
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On–off switch of charge-separated states of pyridine-vinylene-linked porphyrin–C60 conjugates detected by EPR†
Sabrina V.
Kirner‡
a,
Danny
Arteaga‡
b,
Christian
Henkel
a,
Johannes T.
Margraf
ac,
Nuria
Alegret
d,
Kei
Ohkubo
ef,
Braulio
Insuasty
b,
Alejandro
Ortiz
*b,
Nazario
Martín
g,
Luis
Echegoyen
*h,
Shunichi
Fukuzumi
*efi,
Timothy
Clark
 c and
Dirk M.
Guldi
*a
c and
Dirk M.
Guldi
*a
aDepartment of Chemistry and Pharmacy and Interdisciplinary Center for Molecular Materials, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstrasse 3, 91058 Erlangen, Germany
bDepartamento de Química, Facultad de Ciencias Naturales, Universidad del Valle, A.A. 25360 Cali, Colombia
cDepartment of Chemistry and Pharmacy, Computer Chemistry Center, Friedrich-Alexander-University Erlangen-Nürnberg, Nägelsbachstr. 25, 91052 Erlangen, Germany
dDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, 43007, Tarragona, Spain
eDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan
fDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Korea
gDepartamento de Química Orgánica, Facultad de Química, Universidad Complutense 28040, Madrid, Spain
hDepartment of Chemistry, University of Texas at El Paso, El Paso, Texas 79968-0519, USA
iFaculty of Science and Technology, Meijo University and ALCA and SENTAN, Japan Science and Technology Agency (JST), Tempaku, Nagoya, Aichi 468-8502, Japan
First published on 9th July 2015
Abstract
The design, synthesis, and electronic properties of a new series of D–π–A conjugates consisting of free base (H2P) and zinc porphyrins (ZnP) as electron donors and a fullerene (C60) as electron acceptor, in which the two electroactive entities are covalently linked through pyridine-vinylene spacers of different lengths, are described. Electronic interactions in the ground state were characterized by electrochemical and absorption measurements, which were further supported with theoretical calculations. Most importantly, charge-transfer bands were observed in the absorption spectra, indicating a strong push–pull behavior. In the excited states, electronic interactions were detected by selective photoexcitation under steady-state conditions, by time-resolved fluorescence investigations, and by pump probe experiments on the femto-, pico-, and nanosecond time scales. Porphyrin fluorescence is quenched for the different D–π–A conjugates, from which we conclude that the deactivation mechanisms of the excited singlet states are based on photoinduced energy- and/or electron transfer processes between H2P/ZnP and C60, mediated through the molecular spacers. The fluorescence intensity decreases and the fluorescence lifetimes shorten as the spacer length decreases and as the spacer substitution changes. With the help of transient absorption spectroscopy, the formation of charge-separated states involving oxidized H2P/ZnP and reduced C60 was confirmed. Lifetimes of the corresponding charge-separated states, which ranged from ∼400 picoseconds to 165 nanoseconds, depend on the spacer length, the spacer substitution, and the solvent polarity. Interestingly, D–π–A conjugates containing the longest linkers did not necessarily exhibit the longest charge-separated state lifetimes. The distances between the electron donors and the acceptors were calculated by molecular modelling. The longest charge-separated state lifetime corresponded to the D–π–A conjugate with the longest electron donor–acceptor distance. Likewise, EPR measurements in frozen media revealed charge separated states in all the D–π–A conjugates investigated. A sharp peak with g values ∼2.000 was assigned to reduced C60, while a broader, less intense signal (g ∼ 2.003) was assigned to oxidized H2P/ZnP. On–off switching of the formation and decay of the charge-separated states was detected by EPR at 77 K by repeatedly turning the irradiation source on and off.
Introduction
In Nature, photosynthesis is by far the best method to convert solar energy into chemical energy. It involves complex processes based on intramolecular electron/energy transfer reactions between molecular components within photoactive membranes.1–5 In recent decades, photosynthesis has served as an inspiration to design and synthesize new artificial photosynthetic arrays that mimic the function of plants.6 In this context, organic chemists have prepared many artificial photosynthetic systems that have enabled the study of the fundamental chemistry and the reaction mechanisms involved in the biological processes that are responsible for solar energy conversion in nature.7–10The synthesis of molecular architectures consisting of electron donors and acceptors, covalently linked by π-conjugated molecular spacers (D–π–A) is one of the strategies for probing photoinduced electron transfer processes on a molecular level.8,11 The electronic properties of these molecules make them potentially useful in molecular photonics, optoelectronics, nanoscale applications, and in solar energy conversion.12–18 Porphyrins represent an important class of molecular building blocks, which in biological architectures are responsible for oxygen-electron transport, light-to-energy conversion, etc.19 They have been used frequently as electron donors in D–π–A conjugates, mainly because of their ease of synthesis, versatile electrochemical and photochemical properties, and their presence in the naturally occurring chlorophyll.20–22 As a complement, C60 is an excellent electron acceptor because it features low-energy triply degenerate LUMOs and is able to accept up to six electrons.2,23–27 C60 exhibits low reorganization energies upon electron transfer processes, which is essential to obtain ultrafast charge separation and slow charge recombination.28–31 Thus, a wide variety of porphyrin arrays – H2P or ZnP – have been covalently linked to C60 in many different ways and using various wire-like molecular spacers.32–36
It has been observed that the linker in D–π–A conjugates has a profound effect on the rates of the intramolecular photoinduced charge-transfer37 and on the mechanism by which the charge transfer occurs,38,39 which can be via “superexchange”-mediated coherent tunnelling between the electronic states of the D/A pair or via a “hopping” mechanism through localized electronic states on the linker.40
Both charge separation and recombination occur and are defined by the electron-transfer rate constant kET as Ae−βRD–A, where A is the Arrhenius constant, β the damping factor, and RD–A the distance between the electron donors and acceptors. A decreased damping factor therefore means an increase of the distance over which charges can be efficiently transported.41–44β depends primarily on the length of the wire-like molecular spacer, conformational rigidity, and the electronic properties of the electron donors and acceptors.45–48
Several groups have studied π-conjugated oligomers as wire-like molecular spacers connecting the photoactive termini in H2P/C60 and ZnP/C60 conjugates. π-extended spacers such as para-phenylene vinylene (oPPV),49 [2,20]paracyclophaneoligophenylene-vinylene (pCp-oPPV),50–52 oligothiophene (nOT),53 and oligothienylenevinylene (nTV)54,55 are ideal connectors for effective charge transfer from the electron donors to the acceptors with maximum rates and relatively small damping factors.49–55
π-deficient molecular linkers, such as pyridine-vinylene groups have not been investigated so far, so we now report the synthesis and electronic/photophysical properties of a new homologous series of H2P/C60 and ZnP/C60 electron donor–acceptor conjugates bridged covalently by various pyridine-vinylene linkers 15–20a (H2P-n-mC60, ZnP-n-mC60, n = 1–3) and 15–20b (H2P-n-pC60, ZnP-n-pC60, n = 1–3) in addition to their precursors 9–14a (H2P-n-mCHO, ZnP-n-mCHO, n = 1–3) and 9–14b (H2P-n-pCHO, ZnP-n-pCHO, n = 1–3). We have used pyridine-vinylene spacers of different lengths and substitution patterns to conduct a systematic evaluation of the influence of both the length and nature of the linker on intramolecular charge-transfer processes that yield long- and short-lived charge-separated states in solvents of different polarities (Chart 1).
 | ||
| Chart 1 Above: Porphyrin–fullerene conjugates linked by pyridine-vinylene units at meta- and para-positions 15–20a (H2P-n-mC60, ZnP-n-mC60, n = 1–3) and 15–20b (H2P-n-pC60, and ZnP-n-pC60, n = 1–3). Below: Corresponding porphyrin–pyridine-vinylene references 9–14a (H2P-n-mCHO, ZnP-n-mCHO, n = 1–3) and 9–14b (H2P-n-pCHO, ZnP-n-pCHO, n = 1–3). | ||
Results
Synthesis
New photoactive conjugates were prepared using different reactions: cross-coupling reactions, such as the Stille and Heck, condensation reactions, such as the Knoevenagel and Wadsworth–Horner–Emmons, and the 1,3-dipolar cycloaddition reaction. All reactions were conducted under argon and Schlenk conditions. The solvents were first dried by standard procedures such as sodium/benzophenone or CaH2 and freshly distilled before use. Pyridine-vinylene spacers of different lengths 2–6a and 2–6b were synthesized by successive Stille and Heck cross-coupling reactions starting with 6-bromo-2-pyridine-carboxaldehyde 1a, 6-bromo-3-pyridinecarboxaldehyde 1b and 2,6-dibromopyridine 3 as the main building blocks. The synthetic procedures are shown in Scheme 1. Monomers 2a and 2b were prepared using Stille reactions starting from the corresponding bromo-pyridinecarboxaldehydes 1a or 1b and tributyl(vinyl)tin using Pd(PPh3)4 as catalyst and anhydrous toluene as solvent. These monomers were treated with 2,6-dibromopyridine 3 under palladium-catalyzed Heck coupling conditions to give intermediates 4a and 4b, which were subsequently converted to the desired dimers 5a and 5bvia Stille cross-coupling reactions in anhydrous toluene using Pd(PPh3)4 as catalyst. Finally, 6a and 6b were synthesized starting from the previously obtained monomers 2a,b by a Pd(OAc)2 catalyzed double Heck reaction with 2,6-dibromopyridine 3 in dimethylformamide (DMF). For these reactions it was necessary to use an excess of the monomers (2.5 eq.). | ||
| Scheme 1 Synthetic route for the preparation of pyridine-vinylene linkers: top 2–6a and bottom 2–6b. Reagents and conditions: (a) Pd(PPh3)4, tributyl(vinyl)tin, toluene, reflux, 20–22 h, yields 82–86%. (b) 2,6-Dibromopyridine 3, Pd(OAc)2, Bu4NBr, K2CO3, DMF, reflux, 24 h, yields 65–68%. (c) Pd(PPh3)4, tributyl(vinyl)tin, toluene, reflux, 20 h yields 75–80% (d) 2,6-dibromopyridine 3, Pd(OAc)2, Bu4NBr, K2CO3, DMF, reflux, 24 h, yields 62–70%. | ||
The synthetic routes for the preparation of 15–20a and 15–20b involved consecutive multistep procedures, as shown in Scheme 2. In particular, intermediates 9–10a,b (H2P-n-mCHO, H2P-n-pCHO, n = 1–2) and 12–13a,b (ZnP-n-mCHO, ZnP-n-pCHO, n = 1–2) were obtained by coupling of porphyrin precursors 7a,b and pyridine-vinylene linkers 2a,b and 5a,b using DMF as solvent and Pd(OAc)2 as catalyst (see ESI, Scheme S1†). The porphyrin precursors 7a,b and 8a,b were prepared according to previous reports.56–59 The intermediates 11a,b (H2P-3-mCHO, H2P-3-pCHO) and 14a,b (ZnP-3-mCHO, ZnP-3-pCHO) were synthesized using Wadsworth–Horner–Emmons reactions between pyridine-vinylene linkers 6a,b and 8a,b using tetrahydrofuran as solvent (see ESI, Scheme S2†). Finally, the 1,3-dipolar cycloaddition reaction60,61 of azomethine ylides generated in situ in refluxing anhydrous toluene from 9–14a (H2P-n-mCHO, ZnP-n-mCHO, n = 1–3), 9–14b (H2P-n-pCHO, ZnP-n-pCHO), and N-octylglycine to C60 afforded the desired compounds 15–20a (H2P-n-mC60, ZnP-n-mC60, n = 1–3) and 15–20b (H2P-n-pC60, ZnP-n-pC60, n = 1–3) in 30–42% yields (see the ESI† for details).
 | ||
| Scheme 2 Synthesis of new porphyrin–fullerene conjugates: top 15–20a (H2P-n-mC60, ZnP-n-mC60, n = 1-3); bottom 15–20b (H2P-n-pC60, ZnP-n-pC60, n = 1–3). Reagents and conditions: (a) C60, N-octylglycine, toluene, reflux, 4–5 h, yields (15a, 32%); (16a, 35%); (17a, 30%); (18a, 38%); (19a, 34%); (20a, 36%); (b) C60, N-octylglycine, toluene, reflux, 4–5 h, yields (15b, 31%); (16b, 38%); (17b, 37%); (18b, 34%); (19b, 45%); (20b, 42%). | ||
The structures of all the new conjugates were confirmed unambiguously by analytical measurements and spectroscopic techniques including 1H-NMR, 13C-NMR, FT-IR, and MALDI-TOF mass spectrometry. The 1H NMR spectra of 15–20a (H2P-n-mC60, ZnP-n-mC60, n = 1–3) and 15–20b (H2P-n-pC60, ZnP-n-pC60, n = 1–3) exhibit the characteristic 1H-NMR pattern for 2-substituted pyrrolidines with aromatic proton signals due to the porphyrins and pyridine-vinylene spacers. The 13C-NMR spectral data are also in good agreement with the formulated structures. The presence of all the structures was corroborated by MALDI-TOF mass spectrometry, which showed the molecular-ion peak [M + H]+ and the fragment after the loss of C60 [M − C60]+ (see ESI†).
Ground-state interactions
 | ||
| Fig. 1 Left: CVs for porphyrin–fullerene conjugates 15–17a (H2P-n-mC60, n = 1–3) and 15–17b (H2P-n-mC60, n = 1–3) in DCM solutions (0.1 M TBAPF6) at room temperature. Right: CVs for porphyrin–fullerene conjugates 18–20a (ZnP-n-pC60, n = 1–3) and 18–20b (ZnP-n-pC60, n = 1–3) in DCM solutions (0.1 M TBAPF6) at room temperature. Oxidative scans between 0 and 1.25 V; reductive scans between 0 and −2.10 V. | ||
Fig. 1 shows the cyclic voltammograms of the porphyrin–fullerene conjugates 15–20a (H2P-n-mC60, ZnP-n-mC60, n = 1–3) and 15–20b (H2P-n-pC60, ZnP-n-pC60, n = 1–3). In the reductive scan, each conjugate shows the reduction profile of four or five one-electron reversible reduction waves, respectively, corresponding to C60 and porphyrin centred processes. The first, second, and fourth reductions can be assigned to the fulleropyrrolidine-centred processes by comparison with C60. The third and fifth reduction waves are centred on the porphyrin subunit with Zn 18–20a (ZnP-n-mC60, n = 1–3), 18–20b (ZnP-n-pC60, n = 1–3) and without Zn 15–17a (H2P-n-mC60, n = 1–3), 15–17b (H2P-n-pC60, n = 1–3). The reduction potentials for the new electroactive conjugates are cathodically shifted relative to the values for pristine C60, as expected for a monofunctionalized C60.28,62
The oxidative scans show the first and second one-electron reversible oxidation waves corresponding to H2P and ZnP centred processes. H2P derivatives 15–17a (H2P-n-mC60, n = 1–3), 15–17b (H2P-n-pC60, n = 1–3) exhibit one oxidation wave, while the ZnP derivatives 18–20a (ZnP-n-mC60, n = 1–3) and 18–20b (ZnP-n-pC60, n = 1–3) feature two oxidation waves at potentials similar to those observed for the reference tetraphenylporphyrin (TPP).50,52
The electron donor ability of H2P and ZnP within electroactive systems was confirmed by the remarkably low value of their first oxidation potential, between +0.27 and +0.48 V, similar to those found for other compounds. For the reduction processes (shown in Table S1†) the first reduction potential is shifted cathodically by 110–148 mV compared to the first reduction potential of C60, which shows the strong push–pull nature of the electroactive species, allowing the prediction of the formation of a charge-separated state by photoexcitation. The reduction potentials of each series show that the shifts for the different molecular linker lengths are not significant. However, the isomeric 1,3 or 1,4-disubstituted systems (meta or para), do exhibit differences of ∼20 to 27 mV in the cathodic shifts. The 1,4-disubstituted systems 15–20b (H2P-n-pC60, ZnP-n-pC60) display first reduction potentials that are less negative than their 1,3-disubstituted counterparts 15–20a (H2P-n-mC60, ZnP-n-mC60).
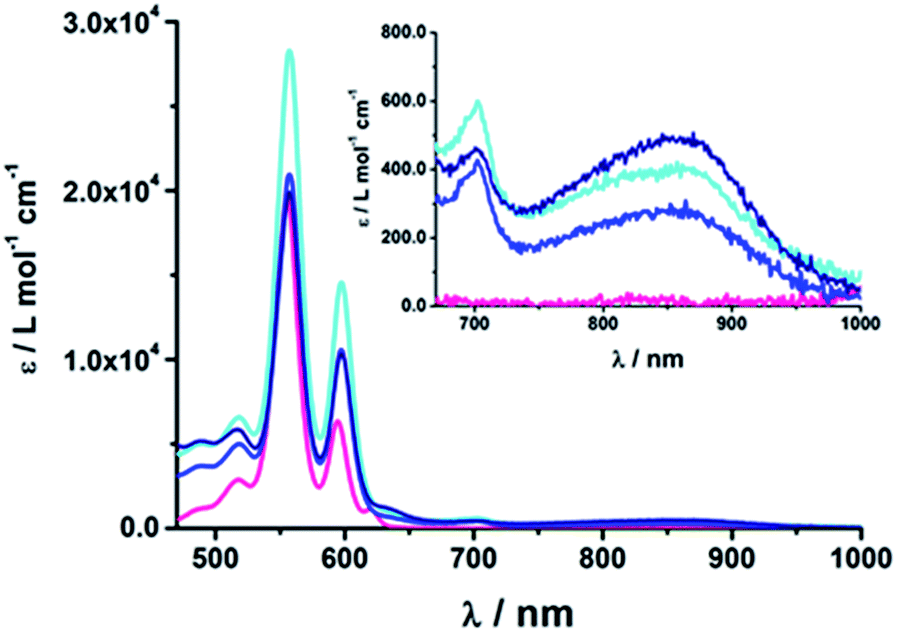 | ||
| Fig. 2 Absorption spectra of (18b) ZnP-1-pC60 (cyan); (19b) ZnP-2-pC60 (blue); (20b) ZnP-3-pC60 (navy) and ZnTPP (pink) as the reference in THF at room temperature. | ||
At first glance, comparing the absorptions of the meta and para linked pyridine–C60 conjugates revealed no particular differences. The more pyridine-vinylene groups are present in the linker, the higher the extinction coefficient between 300 and 400 nm becomes. Thus, the increased absorption in this wavelength range is attributed to the linker.
Excited-state interactions
To gain further insights into the excited-state interactions, both steady state and time-resolved emission spectroscopy (time correlated single photon counting, TCSPC) and time-resolved absorption spectroscopy (transient absorption) were employed. | ||
| Fig. 3 Above: Fluorescence spectra of (15a) H2P-1-mC60 (cyan); (16a) H2P-2-mC60 (blue); (17a) H2P-3-mC60 (navy) and H2TPP (pink) (excitation at 420 nm; OD = 0.04) in THF at room temperature. Below: Fluorescence spectra of (18a) ZnP-1-mC60 (cyan); (19a) ZnP-2-mC60 (blue); (20a) ZnP-3-mC60 (navy) and ZnTPP (pink) (excitation at 420 nm; OD = 0.05) in THF at room temperature. | ||
| Compound | λ max (nm) | Φ F | τ (ns) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Tol | THF | PhCN | Tol | THF | PhCN | Tol | THF | PhCN | |
| H2TPP | 650 | 649 | 651 | 0.10 | 10.0 | ||||
| ZnTPP | 605 | 601 | 607 | 0.040 | 2.0 | ||||
| 15a | 653 | 652 | 655 | 0.008 | 0.003 | 0.002 | 0.5 | 0.22 | 0.16 |
| 15b | 653 | 652 | 655 | 0.014 | 0.007 | 0.005 | 0.8 | 0.27 | 0.26 |
| 16a | 653 | 652 | 655 | 0.036 | 0.029 | 0.026 | 2.3 | 1.9 | 1.7 |
| 16b | 653 | 652 | 655 | 0.061 | 0.051 | 0.047 | 3.4 | 3.0 | 3.0 |
| 17a | 653 | 652 | 655 | 0.064 | 0.048 | 0.040 | 5.6 | 5.8 | 6.0 |
| 17b | 653 | 652 | 655 | 0.112 | 0.099 | 0.095 | 7.3 | 7.1 | 7.1 |
| 18a | 602 | 601 | 610 | 2 × 10−4 | 4.6 × 10−4 | 1 × 10−4 | <0.15 | <0.15 | <0.15 |
| 18b | 608 | 604 | 610 | 2.6 × 10−4 | 3.7 × 10−4 | 4.2 × 10−4 | <0.15 | <0.15 | <0.15 |
| 19a | 610 | 604 | 610 | 0.009 | 0.008 | 0.005 | 0.4 | 0.3 | 0.8 |
| 19b | 611 | 604 | 611 | 0.014 | 0.014 | 0.012 | 0.6 | 0.5 | 0.5 |
| 20a | 610 | 604 | 610 | 0.025 | 0.006 | 0.013 | 0.8 | 0.3 | 0.7 |
| 20b | 610 | 604 | 612 | 0.038 | 0.043 | 0.034 | 1.2 | 1.2 | 1.2 |
Upon excitation at 387 and 420 nm, the differential absorption spectra of 9–11a (H2P-n-mCHO, n = 1–3) and 9–11b (H2P-n-pCHO, n = 1–3) are dominated by features of the H2P singlet excited state,71,72 as shown in Fig. 4, left. This state is formed immediately upon excitation and exhibits maxima at 450, 540, 575, 630, and 670 nm in addition to a broad transient absorption between 1000 and 1150 nm. Additionally observed ground-state bleaching at 420, 520, 550, 590, and 650 nm correspond to the Soret- and Q-band absorptions of H2P, respectively. The porphyrin's singlet excited state is stable during the time scale of our fs transient-absorption setup (7.5 ns, see Fig. 4). The differential absorption spectra of all H2P based references look nearly identical and also resemble the transient absorption spectra of H2TTP closely. Thus, it can be assumed that the pyridine-vinylene linker does not show transients upon photoirradiation. The ZnP-pyridine-vinylene reference compounds give similar results, as shown in Fig. S5.† As for ZnTPP, upon 420 nm excitation the 1*ZnP70,72 state dominates the visible region of the differential absorption spectrum with maxima at 460, 580, and 630 nm and minima at 560 and 600 nm (ground-state bleaching). The singlet excited porphyrin then undergoes intersystem crossing within 2 ns to give the 3*ZnP, which is stable within the 7.5 ns time scale of the system and shows maxima at 480 and 840 nm.
 | ||
| Fig. 4 Above: Differential absorption spectra (visible and near-infrared) observed upon femtosecond flash photolysis (420 nm, 150 nJ) of 9a (H2P-1-mCHO) in THF with time delays between 0 ps (black) and 7.5 ns (wine) at room temperature. Below: Time-absorption profile of the spectra above at 1070 nm, monitoring the deactivation of the porphyrin singlet excited state. | ||
When exciting the H2P–C60 electron donor–acceptor conjugates with a fs-laser pulse at 387 and 420 nm, respectively, the most prominent features of the transient absorption spectra are in the visible region, as seen for the references, those belonging to the H2P singlet excited state, (maxima at ∼450, 540, 575, 630, and 670 nm) and ground-state bleaching at ∼420, 520, 550, 590, and 650 nm; a representative example is shown Fig. 5 and S6 in the ESI.† The features of the 1*H2P, with a broad maximum between 1000 and 1150 nm, can be observed in the NIR region. However, in contrast to the references, the singlet excited state of the porphyrin is shorter lived in the presence of C60. 15a (H2P-1-mC60) and 15b (H2P-1-pC60) exhibit the shortest singlet state lifetimes (hundreds of picoseconds in THF). With increasing length of the linker, the decay of the singlet excited state of the porphyrin becomes slower. In 17b (H2P-3-pC60) the 1*H2P lifetime even exceeds the time scale of our fs setup, as observed for the reference systems that lack C60 (compare Fig. S6†).
 | ||
| Fig. 5 Above: Differential absorption spectra (visible and near-infrared) observed upon femtosecond flash photolysis (387 nm, 200 nJ) of 16a (H2P-2-mC60) in THF with time delays between 0 ps (black) and 7.5 ns (wine) at room temperature. Below: Time-absorption profiles of the 1010 nm decay for (15a) H2P-1-mC60 (cyan); (16a) H2P-2-mC60 (blue) and (17a) H2P-3-mC60 (navy) upon femtosecond flash photolysis (387 nm, 200 nJ) in THF at room temperature, monitoring the charge recombination. | ||
These results also confirm those from the steady-state and time-resolved emission experiments. Furthermore, additional features corresponding to the C60 singlet excited state are observed upon 387 nm excitation, as a 920 nm absorption maximum. The transient characteristics, which correlate with the triplet excited state of C60 at 690 nm are, however, masked by the more intense H2P transients and thus barely visible. Finally, a distinct peak arises in the NIR region of the differential absorption spectra of 15a (H2P-1-mC60) and 15b (H2P-1-pC60) (Fig. S6†) with a maximum at ∼1010 nm. This feature is assigned to the singly reduced fullerene's fingerprint, which is well known from the literature.24,73 Although this signal coincides with the singlet features of H2P, it can be clearly distinguished, since they exhibit comparably short-lived singlets. For 16a (H2P-2-mC60) (Fig. 5), 17a (H2P-3-mC60) and 16b (H2P-2-pC60) (Fig. S6†) the fullerene anion fingerprint cannot be clearly identified in the differential absorption spectra because of the increased signal of the 1*H2P. However, upon closer examination of the NIR region (Fig. 5, above, and Fig. S6†) an individual peak can be discerned at ∼1010 nm. In contrast, for 17b (H2P-3-pC60), the singly-reduced C60 cannot be identified unambiguously, since it is masked by the rather long lived 1*H2P. Therefore, we cannot rule out the formation of a CSS for the latter.
The presence of the C60 anion signature in the transient absorption spectra proves that charge transfer takes place in the conjugates. The associated radical cation transient absorption again overlaps with the porphyrin signatures. However, it can be probed by analyzing the decay kinetics. Even though the triads show the same transients initially, the transient absorption spectra show considerably different lifetimes of their excited states. As discussed above, the singlet excited-state lifetimes increase with the length of the linker. Furthermore, the latter also decrease with solvent polarity, as shown in Table 1. Not only the lifetimes of the singlet excited state vary with the length of the linker and with the solvent polarity; those of the radical ion pair state also do. From a multi-wavelength analysis of the decays of the C60 radical anion and of the H2P radical cation, a lifetime of 1.4 ns was determined for 15a (H2P-1-mC60) in THF. The lifetime of the radical ion pair state changed on varying the solvent. In toluene, for example, a lifetime of 2.0 ns was found for 15a (H2P-1-mC60), while in PhCN the lifetime was only 662 ps, as shown in Table S2 of the ESI.† Even more distinct differences in the radical ion pair lifetime are observed, when different linker lengths are considered, as shown in Fig. 5 (below) for kinetic measurements. Ongoing from one pyridine-vinylene group in 15a (H2P-1-mC60) to two in 16a (H2P-2-mC60), the lifetime increases in THF. Because the 1010 nm decay is not complete for 16a (H2P-2-mC60) within the timescale of 7.5 ns, we turned to EOS fs-measurements and nanosecond transient absorption spectroscopy. Upon excitation of 16a (H2P-2-mC60) with ns laser pulses at either 355 or 420 nm under different conditions, the features of the H2P radical anion are discernable, but are superimposed with those of the C60 triplet excited state. The fingerprint of the C60 radical anion is clearly visible in the near-infrared region (Fig. 6, below). Its decay at 1010 nm, for example, (Fig. 7) fits by a single exponential function to afford a radical ion pair state lifetime of 35 ns for 16a (H2P-2-mC60) in THF. Interestingly, the radical ion pair lifetime in 17a (H2P-3-mC60) is 2.2 ns in THF, appreciably shorter than for 16a (H2P-2-mC60) (Table 2). This behaviour will be discussed further later.74
 | ||
| Fig. 6 Above: Differential absorption spectra (visible) observed upon nanosecond flash photolysis (355 nm, 10 mJ) of 16a (H2P-2-mC60) in THF with time delays between 150 ns (purple) and 2.0 μs (wine) at room temperature under aerobic conditions. Below: Differential absorption spectra (near-infrared) observed upon nanosecond flash photolysis (355 nm, 10 mJ) of 16a (H2P-2-mC60) in THF with time delays between 60 ns (purple) and 2.0 μs (wine) at room temperature under aerobic conditions. | ||
 | ||
| Fig. 7 Above: Time-absorption profile of the 1010 nm decay – Fig. 6 – of 16a (H2P-2-mC60) upon nanosecond flash photolysis (355 nm, 10 mJ) in THF under aerobic conditions, monitoring the charge recombination process. Below: Time-absorption profile of the 1010 nm decay of 16b (H2P-2-pC60) upon nanosecond flash photolysis (355 nm, 10 mJ) in THF under aerobic conditions, monitoring the charge recombination. | ||
| CR | |||
|---|---|---|---|
| CS | fs-setup | ns-setup | |
| 15a | 75 ± 3 ps | 1.4 ± 0.1 ns | |
| 15b | 106 ± 2 ps | 1.3 ± 0.1 ns | |
| 16a | <1 ps | 50 ± 3 ns | 35 ± 9 ns |
| 16b | <1 ps | 58 ± 1 ns | 65 ± 21 ns |
| 17a | <1 ps | 2.2 ± 0.1 ns | |
| 18a | 10 ± 0 ps | 414 ± 2 ps | |
| 18b | <1 ps | 373 ± 1 ps | |
| 19a | <1 ps | 116 ± 19 ns | 98 ± 24 ns |
| 19b | <1 ps | 79 ± 10 ns | 165 ± 41 ns |
| 20a | <1 ps | 1.6 ± 0.1 ns | |
| 20b | <1 ps | 1.4 ± 0.1 ns | |
When probing 16a (H2P-2-mC60) in THF with the EOS fs-setup (Fig. 8, above), the visible region is again dominated by the porphyrin's triplet excited-state features, while the C60 radical anion 1010 nm fingerprint is discernible in the near infrared region. The 1010 nm decay (Fig. 8, below) is best fit by a biexponential function, affording a short-lived component of 4.3 ns attributable to an H2P-centered singlet excited state and a longer lived one of 50 ns assigned to a C60˙− centered charge separation. By comparison with the ns transient absorption measurements, where the laser power is four orders of magnitude higher (1 μJ compared to 10 mJ), we conclude that charge recombination is independent of the applied laser power.
 | ||
| Fig. 8 Above: Differential absorption spectra (visible and near-infrared) observed upon femtosecond flash photolysis (387 nm, 1 μJ) of 16a (H2P-2-mC60) in THF with time delays between 0 ns (black) and 1.0 μs (wine) at room temperature under aerobic conditions. Below: Time-absorption profile of the spectra above at 1010 nm, monitoring the charge recombination. | ||
The same trend is observed for the radical ion pair state lifetimes for the para substituted conjugates, as shown in Table 2. While for 15b (H2P-1-pC60) the radical ion pair state decays with a lifetime of 1.3 ns in THF, that of 16b (H2P-2-pC60) does not decay within the time scale of our femtosecond setup. A lifetime of 65 ± 21 ns was determined for 16b (H2P-2-pC60) in THF in complementary nanosecond experiments, as shown in Fig. 7. It was reassuring that EOS fs-measurements yielded a lifetime of 58 ns (Table 2).75 The formation of the charge-separated states was investigated in order to obtain further insight into the charge-transfer dynamics of the electron donor–acceptor conjugates. The time needed to form the charge separated state upon 387 nm excitation was determined from the rise of the signal corresponding to the fullerene anion, (Table 2). Clear trends can be observed for 15a (H2P-1-mC60) and 15b (H2P-1-pC60). The more polar the solvent, the faster the charge separation process occurs. Furthermore, the charge-separated state is formed more rapidly in 15a (H2P-1-mC60) than for 15b (H2P-1-pC60). For the systems with longer linkers, the electron is transferred to the fullerene in less than 1 ps, so that no further conclusions can be drawn from these results.
The C60 radical anion absorption at 1010 nm can be identified even more clearly for the ZnP–C60 D–A conjugates, since ZnP does not have transients in this region of the spectrum, as shown in Fig. 9 and S7 of the ESI.† Nevertheless, the visible region is again dominated by porphyrin features. To be more precise, maxima at 460, 580, and 620 nm and minima at 420, 560, and 600 nm evolve immediately after the 387 nm laser pulse. These correspond to the ZnP singlet excited state and ground state bleaching, respectively. While for 18a (ZnP-1-mC60) 1*ZnP decays within 400 ps and only weak triplet signatures are discernible, the singlet lifetimes and the intensity of the 3*ZnP peaks (850 nm) increase with increasing length of the linker up to ∼1 ns for 20b (ZnP-3-pC60). Additionally, at 920 nm a transient arises that can be assigned to 1*C60. The same trend as found for the H2P systems was observed for the charge-separated state lifetimes. The shortest CSS lifetime of the ZnP compounds was found for 18b (ZnP-1-pC60) with ∼150 ps in PhCN, while 19a (ZnP-2-mC60) and 19b (ZnP-2-pC60) in THF and toluene do not decay within the 7.5 ns time scale of our fs-setup (Fig. 9, below).
 | ||
| Fig. 9 Above: Differential absorption spectra (visible and near-infrared) observed upon femtosecond flash photolysis (387 nm, 200 nJ) of 19a (ZnP-2-mC60) in THF with time delays between 0 ps (black) and 7.5 ns (wine) at room temperature. Below: Time-absorption profiles of the 1010 nm decay for (18a) ZnP-1-mC60 (cyan); (19a) ZnP-2-mC60 (blue) and (20a) ZnP-3-mC60 (navy) upon femtosecond flash photolysis (387 nm, 200 nJ) in THF at room temperature, monitoring the charge recombination. | ||
Lifetimes of 98 ns for 19a (ZnP-2-mC60) and 165 ns for 19b (ZnP-2-pC60) in THF were determined in complementary ns experiments. In EOS experiments, however, slightly different lifetimes, 116 ns for 19a (ZnP-2-mC60) and 79 ns for 19b (ZnP-2-pC60), were determined. As described for the H2P-conjugates, the CSS lifetime decreases with increasing solvent polarity and the longest lifetimes were determined for the compounds with two pyridine-vinylene groups as linkers rather than of those with the longer linker. All CSS lifetimes determined from multi-wavelength fits either from fs or ns transient absorption experiments are summarized in Tables 2 and S2.†
Analysis of the charge-separation kinetics of the ZnP D–π–A conjugates did not yield a clear trend. For 18a (ZnP-1-mC60) in THF and 18b (ZnP-1-pC60) in toluene, CS takes place within ∼10 ps, while the CSS is formed within 2 ps for both in PhCN. CS is too fast to be monitored with our setup in all other ZnP compounds.
 | ||
| Fig. 10 EPR signals observed under photoirradiation of 15a (H2P-1-mC60) in benzonitrile at 77 K. | ||
Repeated on–off switching of the charge-separated state formation was realized by turning on and off the irradiation source many times, see Fig. 11. The corresponding lifetimes at 77 K are long enough to be detected during the on–off cycling for both series of porphyrin–C60 conjugates (Fig. S13 and S14 in the ESI†). Table 3 lists the CS lifetimes determined from the EPR experiments at 77 K. Due to the time resolution of the available equipment, only lifetimes >200 ms could be detected.
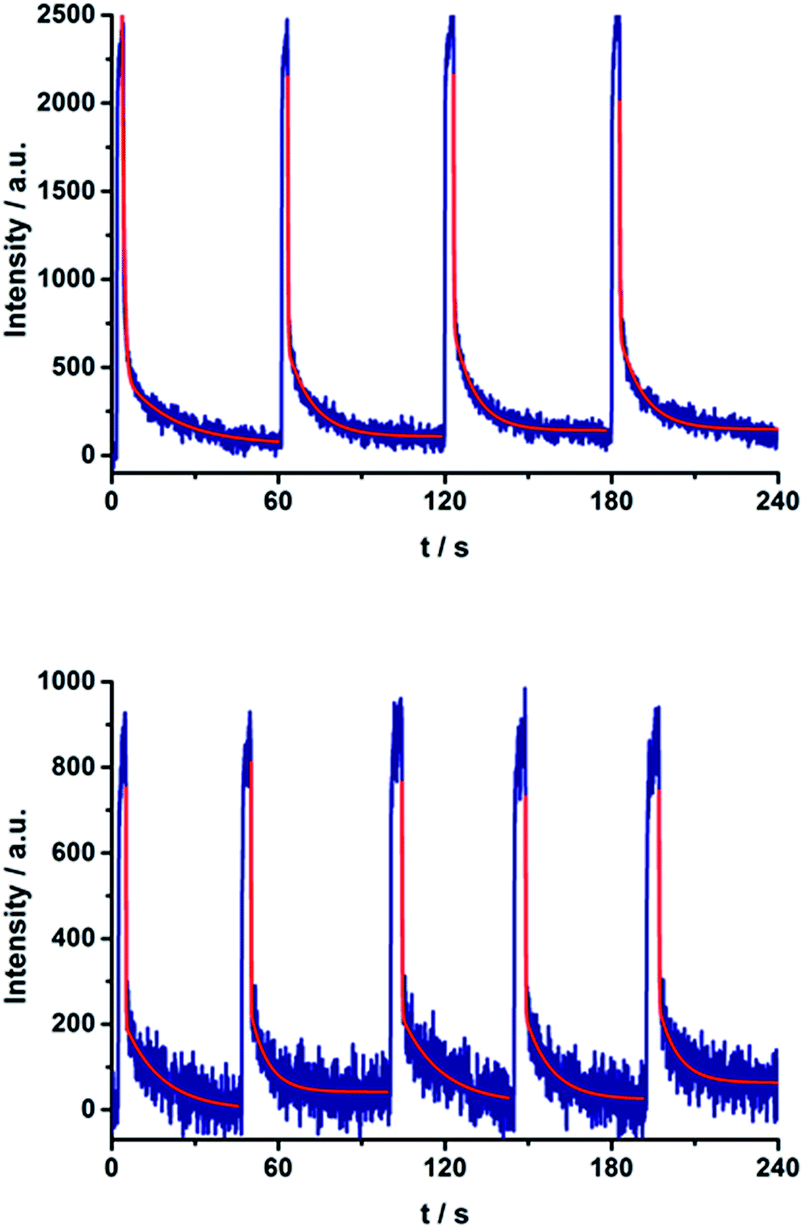 | ||
| Fig. 11 On–off switch of the EPR signal due to charge separation of 17a (top) and 20a (bottom) in PhCN at 77 K by turning on and off the irradiation from a high-pressure mercury lamp. | ||
| Compound | THF | PhCN |
|---|---|---|
| CR | CR | |
| 15a | <200 ms | <200 ms |
| 15b | <200 ms | <200 ms |
| 16a | 440 ± 60 ms | <200 ms |
| 16b | 420 ± 40 ms | 220 ± 40 ms |
| 17a | 510 ± 130 ms | <200 ms |
| 17b | <200 ms | 240 ± 20 ms |
| 18a | 420 ± 40 ms | <200 ms |
| 18b | 260 ± 60 ms | <200 ms |
| 19a | <200 ms | <200 ms |
| 19b | <200 ms | 240 ± 30 ms |
| 20a | <200 ms | <200 ms |
| 20b | 270 ± 30 ms | 260 ± 30 ms |
Molecular modelling
We turned to molecular modelling to investigate the fact that the D–A conjugates with the longest linker do not exhibit the longest lived CSS. Conformational analysis of the free base molecules was performed with the Conformer program76 to determine average electron donor–acceptor distances. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 conformations were determined for each molecule via a Metropolis Monte-Carlo algorithm. Each conformation was optimized with the COMPASS force field.77
000 conformations were determined for each molecule via a Metropolis Monte-Carlo algorithm. Each conformation was optimized with the COMPASS force field.77
The D–A distance for the lowest-energy conformer and the mean distances (including standard deviations) for all conformers within 20 kcal mol−1 of the lowest-energy conformation are given in Table 4. For both the meta- and para- series, the longest molecules do not follow the trend of increased D–A-distance with increased linker length. This is because the longest linkers allow the formation of a porphyrin–C60 van-der-Waals dimer (Fig. 12), which is the lowest energy conformer. Meta isomers, usually display shorter D–A distances and a higher standard deviation (i.e. higher conformational freedom) than their para-equivalents.
| Optimum D–A distance [Å] | Mean D–A distance [Å] | σ [±Å] | |
|---|---|---|---|
| 15a | 9.98 | 12.27 | 1.38 |
| 15b | 18.96 | 18.77 | 0.24 |
| 16a | 11.16 | 11.89 | 1.83 |
| 16b | 19.87 | 20.57 | 1.59 |
| 17a | 6.25 | 8.23 | 3.53 |
| 17b | 6.13 | 6.32 | 0.13 |
 | ||
| Fig. 12 Lowest-energy conformations of (17a) H2P-3-mC60 (left) and (17b) H2P-3-pC60 (right). | ||
To assess whether these results also apply to the metalated system, we compared the dimerisation energy of unsubstituted H2P and Zn porphyrins with C60 using dispersion-corrected density functional theory (DFT) (PBE+TS/DND).78–80 This reveals that the dimer is stabilized by 2.2 kcal mol−1 through metalation. Since the dimer is already the most stable conformation for the free base molecules, the overall picture of the conformational analysis should not change for the metalated case.
Furthermore, the energies of frontier molecular orbitals were calculated for DFT-optimized structures obtained with the MO6 functional.80,81 The frontier orbital energies for all computed structures are summarized in Table S3.† In general, the orbital energies are quite constant, with energy differences on the order of several meV. Energy differences of the HOMO orbitals are observed in the presence of the metal atom, which lead to 40 ± 1 meV stabilization. The larger variations observed for the LUMOs are the result of the linkage between the pyridine and the pyrrolidine: meta-conformations have LUMO energies around 70 ± 5 meV higher than the corresponding para-ones. Analyses of the orbital shapes showed clear electron donor–acceptor interactions. The LUMO is well distributed around the fullerene cage for all the cases (Fig. S9†). The HOMO is mainly localized on the porphyrin and shows a good overlap with the π-orbitals of the benzyl group, which provides electronic coupling with the aromatic chain. The orbitals of the metal atoms showed a high contribution to the electronic distribution of the HOMO around the entire porphyrin, increasing the electronic distribution around it (see Fig. S9 in the ESI†).
Discussion
Table 2 summarizes the radical ion pair state lifetimes for all electron donor–acceptor conjugates in toluene, THF, and PhCN. In brief, several factors influence the charge transfer of the porphyrin–fullerene D–A conjugates. On one hand, the polarity of the solvent affects the CSS lifetimes. In less polar solvents such as toluene, the longest lifetimes are observed. This leads to the assumption that the charge recombination occurs in the inverted region of the Marcus parabola. On the other hand, the length of the linker plays a key role in the electron transfer dynamics. The systems with just one pyridine-vinylene group exhibit the fastest charge recombination. 16a (H2P-2-mC60) and 16b (H2P-2-pC60) feature the longest lived radical ion pairs within the free base porphyrin series, while for the ZnP series 19a (ZnP-2-mC60) and 19b (ZnP-2-pC60) reveal the longest CSS lifetimes. Astonishingly, the triads with the longest linkers do not show the longest CSS lifetimes. Calculations suggest that the flexible linkers lead to shorter through-space distances between the free base porphyrin and the fullerene. The reduced D–A distances and the shorter lifetimes observed for the radical ion pair for 17a (H2P-3-mC60), 17b (H2P-3-pC60), 20a (ZnP-3-mC60) and 20b (ZnP-3-pC60) lead to the conclusion that for this system electron transfer occurs through space rather than through the linker. The compounds with C60 attached to the pyridine in a para-positions yield longer-lived CSS than those with C60 in a meta position. Finally, it should be noted that the longest CS states are formed for 19b (ZnP-2-pC60).Conclusions
We have designed and synthesized a new series of H2P/C60 and ZnP/C60 electron donor–acceptor conjugates, in which the electron donating H2P/ZnP and the electron accepting C60 are linked through a pyrrolidine ring covalently attached to pyridine-vinylene spacers of different lengths. Electrochemical experiments and molecular modelling at the DFT level revealed a strong push–pull nature between the electroactive constituents. Significant interactions were observed in absorption measurements as 1–4 nm red shifts of the H2P/ZnP absorptions. In addition, in the low-energy region of the spectra, charge transfer bands were identified that show considerably stronger interactions for the ZnP conjugates (100–400 cm−1) than for the H2P conjugates (20–40 cm−1). Among the ZnP conjugates, 20b (ZnP-3-pC60) exhibits the strongest coupling between ZnP and C60. Fluorescence assays showed that the H2P/ZnP features depend on the length and the substitution pattern of the pyridine-vinylene spacers. H2P systems generally show stronger fluorescence than ZnP ones. Conjugates with just one pyridine-vinylene unit, that is, 15a, 15b, 18a, and 18b, display the weakest fluorescence and shortest fluorescence lifetimes, while fluorescence quenching is barely detected for the conjugates with three pyridine-vinylene units, 17a, 17b, 20a, and 20b. Importantly, the fluorescence is more intense in the less polar solvent environments, suggesting charge rather than energy transfer. To confirm this, charge transfer was verified using pump–probe experiments. Differential absorption spectra reveal features of oxidized H2P/ZnP and reduced C60 in the visible and in the near-infrared regions, respectively. Kinetic analyses yielded charge-separated state lifetimes between 1.3 ns (15b) and 65 ns (16b) for the H2P conjugates and between 373 ps (18b) and 165 ns (19b) for the ZnP conjugates in THF. In toluene, the H2P/ZnP conjugates generally exhibit longer charge-separated state lifetimes than in THF and benzonitrile. This solvent dependence suggests that charge recombination occurs in the Marcus inverted region. Calculations revealed that conjugates with two pyridine-vinylene units and a para substitution exhibit the longest distances between D and A, ∼21 Å. These findings are perfectly compatible with the charge-separated state lifetimes for 16b and 19b, which were the longest within the series investigated. We infer from a detailed examination of the lowest-energy conformation of the conjugates with the longest spacers that the flexible linkers enable electron donor and acceptor to approach through-space, thus decreasing the effective distance to ∼6–8 Å.Complementary EPR measurements in frozen PhCN and THF confirm the formation of charge-separated states. A sharp peak corresponding to reduced C60 (g ∼ 2.000) was identified and a broader, less intense signal (g ∼ 2.003) was assigned to oxidized H2P/ZnP. Additionally, formation of the charge-separated states was switched on and off repeatedly by turning the irradiation on and off.
Acknowledgements
The authors gratefully acknowledge financial support from COLCIENCIAS, the Universidad del Valle, project PRI-PRIBUS-2011-1067, the Robert A. Welch Foundation for an endowed chair (Grant #AH-0033), the US National Science Foundation (grant CHE-1408865), MINECO of Spain (Projects CTQ2011-24652) and the Comunidad Autónoma de Madrid (FOTOCARBON project S2013/MIT-2841). N.M. thanks the Alexander von Humboldt Foundation. Work in Erlangen was supported by EXC35 “Engineering of Advanced Materials” and the Solar Technologies go Hybrid initiative of the Bavarian State Government. JTM is grateful for a Beilstein Stipendium. Work in Japan was partially supported by ALCA and SENTAN projects from Japan Science and Technology Agency (JST) and Grant-in-Aid (No. 26620154 and 26288037) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) Japan.Notes and references
- H. Imahori, Y. Mori and Y. Matano, J. Photochem. Photobiol., C, 2003, 4, 51 CrossRef CAS.
- M. E. El-Khouly, O. Ito, P. M. Smith and F. D'Souza, J. Photochem. Photobiol., C, 2004, 5, 79 CrossRef CAS PubMed.
- R. Koeppe and N. S. Sariciftci, Photochem. Photobiol., 2006, 5, 1122 RSC.
- J. L. Bahr, G. Kodis, L. de la Garza, S. Lin, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 2001, 123, 7124 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS PubMed.
- N. Martin, L. Sanchez, M. A. Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 198 CrossRef CAS.
- A. Lembo, P. Tagliatesta, D. M. Guldi, M. Wielopolski and M. Nuccetelli, J. Phys. Chem. A, 2009, 113, 1779 CrossRef CAS PubMed.
- S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 22, 4575 RSC.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455 CrossRef CAS PubMed.
- D. K. James and J. M. Tour, Chem. Mater., 2004, 16, 4423 CrossRef CAS.
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 15 CrossRef CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323 CrossRef CAS PubMed.
- J. L. Delgado, P. A. Bouit, S. Filippone, M. A. Herranz and N. Martin, Chem. Commun., 2010, 46, 4853 RSC.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed.
- D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243 CrossRef CAS.
- B. C. Thompson and J. M. Frechet, Angew. Chem., Int. Ed., 2008, 47, 58 CrossRef CAS PubMed.
- S. Fukuzumi and K. Ohkubo, Dalton Trans., 2013, 42, 15846 RSC.
- T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, S. K. Kim, D. Kim, A. Fujimoto, T. Hirakawa and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216 CrossRef CAS PubMed.
- D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC.
- D. Holten, D. F. Bocian and J. S. Lindsey, Acc. Chem. Res., 2002, 35, 57 CrossRef CAS PubMed.
- C. M. Drain, A. Varotto and I. Radivojevic, Chem. Rev., 2009, 109, 1630 CrossRef CAS PubMed.
- R. C. Haddon, L. E. Brus and K. Raghavachari, Chem. Phys. Lett., 1986, 125, 459 CrossRef CAS.
- S. Kirner, M. Sekita and D. M. Guldi, Adv. Mater., 2014, 26, 1482 CrossRef CAS PubMed.
- A. D. J. Haymet, Chem. Phys. Lett., 1985, 122, 421 CrossRef CAS.
- D. M. Guldi, Pure Appl. Chem., 2003, 75, 1069 CrossRef CAS.
- D. M. Guldi and M. Prato, Chem. Commun., 2004, 2517 RSC.
- L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593 CrossRef CAS.
- Q. Xie, E. Perez-Cordero and L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 3978 CrossRef CAS.
- D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695 CrossRef CAS PubMed.
- F. Arias, Q. Xie, L. Echegoyen, Y. Wu, Q. Lu and S. R. Wilson, J. Am. Chem. Soc., 1994, 116, 6388 CrossRef CAS.
- A. Ciammaichella, P. O. Dral, T. Clark, P. Tagliatesta, M. Sekita and D. M. Guldi, Chem.–Eur. J., 2012, 18, 14008 CrossRef CAS PubMed.
- J. Santos, B. M. Illescas, M. Wielopolski, A. M. G. Silva, A. C. Tomé, D. M. Guldi and N. Martín, Tetrahedron, 2008, 64, 11404 CrossRef CAS PubMed.
- D. I. Schuster, K. Li, D. M. Guldi, A. Palkar, L. Echegoyen, C. Stanisky, R. J. Cross, M. Niemi, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2007, 129, 15973 CrossRef CAS PubMed.
- G. Kodis, P. A. Liddell, L. de la Garza, A. L. Moore, T. A. Moore and D. Gust, J. Mater. Chem., 2002, 12, 2100 RSC.
- S.-H. Lee, A. G. Larsen, K. Ohkubo, Z.-L. Cai, J. R. Reimers, S. Fukuzumi and M. J. Crossley, Chem. Sci., 2012, 3, 257 RSC.
- A. Kira, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda, J. K. Park, D. Kim and H. Imahori, J. Am. Chem. Soc., 2009, 131, 3198 CrossRef CAS PubMed.
- F. Giacalone, J. L. Segura, N. Martín and D. M. Guldi, J. Am. Chem. Soc., 2004, 126, 5340 CrossRef CAS PubMed.
- M. R. Wasielewski, W. B. Davis, W. A. Svec and M. A. Ratner, Nature, 1998, 396, 60 CrossRef.
- B. Albinsson, M. P. Eng, K. Pettersson and M. U. Winters, Phys. Chem. Chem. Phys., 2007, 9, 5847 RSC.
- F. G. Brunetti, J. L. López, C. Atienza and N. Martín, J. Mater. Chem., 2012, 22, 4188 RSC.
- M. P. Eng and B. Albinsson, Angew. Chem., Int. Ed., 2006, 45, 5626 CrossRef CAS PubMed.
- K. Pettersson, A. Kyrychenko, E. Ronnow, T. Ljungdahl, J. Martensson and B. Albinsson, J. Phys. Chem. A, 2006, 110, 310 CrossRef CAS PubMed.
- M. U. Winters, K. Pettersson, J. Martensson and B. Albinsson, Chem.–Eur. J., 2005, 11, 562 CrossRef CAS PubMed.
- R. H. Goldsmith, L. E. Sinks, R. F. Kelley, L. J. Betzen, W. Liu, E. A. Weiss, M. A. Ratner and M. R. Wasielewski, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3540 CrossRef CAS PubMed.
- A. S. Lukas, P. J. Bushard, E. A. Weiss and M. R. Wasielewski, J. Am. Chem. Soc., 2003, 125, 3921 CrossRef CAS PubMed.
- E. A. Weiss, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2003, 107, 3639 CrossRef CAS.
- E. A. Weiss, M. J. Tauber, R. F. Kelley, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2005, 127, 11842 CrossRef CAS PubMed.
- G. de la Torre, F. Giacalone, J. L. Segura, N. Martín and D. M. Guldi, Chem.–Eur. J., 2005, 11, 1267 CrossRef CAS PubMed.
- A. Molina-Ontoria, M. Wielopolski, J. Gebhardt, A. Gouloumis, T. Clark, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2011, 133, 2370 CrossRef CAS PubMed.
- M. Wielopolski, A. Molina-Ontoria, C. Schubert, J. T. Margraf, E. Krokos, J. Kirschner, A. Gouloumis, T. Clark, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2013, 135, 10372 CrossRef CAS PubMed.
- S. Wolfrum, J. R. Pinzon, A. Molina-Ontoria, A. Gouloumis, N. Martín, L. Echegoyen and D. M. Guldi, Chem. Commun., 2011, 47, 2270 RSC.
- J. Ikemoto, K. Takimiya, Y. Aso, T. Otsubo, M. Fujitsuka and O. Ito, Org. Lett., 2002, 4, 309 CrossRef CAS PubMed.
- F. Oswald, D. M. Islam, Y. Araki, V. Troiani, R. Caballero, P. de la Cruz, O. Ito and F. Langa, Chem. Commun., 2007, 4498 RSC.
- F. Oswald, D. M. Islam, M. E. El-Khouly, Y. Araki, R. Caballero, P. de la Cruz, O. Ito and F. Langa, Phys. Chem. Chem. Phys., 2014, 16, 2443 RSC.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827 CrossRef CAS.
- C.-H. Lee and J. S. Lindsey, Tetrahedron, 1994, 50, 11427 CrossRef CAS.
- B. J. Littler, Y. Ciringh and J. S. Lindsey, J. Org. Chem., 1999, 64, 2864 CrossRef CAS PubMed.
- M. S. Newman and L. F. Lee, J. Org. Chem., 1972, 37, 4468 CrossRef CAS.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798 CrossRef CAS.
- M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519 CrossRef CAS.
- N. Martín, L. Sánchez, B. Illescas and I. Pérez, Chem. Rev., 1998, 98, 2527 CrossRef PubMed.
- H. Imahori, N. V. Tkachenko, V. Vehmanen, K. Tamaki, H. Lemmetyinen, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2001, 105, 1750 CrossRef CAS.
- N. V. Tkachenko, H. Lemmetyinen, J. Sonoda, K. Ohkubo, T. Sato, H. Imahori and S. Fukuzumi, J. Phys. Chem. A, 2003, 107, 8834 CrossRef CAS.
- E. Krokos, C. Schubert, F. Spänig, M. Ruppert, A. Hirsch and D. M. Guldi, Chem.–Asian J., 2012, 7, 1451 CrossRef CAS PubMed.
- E. Krokos, F. Spänig, M. Ruppert, A. Hirsch and D. M. Guldi, Chem.–Eur. J., 2012, 18, 10427 CrossRef CAS PubMed.
- C. Schubert, M. Wielopolski, L. H. Mewes, G. de Miguel Rojas, C. van der Pol, K. C. Moss, M. R. Bryce, J. E. Moser, T. Clark and D. M. Guldi, Chem.–Eur. J., 2013, 19, 7575 CrossRef CAS PubMed.
- P. G. Seybold and M. Gouterman, J. Mol. Spectrosc., 1969, 31, 1 CrossRef CAS.
- J. D. Megiatto, D. I. Schuster, S. Abwandner, G. D. Miguel and D. M. Guldi, J. Am. Chem. Soc., 2010, 132, 3847 CrossRef CAS PubMed.
- S. V. Kirner, D. M. Guldi, J. D. Megiatto Jr and D. I. Schuster, Nanoscale, 2015, 7, 1145 RSC.
- C. Luo, D. M. Guldi, H. Imahori, K. Tamaki and Y. Sakata, J. Am. Chem. Soc., 2000, 122, 6535 CrossRef CAS.
- T. Kato, T. Kodama, T. Shida, T. Nakagawa, Y. Matsui, S. Suzuki, H. Shiromaru, K. Yamauchi and Y. Achiba, Chem. Phys. Lett., 1991, 180, 446 CrossRef CAS.
- The visible region of the spectrum – Fig. 6 (left) – is dominated by the triplet signature of the porphyrin.
- As mentioned above, for 17b (H2P-3-pC60) the C60˙− fingerprint cannot be identified from the differential absorption spectra, due to the long lived H2P singlet (∼5 ns in EOS experiments). From the weak fluorescence quenching it is assumed that the charge separation is very inefficient. Due to this observation and also due to the short lifetime, the C60˙− peak is overlapped by the porphyrins singlet signal in the transient absorption spectra.
- R. L. C. Akkermans, N. A. Spenley and S. H. Robertson, Mol. Simul., 2013, 39, 1153 CrossRef CAS PubMed.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- B. J. Delley, Chem. Phys., 1990, 92, 508 CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- S. Fukuzumi, H. Mori, T. Suenobu, H. Imahori, X. Gao and K. M. Kadish, J. Phys. Chem. A, 2000, 104, 10688 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and procedures, complete spectroscopic and structural analysis, including Fig. S1–S51, Schemes S1 and S2 and Tables T1–T3. See DOI: 10.1039/c5sc02051d |
| ‡ S. V. Kirner and D. Arteaga contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |