
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNHC-catalysed benzoin condensation – is it all down to the Breslow intermediate?†
Julia
Rehbein
*,
Stephanie-M.
Ruser
and
Jenny
Phan
Organische Chemie, Universität Hamburg, Martin-Luther-King-Platz 6, Germany. E-mail: rehbein@chemie.uni-hamburg.de
First published on 21st July 2015
Abstract
The Breslow catalytic cycle describing the benzoin condensation promoted by N-heterocyclic carbenes (NHC) as proposed in the late 1950s has since then been tried by generations of physical organic chemists. Emphasis has been laid on proofing the existence of an enaminol like structure (Breslow intermediate) that explains the observed umpolung of an otherwise electrophilic aldehyde. The present study is not focusing on spectroscopic elucidation of a thiazolydene based Breslow intermediate but rather tries to clarify if this key-intermediate is indeed directly linked with the product side of the overall reaction. The here presented EPR-spectroscopic and computational data provide a fundamentally different view on how the benzoin condensation may proceed: a radical pair could be identified as a second key-intermediate that is derived from the Breslow-intermediate via an SET process. These results highlight the close relationship to the Cannizarro reaction and oxidative transformations of aldehydes under NHC catalysis.
Introduction
The NHC-catalysed benzoin condensation is not only of synthetic value1,2 but also has challenged generations of physical organic chemists with its mechanistic characteristics.3–5 Above all the Breslow intermediate 5 (Scheme 1) has been a long-hunted phantom which finally could be unveiled in 2012 by Berkessel's fundamental NMR studies5a for a certain type of catalysts. Although one should mention that O-protected Breslow intermediates have been synthesized6b and characterised as intermediates6c of other NHC-catalysed reactions before allowing for some mechanistic interpretation. Berkessel's remarkable study on the other hand provided 1H and 13C NMR data of free enaminols derived of imidazolidinium-based carbenes and aromatic aldehydes. These studies also linked the enaminol to the formation of benzoin. However, the elementary steps as depicted in the catalytic cycle (Scheme 1) still provide room for discussion: is the path of enaminol to benzoin an elementary step? How is the enaminol formed? Since for the thiazolydene catalysed benzoin condensation the only intermediate that had been characterized so far is the primary adduct 4,3b the here presented studies start from there.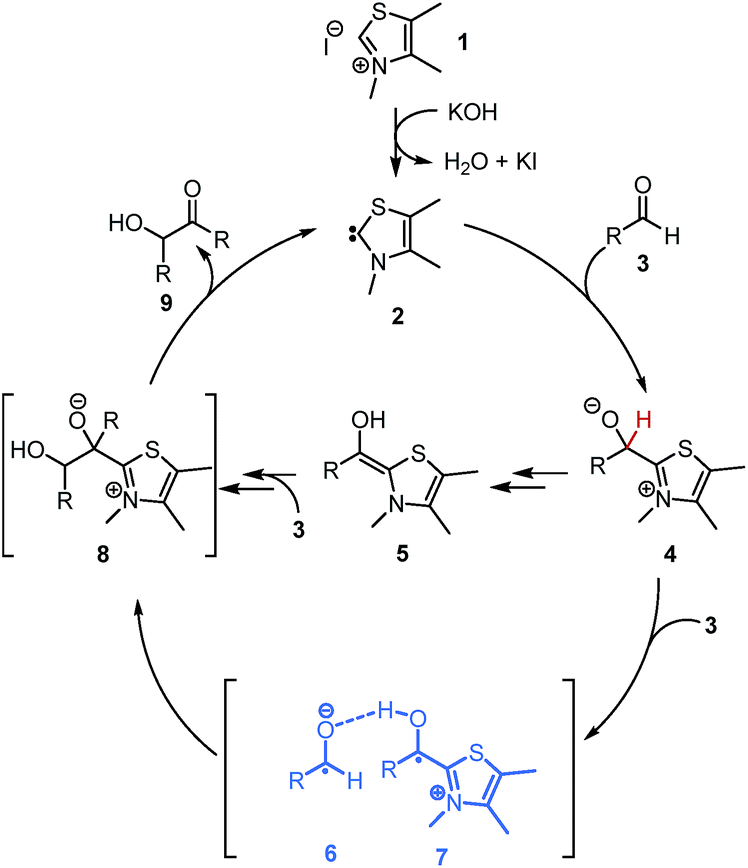 | ||
| Scheme 1 Breslow catalytic cycle and a simplified alternative via Breslow-intermediate-like radical 7. | ||
Driving force for the transformation of 4 (Scheme 1) is the removal of its zwitterionic state. In principle, the charge separation in 4 can be removed by formation of the Breslow intermediate 5 (Scheme 1). The mechanism even of this simple transformation is still not completely understood; in protic polar solvents the formal proton-shift is according to He et al.3a most likely an intermolecular process under assistance by solvent molecules. Under aprotic conditions however, i.e. no Brønsted base and aprotic solvent the proton-shuffle must occur in a different fashion. Here a 1,2-hydride-shift has been discussed but could be excluded based on Berkessel's detailed studies5c which could show that the resulting ketone is unreactive under benzoin condensation conditions.
On second thoughts, one could also envision a homolytic bond cleavage of the discussed C–H bond in 4 (Scheme 1, H in red). Such a homolysis and the resulting radical are not as farfetched as one might think. Carbon centred radicals stabilized by adjacent heterocycles6a,7,8,9,10,11 and heteroatoms12 have been proposed or even characterized in several reactions that involve NHCs: the enzymatic formation of CoAsAc,6 the TEMPO mediated oxidative transformation of aldehydes to carboxylic acid derivatives (Studer et al.)8 and most recently the enantioselective β-hydroxylation of enals by Rovis et al.9a and Webster, Chi and co-workers.9b For understanding how the homolysis of the C–H bond of 4 within the benzoin condensation could take place it is helpful to look at the Cannizzaro reaction13 (Scheme 2). The primary Cannizzaro-adduct 10 of aromatic aldehyde 3 and hydroxide as base reacts with a second molecule 3via a sequence of SET and proton transfer to benzyl alcohol 11 and benzoic acid ester 12. This most often becomes abbreviated to a formal hydride transfer which suits our intuitive ‘polar’ understanding of reactivity. However, the ionic writing of reaction mechanisms may be just the one extreme of the whole electronic continuum of bond cleavage and formation. With a close structural relationship of 10 and 4 it is not unlikely that 4 might undergo similar SET processes.
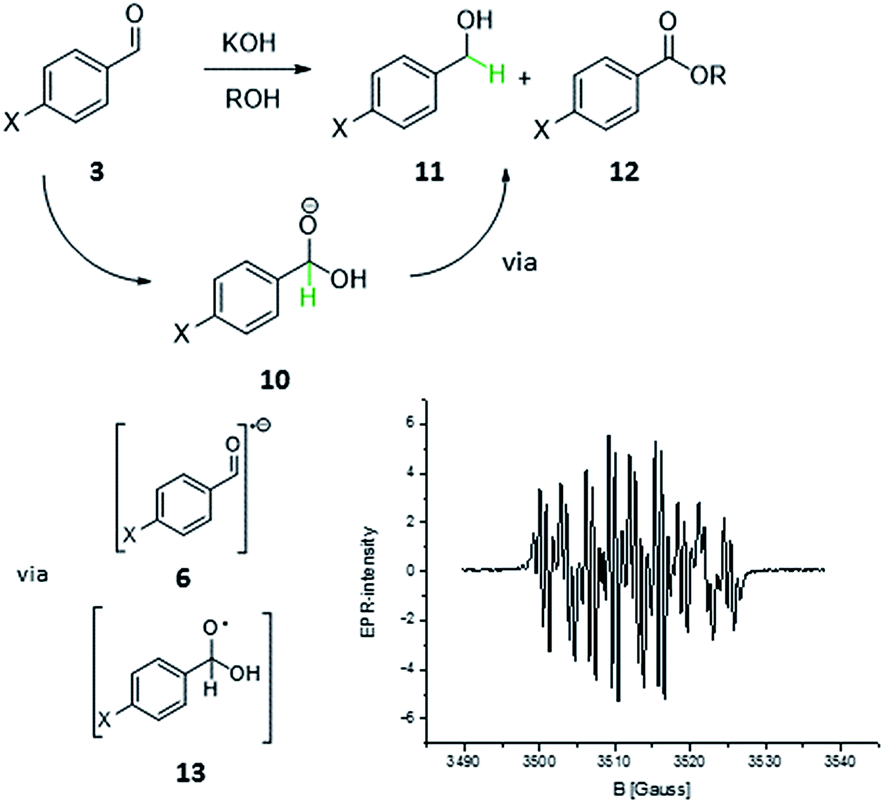 | ||
| Scheme 2 In analogy to the Cannizzaro process: are radical reaction steps in the formation of benzoin possible? Structurally the primary adduct 10 is closely related to 4. The EPR spectrum was recorded of a solution of p-nitro benzaldehyde 3j and KOH (1 eq.) in i-PrOH at room temperature. | ||
A second advantage of the depicted radical pathway (Scheme 1) is its simplicity. The number of necessary elemental steps for the various proton shuffling events in the ionic pathway are reduced.
Results and discussion
Besides this philosophical aspect of Ockham's razor, there is also numeric evidence. Looking at the thermodynamics by simple calculations (Table 1) the C–H bond dissociation energy (BDE) of 4 is surprisingly low, independently of the aldehyde and the catalyst. Connected with a low C–H BDE are the exceptionally high radical stabilization energies (RSEs) of 7 that suggest according to the Bell-Evans-Polanyi-principle (BEP) that the corresponding activation barrier for the formation of 7 should be comparably low. The high stabilization of 7 can be explained by mesomeric effects preferably by the heterocycle, since here the necessary planarity of the π-system is realized best (Fig. 1). In addition the captodative effect14 might contribute to the high stability of 7 as well.|
|
||||
|---|---|---|---|---|
| entry | cmpd | BDE [kcal mol−1] | cmpd | RSEa [kcal mol−1] |
| a In comparison to methyl radical. b Designation of the compounds as follows: number of intermediate as defined in Scheme 1, first letter assigns catalyst, second letter assigns aldehyde. | ||||
| 1 | 4a-a | 36.8 (39.7) [40.5] | 7a-a | -50.1 (−63.9) [-54.6] |
| 2 | 4a-a-H | 72.0 (70.2) [69.9] | 7a-a-H | - 32.9 (−25.6) [-24.8] |
| 3 | 4a-b | 33.8 (36.7) [35.8] | 7a-b | -56.0 (−66.9) [-57.8] |
| 4 | 4a-c | 32.6 (37.7) [38.2] | 7a-c | -57.2 (−65.9) [-56.1] |
| 5 | 4g-a | 54.0 (47.9) [55.6] | 7g-a | -59.6 (−56.9) [-59.2] |
| 6 | 4h-a | 38.3 (42.2) | 7h-a | -66.5 (−62.6) |

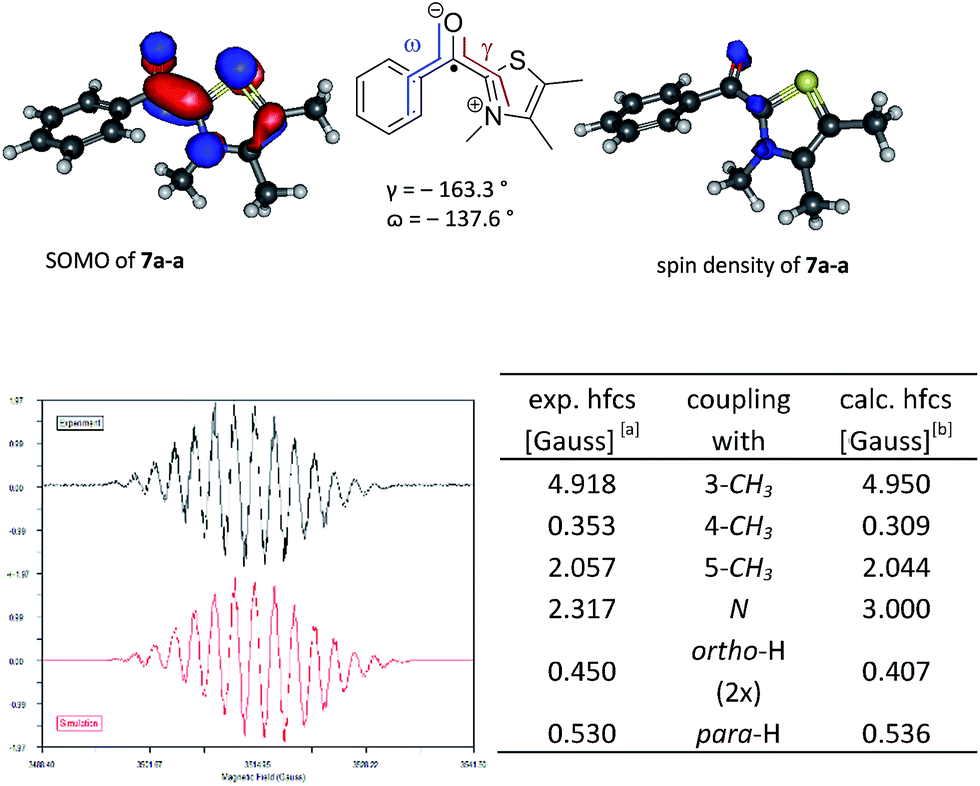 | ||
Fig. 1 Top: Plotted SOMOs and spin densities for the postulated key intermediate 7a-a (B3LYP/6-31G*). Bottom: Experimental EPR spectrum16 and fitted EPR spectrum17 (r² = 0.93) with tabulated hyperfine coupling constants. [a] WINSIM version 0.98.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 [b] B3LYP/EPR-II//B3LYP/6-31G* for C, H, O, N and 6-31G* for S, Br. 17 [b] B3LYP/EPR-II//B3LYP/6-31G* for C, H, O, N and 6-31G* for S, Br. | ||
Subsequently, typical reaction mixtures for the thiazolidene catalysed benzoin condensation in protic polar solvent (1a, KOH as base, i-PrOH, exclusion of light) and prepared under oxygen-free conditions15 were analyzed by CW-EPR spectroscopy. The observation of a strong CW-EPR-signal (Fig. 1) clearly speaks for radicals being present. Moreover, the radical is only formed if all reactants are present and air is vigorously excluded (see ESI†). The questions to be answered next are related to the structure of the radical(s) causing the EPR signal and in a second step the question of their origin and function. To address the first question we used the combination of experiments and computational data.
Led by the hypothesis that the observed EPR-signal corresponds to the suggested structures 7 or [6·7] one would expect that the signal should change in dependence of catalyst 2 and aldehyde 3. Looking at the calculated spin density distribution of 7a-a (Fig. 1 shows 7a-a, for other examples see ESI†) one would expect the strongest dependence of the EPR spectrum on the substituents at the 3- and 5-position of the thiazole. Accordingly, the EPR-spectra resulting from the reaction solutions of 3,4,5-trimethylthiazolium iodide, KOH and various p-substituted benzaldehydes (3a, d-f), propionaldehyde (3h), and 2,2-diphenylcyclopropyl carbaldehyde (3i) showed only minor variations of the EPR-spectra in dependence of the aldehyde (see ESI† for EPR-spectra). On the other hand the experimental EPR-signals changed significantly as a function of the substitution pattern at the thiazolium ring (Fig. 2) supporting the postulated radical structure. Finally, calculated Fermi isotropic coupling constants (uB3LYP/EPRIII for C, H, N, O; 6-31+G* for S) were used as starting points for fitting the observed EPR-spectra and led to fits with r² values ≥0.93 allowing the conclusion that the observed spectra actually belong to [6·7]. It should be noted that in several cases good fits could be obtained with simply using simulated coupling constants of 7 since the radical anions 6 often have similar coupling constants (for an EPR spectrum of 6a see ESI†). That the EPR signal is indeed representing two radical species became apparent when analysing the EPR data derived of the reaction of 1e and 3a. Here the spectra could only be fitted satisfyingly if a 1:1 mixture of 6a and 7e-a were assumed (see Fig. 2 & ESI†).
 | ||
| Fig. 2 EPR spectra derived of PhCHO (3a) in i-PrOH with 0.1 eq. of pre-catalysts 1b (3,4,5-trimethyl thiazolium tetrafluoroborate), 1c (3-benzyl-4,5-dimethyl thiazolium bromide), 1d (3,4-dimethyl-5-(2-hydroxyethyl) thiazolium iodide) and 1e (thiamine hydrochloride) and equimolar amounts of KOH. | ||
Having established the structure of the experimentally observed radicals, the next step was to understand their formation, reactivity and potential role within the catalytic cycle. The simplest hypothesis for the formation of [6·7] is already described in Scheme 1: Another molecule aldehyde 3 acts as an oxidant of 4. The postulated hydrogen atom transfer would benefit of a direct onwards path to benzoin 9 and free carbene 2 by simple recombination of the radical pair. Oxygen for several reasons has been excluded from being the oxidant; samples being prepared in a glove box provided EPR signals similarly to the reaction mixtures prepared under the common preparative benzoin condensation conditions (freeze–pump–thaw procedure).15 Also, opening a degassed reaction sample to air does not increase the radical signal but quenches it within 30 min and led to a different product spectrum consisting mainly of benzoic acid esters.8 Finally, if oxygen alone would cause the formation of 7 one would not observe a constant increase in radical with increasing catalyst load (Fig. 3) or similarly a reduction of EPR-intensity when the samples are diluted (see ESI†).
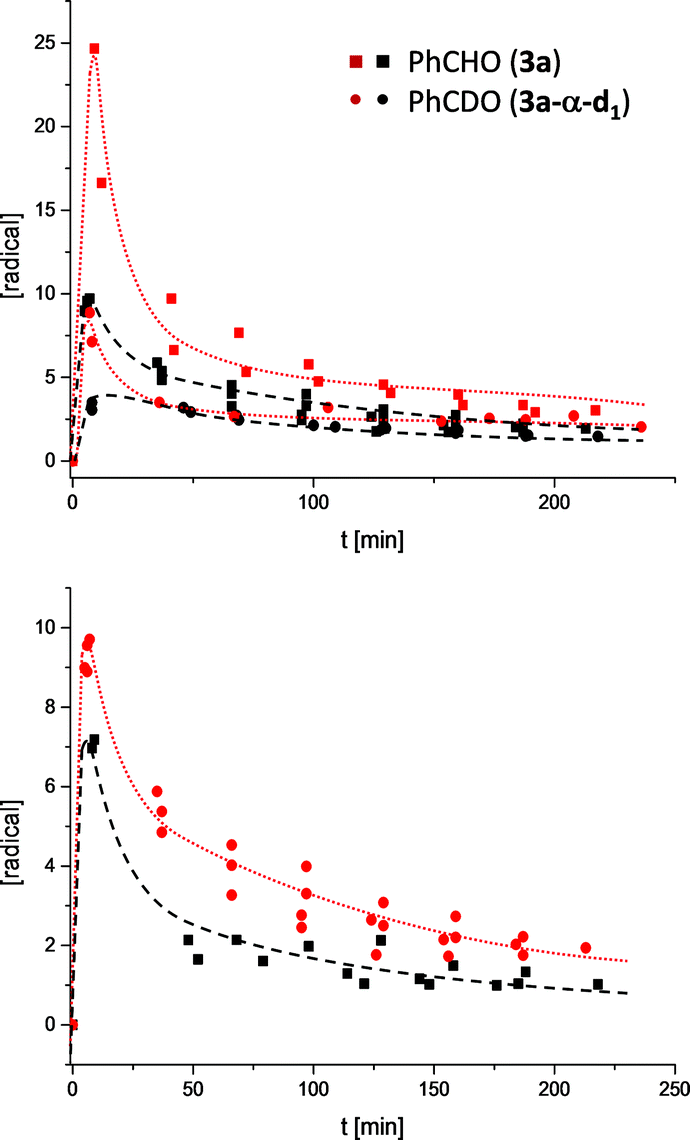 | ||
| Fig. 3 Progress curves of radical under different conditions derived of 2–4 independent runs. Top: in i-PrOD: black symbols 0.4 eq. 1a + KOH and 0.1 M in 3a (square) vs. 0.1 M in 3a-α-d1 (dot); red symbols 1.0 eq. 1a + KOH and 0.1 M 3a (squares) and 0.1 M in 3a-α-d1 (dots). Below: 0.4 eq. 1a + KOH and 0.1 M in 3a in i-PrOD (red), i-PrOH (black). | ||
The associated progress curves (Fig. 3) are characterized by the instantaneous rise in radical concentration and a slower decline, which speaks clearly for a reactive transient. The radicals are not accumulated over time but reach an equilibrium concentration which is proportional to the initial ratio of 1 to 3. Moreover, the shape of the progress curves also exclude a third option for the oxidant, the condensation product 9 itself. Since the highest radical concentrations are observed right in the beginning of the benzoin formation the radicals must originate from early intermediates of the catalytic cycle (Scheme 1). To follow this line of argumentation α-d1-benzaldehyde (3a-α-d1) was prepared and subjected to the standard conditions (1a, KOH, i-PrOD). If the radical is formed from the earliest, and by White and Leeper independently characterized intermediate possible,3bi.e.4, the molecule needs to lose the hydrogen atom marked in red (Scheme 1). Therefore, if 7 is formed out of 4 within a sequence where the hydrogen or proton abstraction is (partially) rate determining one would expect a kinetic isotope effect (KIE).
Indeed, following the radical concentration over time for 3a and 3a-α-d1 (Fig. 3) a normal KIE is observed and the maximum radical concentration for 3a-α-d1 under three different reaction conditions (see ESI†) is on average by a factor of 3.0 ± 0.7 lower than for 3a. Keeping with Ockham's’ razor, the simplest mechanistic scenario rationalizing the observed KIE would be a direct hydrogen atom transfer from 4 to 3 under the formation of [6·7] as depicted in Scheme 1. Interestingly, the KIE of radical formation correlates well with the observed KIE for the overall benzoin formation that has been determined by White and Leeper to be 3.4 in MeOD.3b The only explanation is: benzoin and radicals must stem from mechanisms going through the same rate determining steps. This finding brings up the third question about the function of the radical. The proposed scenario in Scheme 1 describes the radical pair as a productive intermediate. The possibility that the radicals belong to a side reaction can be excluded: All tested reactions that provided an EPR signal produced benzoins and unless oxygen was present only benzoins were formed according to 1H NMR over the course of 3.5 h. Also, based on the reversibility of the benzoin reaction and the law of microscopic reversibility further evidence was found in favour of the proposed connection radical ↔ benzoin:18 Instead of 3a benzoin 9a was subjected to the otherwise same reaction conditions and an EPR spectrum was recorded 10 min19 after benzoin addition (Scheme 3). Not only the occurrence of a radical speaks for the link between benzoin and the proposed radical mechanism, but moreover, the obtained EPR spectrum of the reverse reaction is absolutely identical to the one derived from the forward reaction (Scheme 3).
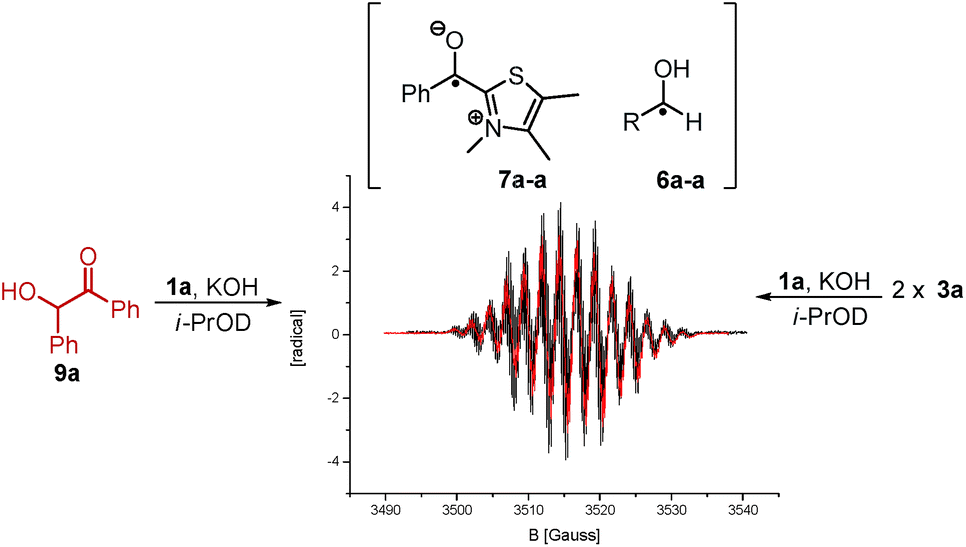 | ||
| Scheme 3 Superimposed EPR spectra obtained from forward (3a) and reverse reaction (9a, 0.2 M in reactant, 0.2 eq. 1a, 0.2 eq. KOH, i-PrOD). The radicals of forward and reverse reaction are identical. | ||
With this information in hand we were intrigued to discover if the proposed radical pathway is an either-or-alternative to the ionic pathway or if both reaction mechanism – ionic and radical – are happening simultaneously. Therefore, conditions established by Berkessel and co-worker for forming a stable Breslow-intermediate5a were used to check if 5 and 7 can be observed at the same time. Samples of 2f and 3a in THF-d8 (Fig. 4) were analysed by 1H NMR to prove that the Breslow-intermediate 5f-a had been formed and an aliquot of the same sample was analysed by CW-EPR spectroscopy. The result is summarized in Fig. 4. Breslow intermediate 5f-a and the corresponding radicals [6a·7f-a] were observed under the same conditions at the same time! In combination with the information about the kinetics and KIEs of radical and benzoin formation, this experiment provokes the question if there are indeed two simultaneous pathways or if both intermediates are actually two (consecutive) intermediates of the same mechanism? The experiment in Fig. 4, however, does not allow for such a conclusion. In order to find out about a possible interconversion between 5 and [6·7] a set of experiments were carried out as described in Fig. 5.
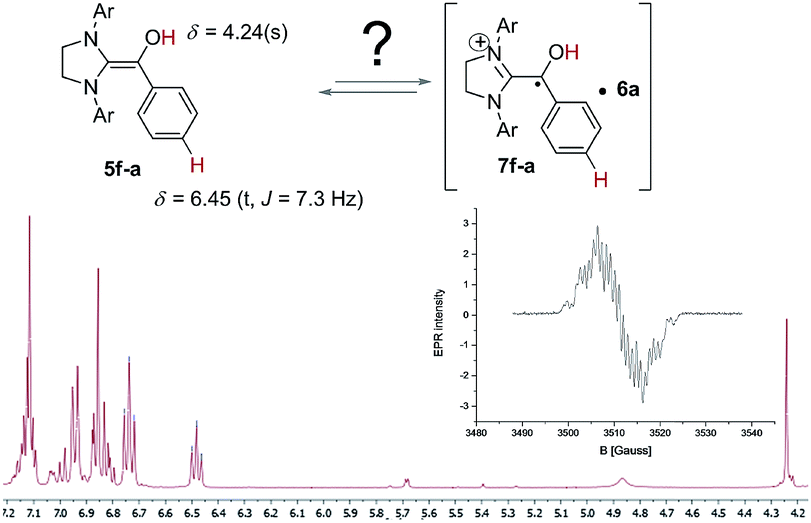 | ||
| Fig. 4 Co-existence of the Breslow intermediate 5f-a and the Breslow-type radical 7f-a in THF-d8. | ||
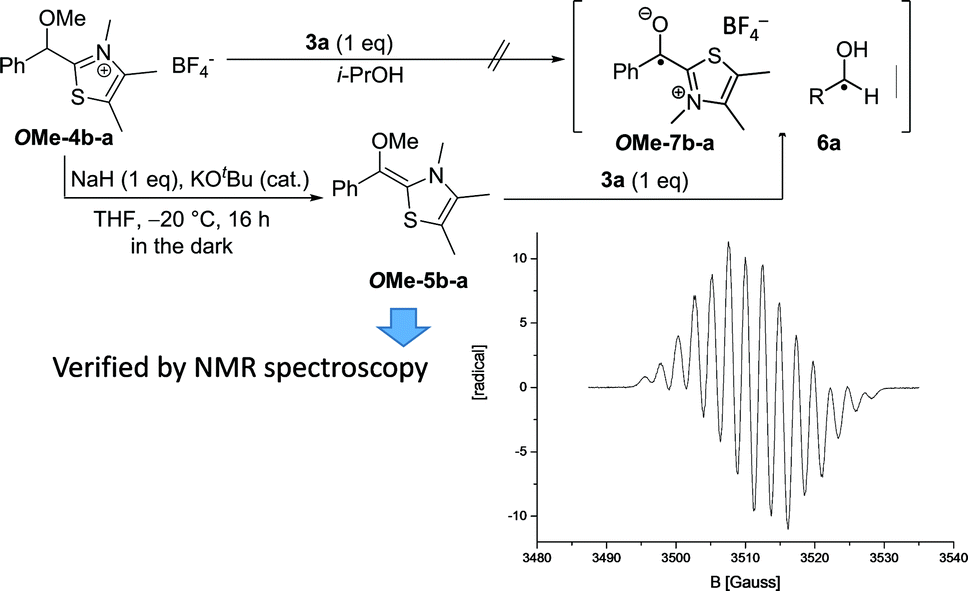 | ||
| Fig. 5 Independent generation of Breslow like radicals OMe-7b-a from OMe-5b-a by addition of 1 eq. 3a. The treatment of OMe-4b-a with 1eq 3a did not provide an EPR signal indicating that at least from the cationic species OMe-4b-a radical formation is not directly possible via a hydrogen atom transfer. | ||
According to a procedure of Mayr and coworkers,6bO-methylated primary adduct OMe-4b-a was quantitatively transformed with stoichiometric amounts of NaH/KOtBu (cat.) in THF into the O-methylated Breslow intermediate OMe-5b-a at −20 °C (Fig. 5). This solution was EPR silent. Only when 3a (1 eq.) was added an EPR-signal emerged spontaneously that was by a factor of >20 more intense than any signal observed at similar substrate concentrations under normal catalysis conditions. The obtained spectrum is besides its reduced hyperfine structure20 identical to the one obtained in the reaction of 3a with 1a and KOH to 9a. This result finally led to a revision of the aforementioned working hypothesis (Scheme 1) and cumulates in a new catalytic cycle (Scheme 4) that incorporates the radical pair as a second key intermediate in the NHC-catalysed benzoin condensation.
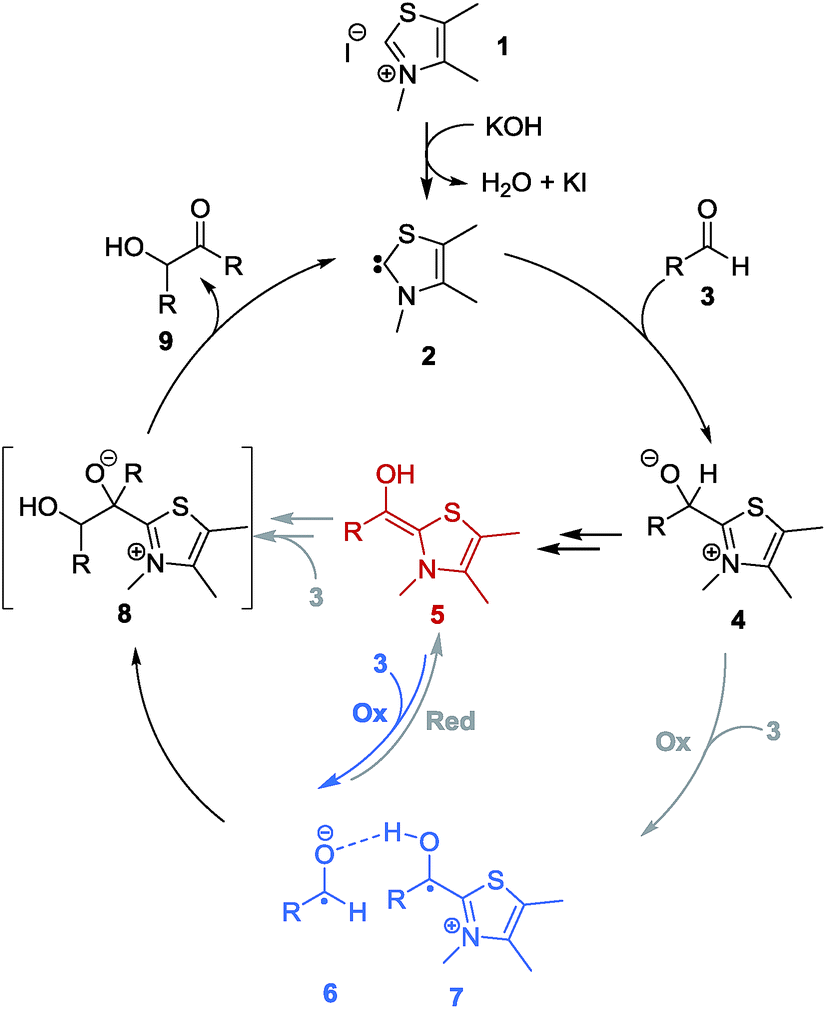 | ||
| Scheme 4 Revised catalytic cycle – under thiazolydene-catalysis benzoin is formed most likely via consecutive ionic and radical steps: Blue arrow represents the newly found SET-process connecting Breslow intermediate 5 with radical pair [6·7] that collapses directly to benzoin 9 and carbene 2via radical recombination. | ||
Evidence that indeed the Breslow intermediate 5 is a very likely precursor of the radical pair [6·7] is found in the Table 2. Listed in column 3 are the peak oxidation potentials obtained from cyclovoltagrams of OMe-5a-a,d,e,f in DMSO as measured by Barletta et al.6a These values correspond very well with the maximal radical concentration observed in the NHCs catalysis of 3a,d,e,f and 1a (+KOH, i-PrOH). The oxidations potentials also correlate with the benzoin conversion obtained after 3 h. The likelihood that such a correlation is achieved by chance cannot be excluded but in combination with the so far presented results this data suggests that under the described conditions the radical pair is involved in the benzoin formation. To which extend will be clarified in future work.
| entry | RCHO | peak potential [mV] | [radical]max | conv. to benzoin [%] |
|---|---|---|---|---|
| 1 | 3e | −47 | 0.6 | 9 |
| 2 | 3d | −33 | 0.8 | 44 |
| 3 | 3a | −12 | 0.8 | 65 |
| 4 | 3f | 22 | 1.6 | 80 |
Conclusions
Radical transients have been observed and characterized for a variety of N-heterocyclic carbenes and aldehydes in the benzoin condensation reaction. The structure of the observed radicals, their kinetic behavior and their instantaneous formation from benzoin under catalysis conditions provided a strong link between radicals and benzoin formation. The subsequently proven coexistence of an imidazolidynium based Breslow intermediate and its corresponding Breslow-like radical raised the question of a potential link between the proposed radical and ionic pathways. The independent generation of the same radical species as before from an O-methylated Breslow intermediate by addition of aldehyde answered this last question and led to the formulation of a new catalytic cycle that unites polar steps and SET–radical recombination steps. The close correlation of oxidation potentials of the O-methylated Breslow intermediates of four different p-substituted benzaldehydes with the observed radical concentrations and benzoin conversion under catalysis conditions strengthened the last conclusion. Moreover, the discovered radical intermediates can be understood as a link between other reactivities that have been exploited for instance in oxidative transformations of aldehydes via NHC-catalysis and also may explain the elusive nature of thiazolium based Breslow intermediates. Further studies that will provide more details on the radical formation processes and its dynamics are underway and will be published in due course.Acknowledgements
Financial support by the FCI, DFG (Emmy-Noether-Stipend) and Fachbereich Chemie, especially C. B. W. Stark is gratefully acknowledged. We wish to thank G. Eggers for measurements of all EPR spectra presented. J. R. thanks U.-P. Apfel (Universität Bochum) for a generous donation of TritSNO.Notes and references
- Review articles: (a) R. Kluger and K. Tittmann, Chem. Rev., 2008, 108, 1797 CrossRef CAS PubMed; (b) N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., 2007, 119, 3046 ( Angew. Chem., Int. Ed. , 2007 , 26 , 2988 ) CrossRef PubMed . Latest Reviews concerning method development and synthetic applications: ; (c) S. de Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem.–Eur. J., 2013, 19, 4664 CrossRef CAS PubMed; (d) A. Grossmann and D. Enders, Angew.Chem., 2012, 124, 320 ( Angew. Chem., Int. Ed. , 2012 , 51 , 314 ) CrossRef PubMed; (e) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC; (f) T. Dröge and F. Glorius, Angew. Chem., 2010, 122, 7094 ( Angew. Chem., Int. Ed. , 2010 , 49 , 6940 ) CrossRef PubMed; (g) J. Douglas, G. Churchill and A. D. Smith, Synthesis, 2012, 2295 CAS; (h) K. Zeitler, Angew. Chem., 2005, 117, 7674 ( Angew. Chem., Int. Ed. , 2005 , 44 , 7506 ) CrossRef PubMed.
- Organocatalysis, selected examples: (a) A. Grossmann and D. Enders, Angew. Chem., 2012, 124, 320 ( Angew. Chem., Int. Ed. , 2012 , 51 , 314 ) CrossRef PubMed; (b) D. Enders and A. A. Narine, J. Org. Chem., 2008, 73, 7857 CrossRef CAS PubMed; (c) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (d) F. Glorius and C. Burstein, Angew. Chem., 2004, 116, 6331 ( Angew. Chem., Int. Ed. , 2004 , 43 , 6205 ) CrossRef PubMed.
- (a) Y. He and Y. Xue, J. Phys. Chem. A, 2011, 115, 1408 CrossRef CAS PubMed; (b) M. J. White and F. J. Leeper, J. Org. Chem., 2001, 66, 5124 CrossRef CAS PubMed; (c) G. L. Barletta, Y. Zou, W. P. Huskey and F. Jordan, J. Am. Chem. Soc., 1997, 119, 2356 CrossRef CAS; (d) Y.-T. Chen, G. L. Barletta, K. Haghjoo, J. T. Cheng and F. Jordan, J. Org. Chem., 1994, 59, 7714 CrossRef CAS.
- (a) R. Breslow, J. Am. Chem. Soc., 1957, 79, 1762 CrossRef CAS; (b) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS.
- (a) A. Berkessel, V. R. Yatham, S. Elfert and J.-M. Neudörfl, Angew. Chem., 2013, 49, 12537 ( Angew. Chem., Int. Ed. , 2013 , 52 , 11158 ) Search PubMed; (b) A. Berkessel, S. Elfert, V. R. Yatham, J.-M. Neudörfl, N. E. Schlörer and J. H. Teles, Angew. Chem., 2012, 124, 12537 ( Angew. Chem., Int. Ed. , 2012 , 51 , 12370 ) CrossRef PubMed; (c) A. Berkessel, S. Elfert, K. Etzenbach-Effers and J. H. Teles, Angew. Chem., 2010, 49, 7120 CrossRef CAS PubMed.
- (a) G. Barletta, A. C. Chung, C. B. Rios, F. Jordan and M. Schlegel, J. Am. Chem. Soc., 1990, 112, 8144 CrossRef CAS; (b) B. Maji and H. Mayr, Angew. Chem., Int. Ed., 2012, 51, 10408 CrossRef CAS PubMed; (c) V. Nair, S. Bindu, V. Sreekumar and N. P. Rath, Org. Lett., 2003, 5, 665 CrossRef CAS PubMed.
- (a) E. Chabriere, X. Vernede, B. Guigliarelli, M. H. Charon, E. C. Hatchikian and J. C. Fontecilla-Camps, Science, 2001, 294, 2559 CrossRef CAS PubMed; (b) S. O. Mansoorabadi, J. Seravalli, C. Furdui, V. Krymov, G. J. Gerfen, T. P. Begley, J. Melnick, S. W. Ragsdale and G. H. Reed, Biochemistry, 2006, 45, 7122 CrossRef CAS PubMed.
- J. Guin, S. de Sarkar, S. Grimme and A. Studer, Angew. Chem., 2008, 120, 8855 ( Angew. Chem., Int. Ed. , 2008 , 47 , 8727 ) CrossRef PubMed.
- (a) N. A. White and T. Rovis, J. Am. Chem. Soc., 2014, 136, 14674 CrossRef CAS PubMed; (b) Y. Zhang, Y. Du, Z. Huang, J. Xu, X. Wu, Y. Wang, M. Wang, S. Yang, R. D. Webster and Y. Robin, J. Am. Chem. Soc., 2015, 137, 2416 CrossRef CAS PubMed.
- Y. Du, Y. Wang, X. Li, Y. Shao, G. Li, R. D. Webster and Y. R. Chi, Org. Lett., 2014, 16, 5678 CrossRef CAS PubMed.
- A. I. Vovk, A. F. Babicheva, V. D. Pokhodenko and A. A. Yasnikov, Dopov. Akad. Nauk Ukr. RSR, Ser. B: Geol., Khim. Biol. Nauki, 1977, 10, 907 Search PubMed.
- Heteroatoms in general are able to stabilize (carbon-centered) radicals by interaction of their free electron pairs with the half-filled p-orbital of the radical center.
- (a) E. C. Ashby, D. T. Coleman III and M. Gamasa, Tetrahedron Lett., 1983, 24, 851 CrossRef CAS; (b) A. Fuentes and J. V. Sinisterra, Tetrahedron Lett., 1986, 27, 2967 CrossRef CAS; (c) A. Fuentes, J. M. Marinas and J. V. Sinisterra, Tetrahedron Lett., 1987, 28, 2947 CrossRef CAS; (d) E. C. Ashby, D. Coleman and M. Gamasa, J. Org. Chem., 1987, 52, 4075 Search PubMed.
- H. Viehe, Z. Janousek, R. Merényi and L. Stella, Acc. Chem. Res., 1985, 18, 148 CrossRef CAS.
- The standard conditions for sample preparation used up to five cycles of the freeze–pump–thaw-procedure (under fine vacuum). Preparation in a glove box was done for matters of comparison for a couple of samples (O2-content < 20 ppm).
- Reaction conditions: 0.4 M 3a, 0.1 eq. 1a, 0.1 eq. KOH in i-PrOH; presented spectrum recorded 40 min after addition of 3a. EPR spectrometer parameters: ν = 9.859 GHz, field resolution 0.049, 10.5 mW, H0 = 3513 G.
- Based on calculated spin densities and Fermi contact couplings (B3LYP/EPR-III//B3LYP/6-31G*) for C, H, O, N and 6-31G* for S, Br. Fitting was done with software WINSIM version 0.98; D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105 CrossRef CAS.
- Intermediate 8 is not stable, as it is residing on a flat plateau with no significant barriers to the forward or reverse reaction as calculations by He et al. could show (see citation 3a). Therefore, one can formulate a direct elemental step involving 9 and [6·7].
- Time delay due to necessary adjustments of the spectrometer.
- The lack of hyperfine structure in the spectrum is caused by the high radical concentration and potentially also by the solvent. The radical concentration was estimated to be in the region of 1.5 m. Estimation was done using a calibration curve with TEMPO in toluene (ESI,† A.2.2.9).
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation data of products, substrates and catalysts, EPR and NMR spectra and progress curves as well as computational details are found. See DOI: 10.1039/c5sc02186c |
| This journal is © The Royal Society of Chemistry 2015 |