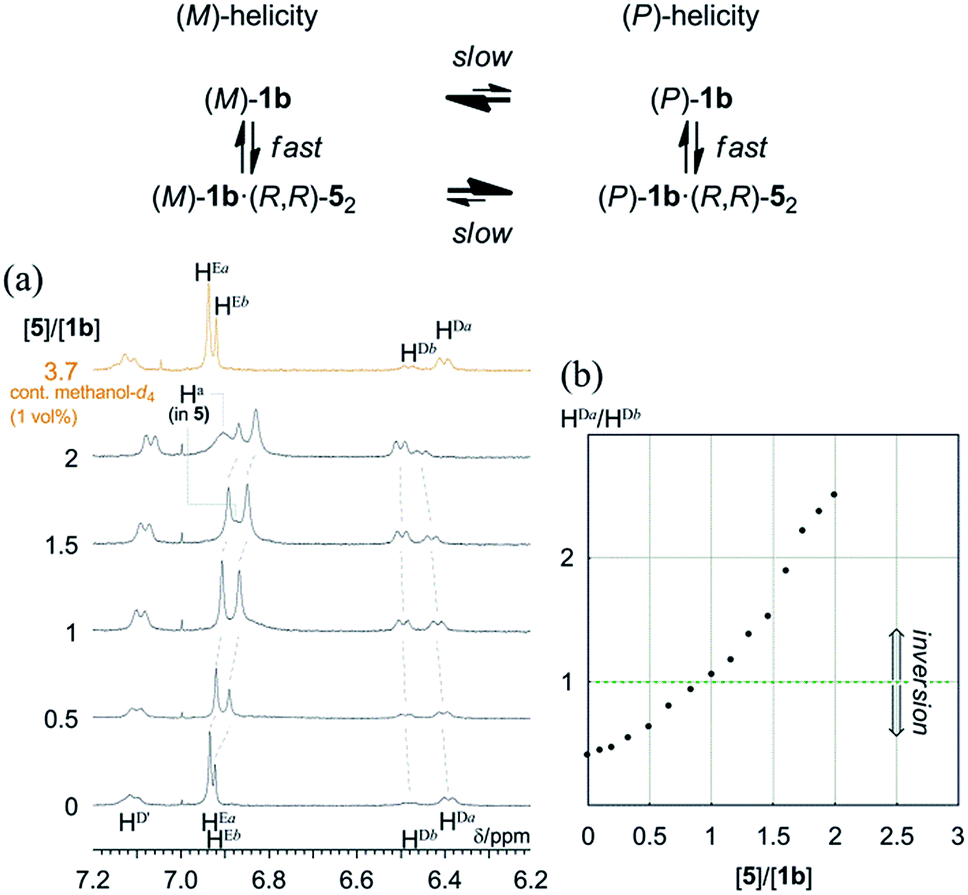 | ||
| Fig. 10 (a) 1H NMR spectra of (R,R,R,R)-1b (1 mM) in the presence of (R,R)-5 (0–2 equiv., and 3.7 equiv. containing 1 vol% methanol-d4). All spectra were measured in CDCl3 at 293 K. (b) Plot of the ratio of the populations of two species with (M)- or (P)-helicity versus equivalents of 5 added, based on phenylene protons HDa and HDb. | ||
Conclusions
We have demonstrated the control of the screw-sense preference of dynamic double helices, which were generated in a macrocycle through conrotatory twisting about a C2 axis toward (M)- or (P)-helicity. The dynamic chirality was controlled to prefer a particular screw sense through the supramolecular transmission of external chirality upon 1:2 complexation with a chiral ditopic guest. The attachment of internal chirality also led to a screw-sense preference through the intramolecular transmission of chirality. Not only did we observe such simple biasing of the helical preference for a particular screw sense of a dynamic double-helix structure, we also demonstrated that the preferred screw sense was reversed upon 1:2 complexation with an achiral guest, as well as chiral guests. Through the systematic modulation of a spacing group, which was inserted between two strands, we identified multifarious inversions of a screw-sense preference upon complexation, which included inversion triggered by an achiral guest, and stereospecific inversion triggered only by a particular enantiomeric guest. Finally, we successfully monitored the inversion of the screw-sense preference in a quantitative manner using 1H NMR spectroscopy.
Acknowledgements
We thank Dr Yasuhiro Kumaki (Hokkaido University) for 2D ROESY/EXSY measurements.Notes and references
- M. Albrecht, Chem. Rev., 2001, 101, 3457 CrossRef CAS PubMed; D. Haldar and C. Schmuck, Chem. Soc. Rev., 2009, 38, 363 RSC; Y. Furusho and E. Yashima, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5195 CrossRef; D. S. Su, Angew. Chem., Int. Ed., 2011, 50, 4747 CrossRef PubMed.
- H. Goto, H. Katagiri, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7176 CrossRef CAS PubMed; D. Haldar, H. Jiang, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 5483 CrossRef PubMed; Q. Gan, Y. Ferrand, N. Chandramouli, B. Kauffmann, C. Aube, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2012, 134, 15656 CrossRef PubMed; H. Yamada, Z.-Q. Wu, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2012, 134, 9506 CrossRef PubMed; P. Prabhakaran, G. Priya and G. J. Sanjayan, Angew. Chem., Int. Ed., 2012, 51, 4006 CrossRef PubMed; Y. Shinozaki, G. Richards, K. Ogawa, A. Yamano, K. Ohara, K. Yamaguchi, S. Kawano, K. Tanaka, Y. Araki, T. Wada and J. Otsuki, J. Am. Chem. Soc., 2013, 135, 5262 CrossRef PubMed; M. L. Singleton, G. Pirotte, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 13140 CrossRef PubMed; M. Miyagawa, W. Ichinose and M. Yamaguchi, Chem.–Eur. J., 2014, 20, 1272 CrossRef PubMed.
- V. L. Malinovskii, F. Samain and R. Häner, Angew. Chem., Int. Ed., 2007, 46, 4464 CrossRef CAS PubMed; T. Sugimoto, T. Suzuki, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2007, 129, 270 CrossRef PubMed; T. Maeda, Y. Furusho, S.-I. Sakurai, J. Kumaki, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2008, 130, 7938 CrossRef PubMed.
- A. Rajca, A. Andrej, S. Rajca and R. Shoemaker, Angew. Chem., Int. Ed. Engl., 1997, 36, 488 CrossRef CAS; M. J. Marsella, I. T. Kim and F. Tham, J. Am. Chem. Soc., 2000, 122, 974 CrossRef.
- D. E. Fenton, R. W. Matthews, M. McPartlin, B. P. Murphy, I. J. Scowen and P. A. Tasker, J. Chem. Soc., Chem. Commun., 1994, 1391 RSC; R. W. Matthews, M. McPartlin and I. J. Scowen, Chem. Commun., 1996, 309 RSC.
- J. M. Fox, D. Lin, Y. Itagaki and T. Fujita, J. Org. Chem., 1998, 63, 2031 CrossRef CAS; M. Mascal, C. J. Moody, A. I. Morrell, A. M. Z. Slawin and D. J. Williams, J. Am. Chem. Soc., 1993, 115, 813 CrossRef; M. Mascal, I. G. Wood, M. J. Begley, A. S. Batsanov, T. Walsgrove, A. M. Z. Slawin, D. J. Williams, A. F. Drake and G. Siligardi, J. Chem. Soc., Perkin Trans. 1, 1996, 2427 RSC; D. L. An, T. Nakano, A. Orita and J. Otera, Angew. Chem., Int. Ed., 2002, 41, 171 CrossRef PubMed; F. Lin, H.-Y. Peng, J.-X. Chen, D. T. W. Chik, Z. Cai, K. M. C. Wong, V. W. W. Yam and H. N. C. Wong, J. Am. Chem. Soc., 2010, 132, 16383 CrossRef PubMed; H. Maeda, T. Nishimura, R. Akuta, K. Takaishi, M. Uchiyama and A. Muranaka, Chem. Sci., 2013, 4, 1204 RSC.
- Y. Nakatani, Y. Furusho and E. Yashima, Org. Biomol. Chem., 2013, 11, 1614 Search PubMed.
- Dynamic helical molecules based on terephthalamides and related compounds can be classified into three categories: (1) a molecule undergoes a change in conformation from a non-helical form to dynamic helical forms upon complexation with a chiral guest, and prefers a particular sense in a complexed state due to the supramolecular transmission of chirality in the guest;9a (2) a molecule adopts dynamic helical forms, and prefers a particular sense when it forms a complex with a chiral guest due to the supramolecular transmission of chirality in the guest,9b or in a complexed9c/uncomplexed9d state due to the intramolecular transmission of chirality associated with the molecule (no change in the helical preference even upon complexation with a guest); and (3) a molecule adopts a non-helical form to which no local chirality is transferred either supramolecularly or intramolecularly, but undergoes a change in conformation from a non-helical form to dynamic helical forms upon complexation with a chiral guest only when a chiral auxiliary is attached to the host.9e The present macrocyclic system cannot be classified into any of the above categories, and should be classified into a new category for which we recently reported a qualitative prototype with a smaller series of macrocycles based on dynamic figure of eight chirality9f.
- (a) R. Katoono, H. Kawai, K. Fujiwara and T. Suzuki, J. Am. Chem. Soc., 2009, 131, 16896 CrossRef CAS PubMed; (b) R. Katoono, K. Kusaka, K. Fujiwara and T. Suzuki, Chem.–Asian J., 2014, 9, 3182 CrossRef CAS PubMed; (c) R. Katoono, K. Kusaka, S. Kawai, Y. Tanaka, K. Hanada, T. Nehira, K. Fujiwara and T. Suzuki, Org. Biomol. Chem., 2014, 12, 9532 RSC; (d) R. Katoono, H. Kawai, K. Fujiwara and T. Suzuki, Chem. Commun., 2008, 4906 RSC; (e) R. Katoono, K. Fujiwara and T. Suzuki, Chem. Commun., 2014, 50, 5438 RSC; (f) R. Katoono, Y. Tanaka, K. Kusaka, K. Fujiwara and T. Suzuki, J. Org. Chem., 2015, 80, 7613–7625 CrossRef CAS PubMed.
- (a) E. Yashima, T. Matsushima and Y. Okamoto, J. Am. Chem. Soc., 1995, 117, 11596 CrossRef CAS; (b) Y. Inai, N. Ousaka and T. Okabe, J. Am. Chem. Soc., 2003, 125, 8151 CrossRef CAS PubMed; (c) Y. Inai and H. Komori, Biomacromolecules, 2004, 5, 1231 CrossRef CAS PubMed.
- (a) M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860 CrossRef CAS PubMed; (b) O. Pieroni, F. Matera and F. Ciardelli, Tetrahedron Lett., 1972, 13, 597 CrossRef; (c) H. Jiang, C. Dolain, J.-M. Léger, H. Gornitzka and I. Huc, J. Am. Chem. Soc., 2004, 126, 1034 CrossRef CAS PubMed; (d) V. Percec, E. Aqad, M. Peterca, J. G. Rudick, L. Lemon, J. C. Ronda, B. B. De, P. A. Heiney and E. W. Meijer, J. Am. Chem. Soc., 2006, 128, 16365 CrossRef CAS PubMed; (e) D. Pijper and B. L. Feringa, Angew. Chem., Int. Ed., 2007, 46, 3693 CrossRef CAS PubMed; (f) A. M. Kendhale, L. Poniman, Z. Dong, K. Laxmi-Reddy, B. Kauffmann, Y. Ferrand and I. Huc, J. Org. Chem., 2011, 76, 195 CrossRef CAS PubMed; (g) J. Solà, G. A. Morris and J. Clayden, J. Am. Chem. Soc., 2011, 133, 3712 CrossRef PubMed; (h) M. D. Poli, L. Byrne, R. A. Brown, J. Solà, A. Castellanos, T. Boddaert, R. Wechsel, J. D. Beadle and J. Clayden, J. Org. Chem., 2014, 79, 4659 CrossRef PubMed; (i) M. D. Poli and J. Clayden, Org. Biomol. Chem., 2014, 12, 836 RSC; (j) B. A. F. le Bailly, L. Byrne, V. Diemer, M. Foroozandeh, G. A. Morris and J. Clayden, Chem. Sci., 2015, 6, 2313 RSC.
- B. I. Kharisov, P. E. Martínez, V. M. Jiménez-Pérez, O. V. Kharissova, B. N. Martínez and N. Pérez, J. Coord. Chem., 2010, 63, 1 CrossRef CAS; B. H. Northrop, H.-B. Yang and P. J. Stang, Chem. Commun., 2008, 5896 RSC.
- (a) H. A. Staab, H. Mack and A. Nissen, Chem. Ber., 1972, 105, 2310 CrossRef CAS; (b) Y. Ruan, B.-Y. Wang, J. M. Erb, S. Chen, C. M. Hadad and J. D. Badjić, Org. Biomol. Chem., 2013, 11, 7667 RSC; (c) M. Greenwald, D. Wessely, I. Goldberg and Y. Cohen, New J. Chem., 1999, 337 RSC; (d) P. Comba, A. Fath, T. W. Hambley, A. Kühner, D. T. Richens and A. Vielfort, Inorg. Chem., 1998, 37, 4389 CrossRef CAS PubMed; (e) R.-P. Hummel, C. Toniolo and G. Jung, Angew. Chem., Int. Ed. Engl., 1987, 26, 1150 CrossRef; (f) K. Ute, Y. Fukunishi, S. K. Jha, K.-S. Cheon, B. Muñoz, K. Hatada and M. M. Green, Macromolecules, 1999, 32, 1304 CrossRef CAS.
- We estimated the activation energy at the coalescence temperature (ΔG≠ = 71 kJ mol−1 at 358 K) based on the coalescence of diastereotopic protons HF and HF′ (Δδ = 160 Hz) in dimethylsulfoxide-d6.16 Similar observations were noted with 2a and 3a, measured in chloroform-d (Fig. S1†), and showed dynamic interconversion between two equivalent forms in each solution. The activation energies were estimated to be 49 and 58 kJ mol−1 for 2a (Tc = 257 K, Δδ = 236 Hz) and 3a (Tc = 305 K, Δδ = 384 Hz), respectively.
- H. Sato, Y. Mori, T. Kitazawa and A. Yamagishi, Dalton Trans., 2013, 42, 232 RSC; H. T. N. Tran and H. Falk, Monatsh. Chem., 2002, 133, 1231 CrossRef CAS; T. Kimura, M. Hanzawa, K. Tsujimura, T. Takahashi, Y. Kawai, E. Horn, T. Fujii, S. Ogawa and R. Sato, Bull. Chem. Soc. Jpn., 2002, 75, 817 CrossRef; T. Mizutani, S. Yagi, A. Honmaru and H. Ogoshi, J. Am. Chem. Soc., 1996, 118, 5318 CrossRef; V. M. Tormyshev, A. M. Genaev, G. E. Sal'nikov, O. Yu. Rogozhnikova, T. I. Troitskaya, D. V. Trukhin, V. I. Mamatyuk, D. S. Fadeev and H. J. Halpern, Eur. J. Org. Chem., 2012, 623 CrossRef PubMed; A. Martinez, L. Guy and J.-P. Dutasta, J. Am. Chem. Soc., 2010, 132, 16733 CrossRef PubMed; C. Dolain, H. Jiang, J.-M. Léger, P. Guionneau and I. Huc, J. Am. Chem. Soc., 2005, 127, 12943 CrossRef PubMed; Z. Dong, S. Bai, G. P. A. Yap and J. M. Fox, Chem. Commun., 2011, 47, 3781 RSC; K. Ute, K. Hirose, H. Kashimoto, H. Nakayama, K. Hatada and O. Vogl, Polym. J., 1993, 25, 1175 CrossRef; Y. Ito, T. Miyake, S. Hatano, R. Shima, T. Ohara and M. Suginome, J. Am. Chem. Soc., 1998, 120, 11880 CrossRef.
- M. Pons and O. Millet, Prog. Nucl. Magn. Reson. Spectrosc., 2001, 38, 267 CrossRef CAS.
- We confirmed a similar dynamic interconversion between two diastereomeric forms in solutions of (R,R,R,R)-2b and (R,R,R,R)-3b (Fig. S1†). The populations of two non-equivalent species at the coalescence temperature were calculated from a plot of lnK versus 1/T (Fig. S2†), where K is the equilibrium constant between the two diastereomeric forms, obtained at lower temperatures. We estimated the activation energies for the dynamic interconversion to be 68 kJ mol−1 (Tc = 320 K, Δδ = 36 Hz) for (R,R,R,R)-1b and 57 kJ mol−1 (Tc = 273 K, Δδ = 40 Hz) for (R,R,R,R)-2b.
- A conformational search for (R,R,R,R)-1b predicted that a double-helix form with (M)-helicity was more stable (rel. 0 kJ mol−1) than the other form with (P)-helicity (+2.65 kJ mol−1) (Fig. S3†). We found a similar diastereomeric pair of double-helix forms with non-equivalent energies, regarding (R,R,R,R)-2b [rel. 0 kJ mol−1 for (M)-2b and +2.93 kJ mol−1 for (P)-2b] and (R,R,R,R)-3b [rel. 0 kJ mol−1 for (M)-3b and +1.48 kJ mol−1 for (P)-3b].
- An equivalent mixture of (R,R)-5 and (S,S)-5 was used for complexation with 1a, 2a, or 3a in NMR experiments.
- M. Suginome, S. Collet and Y. Ito, Org. Lett., 2002, 4, 351 CrossRef CAS PubMed.
- H. Hayasaka, T. Miyashita, M. Nakayama, K. Kuwada and K. Akagi, J. Am. Chem. Soc., 2012, 134, 3758 CrossRef CAS PubMed.
- J. W. Canary, S. Mortezaei and J. Liang, Chem. Commun., 2010, 46, 5850 RSC.
- M. Fujiki, J. Am. Chem. Soc., 2000, 122, 3336 CrossRef CAS; H.-Z. Tang, P. D. Boyle and B. M. Novak, J. Am. Chem. Soc., 2005, 127, 2136 CrossRef PubMed; J. Gregoliński and J. Lisowski, Angew. Chem., Int. Ed., 2006, 45, 6122 CrossRef PubMed; Y. Imada and A. Abe, Polymer, 2010, 51, 6227 CrossRef; M. Peterca, M. R. Imam, C.-H. Ahn, V. S. K. Balagurusamy, D. A. Wilson, B. M. Rosen and V. Percec, J. Am. Chem. Soc., 2011, 133, 2311 CrossRef PubMed; H. Miyake, M. Ueda, S. Murota, H. Sugimoto and H. Tsukube, Chem. Commun., 2012, 48, 3721 RSC.
- Y. Inai, Y. Kurokawa and N. Kojima, J. Chem. Soc., Perkin Trans. 2, 2002, 1850 RSC; X. Jiang, Y.-K. Lim, B. J. Zhang, E. A. Opsitnick, M.-H. Baik and D. Lee, J. Am. Chem. Soc., 2008, 130, 16812 CrossRef CAS PubMed; L. Ma, R. Jin, Z. Bian, C. Kang, Y. Chen, J. Xu and L. Gao, Chem.–Eur. J., 2012, 18, 13168 CrossRef PubMed; N. Ousaka, Y. Takeyama and E. Yashima, Chem. Sci., 2012, 3, 466 RSC; T. Ogoshi, T. Akutsu, D. Yamafuji, T. Aoki and T. Yamagishi, Angew. Chem., Int. Ed., 2013, 52, 8111 CrossRef PubMed.
- S. Yano, M. Nakagoshi, A. Teratani, M. Kato, T. Onaka, M. Iida, T. Tanase, Y. Yamamoto, H. Uekusa and Y. Ohashi, Inorg. Chem., 1997, 36, 4187 CrossRef CAS; V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, Org. Lett., 2002, 4, 169 CrossRef PubMed; K. Maeda, H. Mochizuki, M. Watanabe and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7639 CrossRef PubMed; R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926 CrossRef PubMed; I. Otsuka, R. Sakai, R. Kakuchi, T. Satoh and T. Kakuchi, Eur. Polym. J., 2008, 44, 2971 CrossRef; J. Suk, V. R. Naidu, X. Liu, M. S. Lah and K.-S. Jeong, J. Am. Chem. Soc., 2011, 133, 1393 Search PubMed; S. Yamamoto, H. Iida and E. Yashima, Angew. Chem., Int. Ed., 2013, 52, 6849 CrossRef PubMed; N. Ousaka, Y. Takeyama and E. Yashima, Chem.–Eur. J., 2013, 19, 4680 CrossRef PubMed.
- As a control experiment, we examined a monotopic guest (R)-15,9a which resulted in no change in the spectrum (Fig. S6†).
- When we gradually added up to two equivalents of 4 to a solution of (R,R,R,R)-3b, the original Cotton effects were stepwise-modulated to show different effects. As we continued to add 4, the spectrum changed to show some impaired Cotton effects, close to a pseudo mirror-image (Fig. S7†). We considered that this discontinuous modulation was the result of the generation of an intermediary 1:1 complex. As 1:2 complexation proceeded, the once-modulated Cotton effects disappeared and changed to ultimately mostly-reversed effects. This result might show that the internal chirality was impaired in a complexed state in this macrocycle (3b).
- J. Solà, S. P. Fletcher, A. Castellanos and J. Clayden, Angew. Chem., Int. Ed., 2010, 49, 6836 CrossRef CAS PubMed; C. Bonnot, E. Aubert, N. Banerji, J. Lacour, E. Espinosa and J.-C. Chambron, Chem.–Eur. J., 2010, 16, 5706 CrossRef PubMed; S. Akine, S. Hotate and T. Nabeshima, J. Am. Chem. Soc., 2011, 133, 13868 CrossRef PubMed; S. Akine, S. Sairenji, T. Taniguchi and T. Nabeshima, J. Am. Chem. Soc., 2013, 135, 12948 CrossRef PubMed; J. Brioche, S. J. Pike, S. Tshepelevitsh, I. Leito, G. A. Morris, S. J. Webb and J. Clayden, J. Am. Chem. Soc., 2015, 137, 6680 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, UV and CD spectroscopic data, energy-minimized structures and experimental details of new compound preparation. CCDC 1404043. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02614h |
| This journal is © The Royal Society of Chemistry 2015 |