
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCycloaddition of cyclobutenone and azomethine imine enabled by chiral isothiourea organic catalysts†
Bao-Sheng
Li‡
a,
Yuhuang
Wang‡
a,
Zhichao
Jin
a and
Yonggui Robin
Chi
*ab
aDivision of Chemistry & Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, Singapore, 637371, Singapore. E-mail: robinchi@ntu.edu.sg
bState Key Laboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Guizhou University, Guiyang, 550025, China
First published on 20th July 2015
Abstract
The addition of an organic catalyst to the ketone moiety of a γ-mono-chloride substituted cyclobutenone destroys its stable, conjugated and nearly planar structure. The C–C bond in the resulting less stable anionic oxy-substituted non-planar intermediate is then activated. The breaking of one C–C single bond leads to a catalyst-bound intermediate that undergoes α-carbon selective reactions with azomethine imines to afford nitrogen-containing heterocyclic compounds with excellent diastereo- and enantio-selectivities. Our organocatalytic approach provides a new reaction pattern for C–C bond activation of cyclobutenones that is unavailable with transition metal catalysis. In addition, the present study with isothioureas as the organocatalysts expands the potential in using organocatalysts for C–C bond breaking and selective reactions.
Introduction
The direct breakage of carbon–carbon (C–C) single bonds1 provides unique opportunities for organic synthesis. Cyclobutane derivatives,2,3 especially cyclobutenones,4 are privileged synthons because a release of the ring strain via carbon–carbon bond breakage can provide versatile reactive intermediates. The thermal four-electron electrocyclic cleavage of cyclobutenones (typically at 100 °C or above) can generate vinyl ketene intermediates that participate in a number of reactions, such as the benzannulation first reported by Danheiser in 1984.5 To achieve better reaction control and diversity, transition metal catalysts have been developed to activate cyclobutenones and modulate the subsequent reactions (Scheme 1a).4 Typically, the transition metal catalysis process is initiated by oxidative addition of a transition metal catalyst to the C–C bond of cyclobutenones. It has been observed that both chemo- and stereo-selectivities are difficult to control in these otherwise elegant reactions, likely due to the high reactivity of the metal catalyst and the metal-bound intermediates.6,7 | ||
| Scheme 1 Organocatalytic carbon–carbon activation of cyclobutenones. | ||
We're interested in using organic catalysts to initiate selective and efficient reactions. It has been observed that the C–C bond of cyclobutenones (and cyclobutenes) can be weakened by destroying their nearly planar conjugated structures via substitutions, as indicated by computational findings from Houk and co-workers.8a,8b Usually, an “outward” rotation of the thermal electrocyclic ring-opening process is favorable when a substituent is installed at the C4 carbon of cyclobutenone.8 In particular, studies from Baldwin suggested a preference for outward rotation of the chlorine substituent at the C4 carbon of cyclobutenone.8c By taking advantage of this intrinsic property of cyclobutenone, we hypothesized that addition of an organic catalyst to the ketone moiety of cyclobutenone might achieve the activation of the carbon–carbon single bond via formation of a non-planar intermediate that leads to the breakage of its C–C bond for further reactions (Scheme 1b).9 This reaction would constitute a highly efficient approach in organocatalysis, in which all atoms of the substrate end up in the product (atom economy) and no overall redox process is involved (redox economy). Recently, we reported the addition of an N-heterocyclic carbene catalyst to the ketone moiety of cyclobutenones to initiate highly enantioselective formal [4 + 2] reactions, in which the γ-carbon of cyclobutenone reacted as a nucleophilic carbon.9 Here, we report highly selective [3 + 2] reactions by using an isothiourea organic catalyst to activate and modulate the reactivities/selectivities of mono-chloride substituted cyclobutenones (Scheme 1c).
Results and discussion
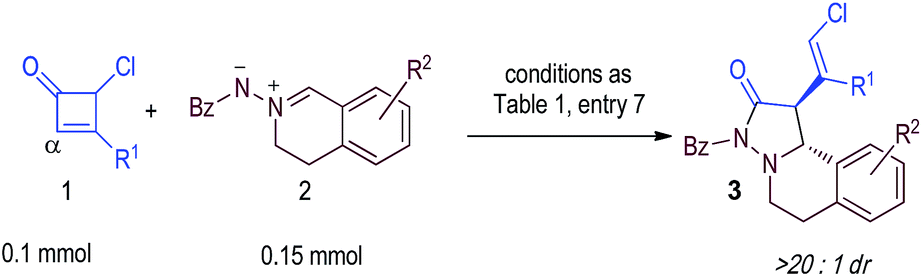
Under the catalysis of isothiourea with cyclobutenone 1a and azomethine imine102a as the substrate, the reaction exclusively took place on the α-carbon of the cyclobutenone via a formal [2 + 3] process. Our result is consistent with an earlier report by Smith that the reaction of the isothiourea-generated dienolate intermediate is α-carbon selective.12e Notably, Studer has recently reported the [2 + 3] cycloaddition of azomethine imines with enolates prepared using an isothiourea catalyst.10d Briefly, in our reaction, nucleophilic 1,2-addition of the isothiourea catalyst to the ketone moiety of cyclobutenone 1a generates intermediate I (the conjugated structure is broken and an anionic oxy-substituted intermediate I is formed) that undergoes subsequent C–C bond cleavage to form intermediate II. The anionic oxy-substituent of I likely accelerates the electrocyclic ring opening process.8d The α-carbon of the vinyl enolate11,12 selectively reacts with azomethine imine 2a to form product 3a with excellent enantioselectivity and diastereoselectivity. The intermediate II likely adopts an s-cis diene configuration, and the cis-configuration of Cl and Ph substituents on the γ- and β-carbon of our product 3a suggests that intermediate II adopts an “outward” configuration.8 Previous studies from Houk and Baldwin have suggested that the ring-opening of cyclobutene led to an intermediate similar to II with an “outward” configuration.8 It is noteworthy that isothiourea as a Lewis base catalyst has not been exploited for the activation of cycloketones.We started by using cyclobutenone 1a and azomethine imine 2a as the model substrates (Table 1). We first examined cinchona alkaloid nucleophilic catalysts and found that no product was obtained with either A or B as the catalysts (entries 1 and 2).13 We next evaluated chiral guanidines, organocatalysts previously explored by Corey,14 Tan15 and others.16 We were delighted to find that the proposed product 3a could be formed in moderate yields (entries 3 and 4), although attempts to obtain enantioselectivity using guanidine catalysts were unsuccessful. Encouraged by these results, we subsequently studied isothioureas, analogs of guanidines, as the organic catalysts. Notably, pioneering studies in using isothioureas as enantioselective organic catalysts have been reported by Birman,17 Smith18 and others.19,20 Here, we found that when isothiourea E17 was used as the catalyst with CHCl3 as the solvent at room temperature, 3a could be obtained in 62% yield and with a promising 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 er (entry 5). The use of catalyst F17 led to 3a in similar yield with improved er (96:4) under otherwise identical conditions (entry 6). Finally, we found that the use of Et3N as an additive could slightly (and consistently) improve the enantioselectivity (97:3) and yield (67%) (entry 7). Decreasing the catalyst loading to 10 mol% gave the product 3a in lower yield (38%) without an apparent change in enantioselectivity (entry 7). Finally, we compared NHC catalysts that were used in our earlier [4 + 2] reactions (entries 8–10).9 These carbene catalysts could lead to products in low yields but with nearly no enantioselectivity.
:15 er (entry 5). The use of catalyst F17 led to 3a in similar yield with improved er (96:4) under otherwise identical conditions (entry 6). Finally, we found that the use of Et3N as an additive could slightly (and consistently) improve the enantioselectivity (97:3) and yield (67%) (entry 7). Decreasing the catalyst loading to 10 mol% gave the product 3a in lower yield (38%) without an apparent change in enantioselectivity (entry 7). Finally, we compared NHC catalysts that were used in our earlier [4 + 2] reactions (entries 8–10).9 These carbene catalysts could lead to products in low yields but with nearly no enantioselectivity.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Additive | 3a yieldc (%) | 3a er |
| a All reactions of 1a (0.10 mmol, 17.8 mg) with 2a (0.15 mmol, 38 mg) were carried out in the presence of catalyst (20 mol%; 20 mol% Cs2CO3 was added for G–I) in CHCl3 (1.0 mL) for 3 days. b Et3N (1.0 mmol, 14.0 μL) was added. c Isolated yield. d F (10 mol%) was used. e er of 3a was determined by chiral HPLC analysis. | ||||
| 1 | A | — | 0 | — |
| 2 | B | — | 0 | — |
| 3 | C | — | 48 | 50:50 |
| 4 | D | — | 38 | 50:50 |
| 5 | E | — | 62 | 85:15 |
| 6 | F | — | 65 | 96:4 |
| 7 | F | Et3Nb | 67 (38) |
97:3 |
| 8 | G | Cs2CO3 | 35 | 50:50 |
| 9 | H | Cs2CO3 | 40 | 52:46 |
| 10 | I | Cs2CO3 | 42 | 52:46 |

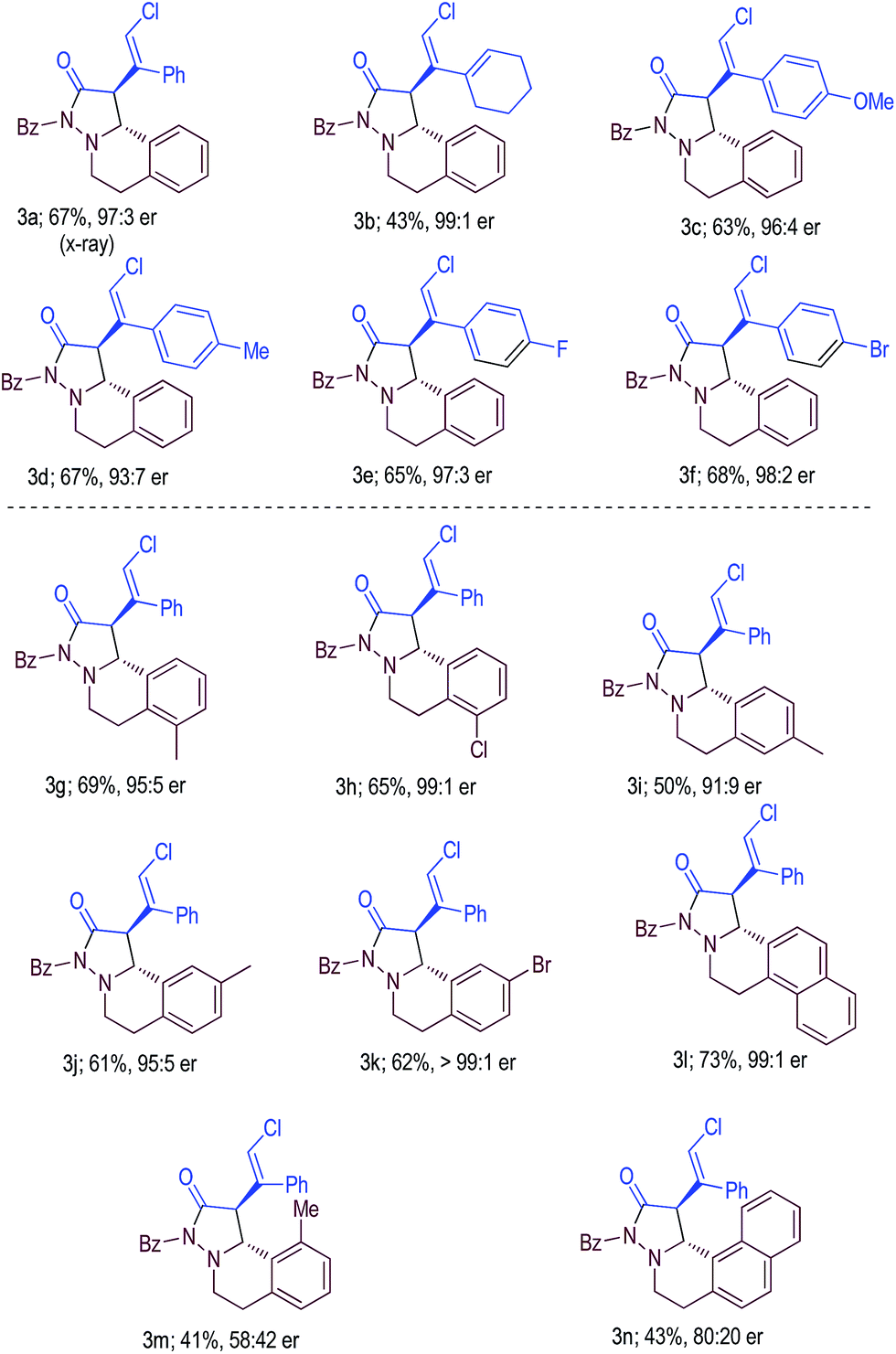
With acceptable conditions in hand, we next evaluated the scope of the asymmetric reaction by first varying the substituents at the β-carbon of cyclobutenone substrate 1 (Table 2, 3a–f). Replacing the Ph substituent in 1a with an alkyl unit could give the desired product 3b with excellent enantioselectivity, albeit with a lower yield. Electron-donating and withdrawing substituents on the Ph group of 1a were also well tolerated, giving products 3c–f in good yields and excellent enantioselectivities. Azomethine imine substrates were then examined (3g–n). It appeared that sterically hindered substrates (3m, 3n) led to lower yields and er values under the current reaction conditions. The absolute configurations of the products were confirmed via X-ray diffraction of product 3a.21
Notably, when the chlorine atom of substrate 1 was changed to a proton substituent or when γ,γ′-di-chloride substituted cyclobutenone was used, no cycloaddition products were obtained under our reaction conditions. In these cases, the cyclobutenone substrates remained unreacted. These results are constituent with Houk's computational findings that cyclobutenones bearing a mono-substituent at the C4 position are more reactive (less stable). When the chlorine atom of substrate 1 was changed to a methyl substituent, the methyl substituted substrate was unreactive under our catalytic conditions. It appears that with a methyl substituent the ketone moiety is not reactive enough (likely due to electronic reasons). This result is different from our early NHC-catalyzed reactions,9 likely because isothiourea organic catalysts are less nucleophilic than NHC catalysts.22
 | (1) |
 | (2) |
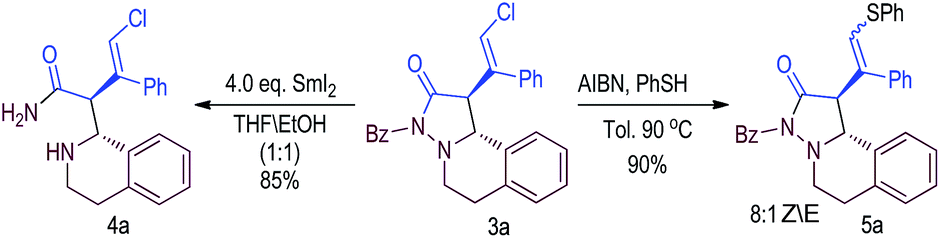
The catalytic reaction can be carried out on a gram scale without loss of yield and selectivity (eqn (1)).23 As a technical note, the catalyst (F) could be recovered (via SiO2 column chromatography) and reused without loss of reaction efficiency and selectivity. The catalytic product 3a from our reaction could readily undergo further transformations to give nitrogen-containing heterocyclic compounds (eqn (2)).24 For example, the N–N bond in 3a could be cleaved in the presence of SmI2 with ethanol as the solvent to give product 4a. The vinyl chloride unit in 3a is a widely used functional group in organic synthesis. Here, we show that the chloride atom in 3a can be substituted by a sulfa substituent to give product 5a.
Conclusions
In summary, we have developed a new C–C bond activation of cyclobutenones enabled by an isothiourea organocatalyst. The catalytically generated intermediate undergoes an α-carbon selective reaction with azomethine imines to afford nitrogen-containing heterocyclic compounds with excellent diastereo- and enantio-selectivities. Our approach offers new reaction modes that are not readily available with transition metal catalysis. It also expands the potential in using organocatalysts for C–C bond breaking and selective reactions.Acknowledgements
Generous financial support for this work is provided by the Singapore National Research Foundation, Ministry of Education, Nanyang Technological University (NTU), and China's Thousand Talent Plan, National Natural Science Foundation of China (No. 21132003; No. 21472028), and Guizhou University. We thank Dr Yongxin Li (NTU) for assistance with X-ray structure analysis.Notes and references
- For reviews on C–C bond activation: (a) R. H. Crabtree, Chem. Rev., 1985, 85, 245 CrossRef CAS; (b) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (c) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (d) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (e) D. Nečas and M. Kotora, Curr. Org. Chem., 2007, 11, 1566 CrossRef; (f) M. Tobisub and N. Chatani, Chem. Soc. Rev., 2008, 37, 300 RSC; (g) C. Aïssa, Synthesis, 2011, 21, 3389 CrossRef; (h) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed.
- Recent selected reviews: (a) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485 CrossRef CAS PubMed; (b) T. J. Snape, Chem. Soc. Rev., 2007, 36, 1823 RSC; (c) K. Prantz and J. Mulzer, Chem. Rev., 2010, 110, 3741 CrossRef CAS PubMed; (d) E. Leemans, M. D'hooghe and N. de Kimpe, Chem. Rev., 2011, 111, 3268 CrossRef CAS PubMed; (e) Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523 CrossRef CAS PubMed.
- For reactions involving ring-opening of cyclobutane alcohol: (a) T. Nishimura and S. Uemura, J. Am. Chem. Soc., 1999, 121, 11010 CrossRef CAS; (b) B. M. Trost and T. Yasukata, J. Am. Chem. Soc., 2001, 123, 7162 CrossRef CAS; (c) T. Seiser and N. Cramer, Angew. Chem., Int. Ed., 2008, 47, 9294 CrossRef CAS PubMed; (d) E. Zhang, C.-A. Fan, Y.-Q. Tu, F.-M. Zhang and Y.-L. Song, J. Am. Chem. Soc., 2009, 131, 14626 CrossRef CAS PubMed.
- (a) D. Bellus and B. Ernstn, Angew. Chem., Int. Ed., 1988, 27, 797 CrossRef PubMed; (b) T. Xu and G. Dong, Angew. Chem., Int. Ed., 2012, 51, 7567 CrossRef CAS PubMed; (c) P.-H. Chen, T. Xu and G. Dong, Angew. Chem., Int. Ed., 2014, 53, 1674 CrossRef CAS PubMed; (d) A. Cammers-Goodwi, J. Org. Chem., 1993, 58, 7619 CrossRef; (e) M. Murakami, Y. Miyamoto and Y. Ito, J. Am. Chem. Soc., 2001, 123, 6441 CrossRef CAS; (f) N. A. Magomedov, P. L. Ruggiero and Y. Tang, J. Am. Chem. Soc., 2004, 126, 1624 CrossRef CAS PubMed.
- (a) R. L. Danheiser and S. K. Gee, J. Org. Chem., 1984, 49, 1672 CrossRef CAS; (b) S. T. Perri, L. D. Foland, O. H. W. Decker and H. W. Moore, J. Org. Chem., 1986, 51, 3067 CrossRef CAS; (c) X. Y. Mak, A. L. Crombie and R. L. Danheiser, J. Org. Chem., 2011, 76, 1852 CrossRef CAS PubMed.
- For asymmetric C–C activation under metal-catalysis: (a) T. Matsuda, M. Shigeno and M. Murakami, J. Am. Chem. Soc., 2007, 129, 12086 CrossRef CAS PubMed; (b) C. Nájera and J. M. Sansano, Angew. Chem., Int. Ed., 2009, 48, 2452 CrossRef PubMed; (c) C. Winter and N. Krause, Angew. Chem., Int. Ed., 2009, 48, 2460 CrossRef CAS PubMed; (d) T. Seiser and N. Cramer, Org. Biomol. Chem., 2009, 7, 2835 RSC; (e) T. Xu, H. M. Ko, N. A. Savage and G. Dong, J. Am. Chem. Soc., 2012, 134, 20005 CrossRef CAS PubMed; (f) T. Seiser and N. Cramer, Chem.–Eur. J., 2010, 16, 3383 CrossRef CAS PubMed; (g) L. Souillart, E. Parker and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 3001 CrossRef CAS PubMed.
- H. M. Ko and G. Dong, Nat. Chem., 2014, 6, 789 CrossRef PubMed.
- (a) S. E. Niwayama, A. Kallel, C. Sheu and K. N. Houk, J. Org. Chem., 1996, 61, 2517 CrossRef CAS; (b) W. Kirmse, N. G. Rondan and K. N. Houk, J. Am. Chem. Soc., 1984, 106, 7989 CrossRef CAS; (c) J. E. Baldwin and M. C. McDanie, J. Am. Chem. Soc., 1968, 90, 6118 CrossRef CAS; (d) M. Murakami, Y. Miyamoto and Y. Ito, J. Synth. Org. Chem., Jpn., 2002, 60, 1049 CrossRef CAS.
- (a) B.-S. Li, Y. Wang, Z. Jin, P. Zheng, R. Ganguly and Y. R. Chi, Nat. Commun., 2015, 6, 6027 CrossRef PubMed.
- For related organocatalytic asymmetric [3 + 2] cycloaddition reactions: (a) T. Hashimoto, Y. Maeda, M. Omote, H. Nakatsu and K. Maruoka, J. Am. Chem. Soc., 2010, 132, 4076 CrossRef CAS PubMed; (b) T. Hashimoto, M. Omote and K. Maruoka, Angew. Chem., Int. Ed., 2011, 50, 3489 CrossRef CAS PubMed; (c) T. Hashimoto, M. Omote and K. Maruoka, Angew. Chem., Int. Ed., 2011, 50, 8952 CrossRef CAS PubMed; (d) L. Hesping, A. Biswas, C. G. Daniliuc, C. Mück-Lichtenfeld and A. Studer, Chem. Sci., 2015, 6, 1252 RSC.
- For γ-activation of NHC catalysis, see: (a) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617 CrossRef CAS PubMed; (b) J. Mo, X. Chen and Y. R. Chi, J. Am. Chem. Soc., 2012, 134, 8810 CrossRef CAS PubMed; (c) X. Chen, S. Yang, B.-A. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 11134 CrossRef CAS PubMed; (d) Y.-M. Zhao, M. S. Cheung, Z. Lin and J. Sun, Angew. Chem., Int. Ed., 2012, 51, 10359 CrossRef CAS PubMed; (e) X.-Y. Chen, F. Xia, J.-T. Cheng and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 10644 CrossRef CAS PubMed.
- For vinyl enolate generated from ammonium intermediate, see: (a) P. S. Tiseni and R. Peters, Angew. Chem., Int. Ed., 2007, 46, 5325 CrossRef CAS PubMed; (b) P. S. Tiseni and R. Peters, Org. Lett., 2008, 10, 2019 CrossRef CAS PubMed; (c) L. Shen, P. Shao and S. Ye, Adv. Synth. Catal., 2011, 353, 1943 CrossRef CAS PubMed; (d) L. Shen, L. Sun and S. Ye, J. Am. Chem. Soc., 2011, 133, 15894 CrossRef CAS PubMed; (e) L. C. Morrill, S. M. Smith, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1640 CrossRef CAS PubMed. For recent review of covalent bond catalysis, see: (f) M. C. Holland and R. Gilmour, Angew. Chem., Int. Ed, 2015, 54, 3862 CrossRef CAS PubMed.
- (a) M. A. Calter, J. Org. Chem., 1996, 61, 8006 CrossRef CAS; (b) S. Vellalath, K. N. van and D. Romo, Angew. Chem., Int. Ed., 2013, 52, 13688 CrossRef CAS PubMed.
- E. J. Corey and M. J. Grogan, Org. Lett., 1999, 1, 157 CrossRef CAS.
- (a) W. Ye, D. Leow, S. L. M. Goh, C.-T. Tan, C.-H. Chian and C.-H. Tan, Tetrahedron Lett., 2006, 47, 1007 CrossRef CAS PubMed; (b) J. Wang, J. Chen, C. W. Kee and C.-H. Tan, Angew. Chem., Int. Ed., 2012, 51, 2382 CrossRef CAS PubMed.
- (a) M. Terada, M. Nakano and H. Ube, J. Am. Chem. Soc., 2006, 128, 16044 CrossRef CAS PubMed; (b) M. Terada, M. Nakano and H. Ube, J. Am. Chem. Soc., 2007, 129, 14112 CrossRef CAS PubMed; (c) T. Misaki, K. Kawano and T. Sugimura, J. Am. Chem. Soc., 2011, 133, 5695 CrossRef CAS PubMed.
- V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351 CrossRef CAS PubMed.
- (a) C. Joannesse, C. P. Johnston, C. Concellón, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914 CrossRef CAS PubMed; (b) L. C. Morrill, J. Douglas, T. Lebl, A. M. Z. Slawin, D. J. Fox and A. D. Smith, Chem. Sci., 2013, 4, 4146 RSC; (c) E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193 RSC; (d) D. Belmessieri, L. C. Morrill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2710 CrossRef PubMed.
- (a) M. S. Kerr, J. Read de Alaniz and T. Rovis, J. Org. Chem., 2005, 70, 5725 CrossRef CAS PubMed; (b) M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418 CrossRef CAS PubMed; (c) H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2007, 129, 13796 CrossRef CAS PubMed; (d) H. U. Vora, J. R. Moncecchi, O. Epstein and T. Rovis, J. Org. Chem., 2008, 73, 9727 CrossRef CAS PubMed.
- (a) G. Liu, M. E. Shirley, K. N. van, R. L. McFarlin and D. Romo, Nat. Chem., 2013, 5, 1049 CrossRef CAS PubMed; (b) R. W. T. Clark, M. Deaton, Y. Zhang, M. Moore and S. L. Wiskur, Org. Lett., 2013, 15, 6132 CrossRef CAS PubMed.
- ESI†.
- (a) L. Candish, Y. Nakano and D. W. Lupton, Synthesis, 2014, 46, 1823 CrossRef; (b) B. Maji, C. Joannesse, T. A. Nigst, A. D. Smith and H. Mayr, J. Org. Chem., 2011, 76, 5104 CrossRef CAS PubMed; (c) B. Maji, M. Breugst and H. Mayr, Angew. Chem., Int. Ed., 2011, 50, 6915 CrossRef CAS PubMed; (d) H. Mayr, S. Lakhdar, B. Maji and A. R. Ofial, Beilstein J. Org. Chem., 2012, 8, 1458 CrossRef CAS PubMed.
- ESI†.
- (a) W. A. Gallimore and P. J. Scheuer, J. Nat. Prod., 2000, 63, 1422 CrossRef CAS PubMed; (b) K. E. Milligan, B. Márquez, R. T. Williamson, M. Davies-Coleman and W. H. Gerwick, J. Nat. Prod., 2000, 63, 965 CrossRef CAS PubMed; (c) S. Suntornchaswej, K. Suwanborirux, K. Koga and M. Isobe, Chem.–Asian J., 2007, 2, 114 CrossRef PubMed; (d) K. L. Malloy, T. L. Suyama, N. Engene, H. Debonsi, Z. Cao, T. Matainaho, C. Spadafora, T. F. Murray and W. H. Gerwick, J. Nat. Prod., 2012, 75, 60 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data and experimental procedures. CCDC 1041242. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01972a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |