
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecule antagonists of cell-surface heparan sulfate and heparin–protein interactions†
Ryan J.
Weiss
a,
Philip L. S. M.
Gordts
b,
Dzung
Le
cd,
Ding
Xu
e,
Jeffrey D.
Esko
bd and
Yitzhak
Tor
*ad
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, CA 92093-0358, USA. E-mail: ytor@ucsd.edu
bCellular and Molecular Medicine, University of California, San Diego, La Jolla, CA 92093-0687, USA
cDepartment of Medicine, University of California, San Diego, La Jolla, CA 92093-0612, USA
dGlycobiology Research and Training Center, University of California, San Diego, La Jolla, CA 92093-0687, USA
eDepartment of Oral Biology, University at Buffalo, Buffalo, NY 14260-1660, USA
First published on 29th July 2015
Abstract
Surfen, bis-2-methyl-4-amino-quinolyl-6-carbamide, was previously reported as a small molecule antagonist of heparan sulfate (HS), a key cell-surface glycosaminoglycan found on all mammalian cells. To generate structure–activity relationships, a series of rationally designed surfen analogs was synthesized, where its dimeric structure, exocyclic amines, and urea linker region were modified to probe the role of each moiety in recognizing HS. An in vitro assay monitoring inhibition of fibroblast growth factor 2 binding to wild-type CHO cells was utilized to quantify interactions with cell surface HS. The dimeric molecular structure of surfen and its aminoquinoline ring systems was essential for its interaction with HS, and certain dimeric analogs displayed higher inhibitory potency than surfen and were also shown to block downstream FGF signaling in mouse embryonic fibroblast cells. These molecules were also able to antagonize other HS–protein interactions including the binding of soluble RAGE to HS. Importantly, selected molecules were shown to neutralize heparin and other heparinoids, including the synthetic pentasaccharide fondaparinux, in a factor Xa chromogenic assay and in vivo in mice. These results suggest that small molecule antagonists of heparan sulfate and heparin can be of therapeutic potential for the treatment of disorders involving glycosaminoglycan–protein interactions.
Introduction
Heparan sulfate proteoglycans (HSPGs) are expressed on virtually all animal cells and in the extracellular matrix. Each HSPG consists of a core protein with one or more covalently attached linear heparan sulfate (HS) chains composed of alternating glucosamine and uronic acids that are heterogeneously N- and O-sulfated (Fig. 1). These complex cell surface carbohydrates regulate important biological processes including cell proliferation and motility, development, and organ physiology through their interaction with a large number of matrix proteins and growth factors.1–3 Sometimes these processes go awry, for example in cancer, inflammation and neurodegenerative disorders.4,5 Thus, much interest exists in agents controlling HS–protein interactions as both therapeutics and research tools.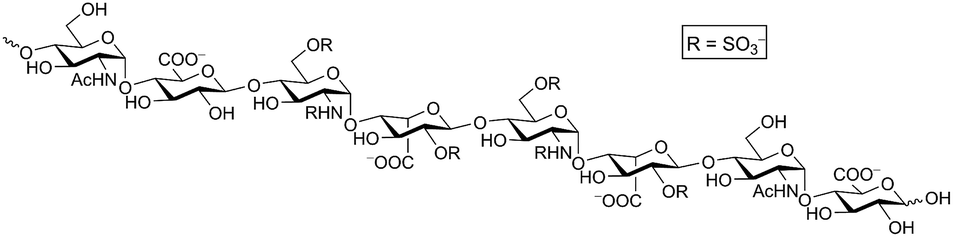 | ||
| Fig. 1 Heparan sulfate. A representative octasaccharide segment as expressed on wild-type CHO-K1 cells is shown. | ||
The biological function of HSPGs is largely determined through interactions of the HS chains with protein ligands. Multiple agents for antagonizing HS–protein interactions have been developed including heparin mimetic compounds6,7 and metabolic inhibitors that alter its biosynthesis.8,9 Other approaches use proteins,10 polypeptides,11,12 foldamers,13,14 or small molecule antagonists15–18 containing positively charged residues that bind to the negatively charged carboxylate or sulfate groups on HS. The accessibility and versatility of small molecules that antagonize HS–protein interactions make them particularly attractive as potential research tools and therapeutic agents.
We previously identified surfen (1) in the National Cancer Institute small molecule Diversity Set as an antagonist of HS and heparin (Fig. 2).19 Binding of surfen to heparin blocks its degradation by bacterial lyases and prevents its ability to activate antithrombin. In cell culture, surfen inhibits angiogenesis by preventing the interaction of HS with fibroblast growth factor 2 (FGF2) and inhibits type I Herpes simplex virus-infection. Surfen also blocks the enhancement of HIV-1 infection by amyloid fibrils found in semen,20,21 alters murine T-cell activation and proliferation,22 and blocks C5a receptor binding,23 suggesting that some of these processes might also depend on HS–protein interactions. Surfen also acts independently of HS, as a trypanocidal agent24 and as an inhibitor of the protease activity of anthrax lethal factor.25 These diverse properties of surfen suggest the possibility of designing more selective and potent analogs to target HS–protein interactions.
 | ||
| Fig. 2 Structures of surfen (1) and its analogs. Modifications highlighted in red and green. | ||
To probe the interactions of surfen with HS, identify the key molecular determinants responsible for its activity, and potentially discover more potent antagonists, a small set of analogs was synthesized and studied. A structure–activity relationship was established through quantifying their ability to inhibit cell surface HS–FGF2 binding in vitro. Certain analogs proved to be more potent antagonists of HS and were shown to inhibit other types of HS–protein interactions, including cell surface binding of soluble RAGE and neutralization of the anticoagulant activity of unfractionated heparin and low molecular weight heparins (LMWHs). Importantly, we found that surfen analogs were able to neutralize both in vitro and in vivo the synthetic pentasaccharide fondaparinux, for which no antidote exists.26 These observations imply that small molecule antagonists of HS can potentially be of therapeutic value and can serve as tools for chemical biologists interested in probing HS-dependent cellular processes.
Methodology
Derivative design
Surfen is a symmetric small molecule (MW 372) consisting of two quinoline moieties linked together through a urea. The quinoline rings are functionalized with a methyl group at the 2-position and an exocyclic amine at the 4-position. It has been previously proposed that the exocyclic amines and urea linker region of surfen could interact electrostatically with the anionic carboxylate and sulfate moieties of HS or through hydrogen bonding.19 It is also possible that the distance between the aminoquinoline moieties and their orientation are essential for its biological activity. To examine these possibilities, we synthesized a series of surfen analogs (Fig. 2).“Monomeric” versions of surfen, containing a single quinoline moiety (2, 3), were synthesized to probe the importance of the dimeric structure of surfen for its biological activity. Here we refer to these compounds as hemisurfen (2) and acetyl-hemisurfen (3). A thiocarbonyl analog of surfen (4), referred to as “thio surfen” in this study, was made with a thiourea core, a modification that replaces oxygen with larger, less electronegative sulfur. This substitution should primarily affect the hydrogen bonding capability of the linker region. To investigate the importance of the 4-aminopyridine fragment within the aminoquinoline moieties in binding HS, the exocyclic amines were replaced by methoxy groups (5) or removed altogether (6). For simplicity, these compounds are referred to here as “methoxy surfen” and “deaminated surfen”, respectively. To assess the significance of the distance between the aminoquinoline moieties, the linker between the two heterocycles was extended (7–12) which also increased the hydrophobicity of this region. Additionally, the urea group was substituted with two amide bonds. A compound with a glycol-like linker region (13), trivially named here “diglycolyl surfen”, was synthesized to probe whether increasing the hydrophilicity of the extended linker would impact its interaction with HS.
Synthesis
The core heterocycle in surfen, 4,6-diamino-2-methylquinoline (14), was synthesized as previously reported and was used in the synthesis of surfen analogs 2–4 and 7–13 (Schemes 1 and 2).27,28 4-Aminoacetanilide (15) was condensed with ethyl acetoacetate to give ethyl-β-(p-acetamidophenylamino) crotonate (16). This ethyl ester intermediate then underwent thermal cyclization in Dowtherm A to yield 6-acetamide-4-hydroxy-2-methylquinoline (17). The hydroxyquinoline intermediate was then methylated with dimethyl sulfate to yield the methoxy derivative (18). This compound then underwent a two-step reaction to yield 4,6-diamino-2-methylquinoline (14) through a 6-acetamido-4-aminoquinaldine intermediate (19). This building block was used in the synthesis of specific surfen analogs (4–13) (Schemes 2 and 3).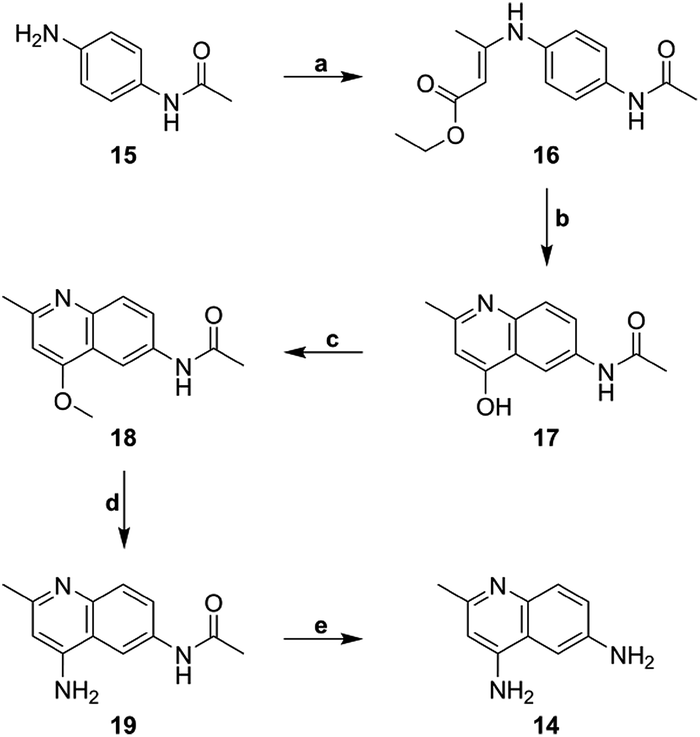 | ||
| Scheme 1 Synthesis of 4,6-diamino-2-methylquinoline building block (14) (a) ethyl acetoacetate, MeOH, reflux, (b) Dowtherm A, 265 °C, (c) dimethyl sulfate, toluene, 120 °C, (d) ammonium acetate, 135 °C, (e) 37% HCl, 90 °C. | ||
 | ||
| Scheme 2 Synthesis of surfen derivatives (a) (X = NH2) potassium cyanate, 10% acetic acid, H2O, rt (b) (X = NH2) acetyl chloride, acetic acid, rt (c) (X = NH2) thiophosgene, DMF, rt (d) (X = OCH3) triphosgene, 1,4-dioxane, acetic acid, rt (e) (X = H) triphosgene, 1,4-dioxane, acetic acid, rt (f) (X = NH2) oxalyl chloride, acetic acid, rt (g) (X = NH2) malonyl chloride, acetic acid, rt (h) (X = NH2) succinyl chloride, acetic acid, rt (i) (X = NH2) glutaryl chloride, acetic acid, rt (j) (X = NH2) adipoyl chloride, acetic acid, rt (k) (X = NH2) pimeloyl chloride, acetic acid, rt (l) (X = NH2) diglycolyl chloride, acetic acid, rt. | ||
 | ||
| Scheme 3 Synthesis of 5 (a) 37% HCl, 90 °C, (b) triphosgene, 1,4-dioxane, acetic acid, rt. | ||
1,3-Bis(2-methylquinolin-6-yl)urea (deaminated surfen) (6) was synthesized by reacting triphosgene with the commercially available 6-amino-2-methylquinoline in acetic acid. Furthermore, compounds 7–13 were synthesized using their respective diacid chlorides in acetic acid at room temperature. These compounds (7–13) were named according to the diacid chloride used in their synthesis. This procedure was adapted from a previous report.23 4-Amino-2-methyl-6-quinolyl-urea (hemisurfen) (2) was prepared using potassium cyanate, 10% acetic acid, and water followed by recrystallization from water. Finally, 6-acetamido-4-aminoquinaldine (acetyl-hemisurfen) (3) was synthesized by reacting 4,6-diaminoquinaldine with acetic chloride in acetic acid, and N,N′-bis-(4-amino-2-methyl-6-quinolyl)-thiourea (thio surfen) (4) was synthesized using thiophosgene in DMF. Hemisurfen and acetyl-hemisurfen were recrystallized from water, while all other final products were recrystallized from hot DMF by the addition of diethyl ether. To synthesize the methoxy analog 1,3-bis(4-methoxy-2-methyl-quinolin-6-yl)urea (methoxy surfen) (5), 6-acetamido-4-methoxyquinaldine (18) was deprotected with 37% HCl in H2O to form 6-amino-4-methoxyquinaldine (20). This compound was then reacted with triphosgene to yield 5 (Scheme 3).
Surfen (bis-2-methyl-4-amino-quinolyl-6-carbamide) was previously obtained from the National Cancer Institute as a hydrochloride salt (NCI 12155). All compounds were therefore converted to their hydrochloride salts for use in biological assays. The hydrochloride products were precipitated from an appropriate solvent using 4 M HCl in 1,4-dioxane (Scheme S1†). The products were analyzed using 1H NMR, 13C NMR, and ESI-MS. An X-ray crystal structure of one of the analogs (13) confirmed that these molecules are doubly protonated on their aminoquinoline ring systems (Fig. 3d). Furthermore, the X-ray structures of 1 and 13 displayed syn orientations in regards to their quinoline ring systems (Fig. 3a and d), while the crystal structures of oxalyl and adipoyl surfen displayed anti orientations (Fig. 3b and c). These structures suggest that surfen analogs within this collection could present diverse molecular configurations that could affect their interactions with the anionic subunits of HS.
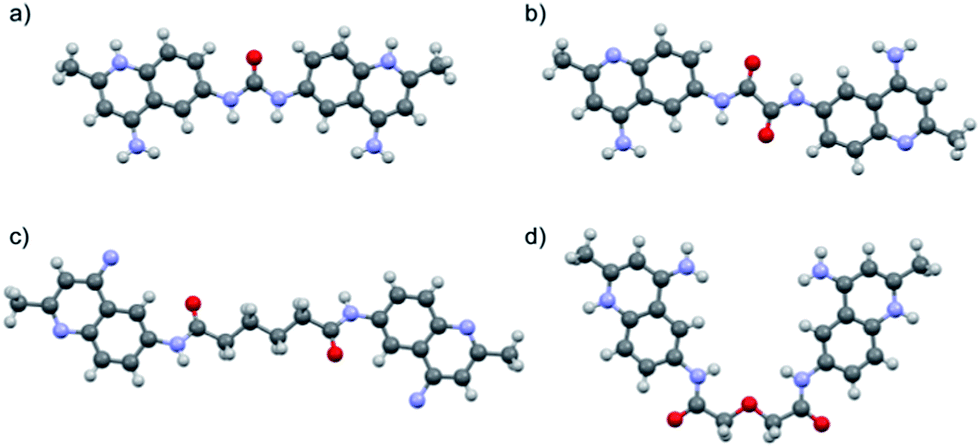 | ||
| Fig. 3 X-ray crystal structures of (a) surfen·2CF3COOH (1), (b) oxalyl surfen (7), (c) adipoyl surfen (11), and (d) diglycolyl surfen·2HCl (13). Counterions and solvent molecules omitted for clarity (see ESI† for additional information). | ||
Results
Inhibition of HS binding
To determine the potency of surfen and its analogs as HS antagonists, their ability to inhibit the binding of FGF2 was quantified in vitro.19 CHO cells express very low levels of FGF receptors (FGFRs), thus binding primarily occurs through cell surface HSPGs. Each surfen analog was pre-incubated with CHO cells. Biotinylated FGF2 was then added, followed by the addition of fluorescently-tagged streptavidin thus forming a conjugate with cell-bound biotin-FGF2. Flow cytometry was utilized to monitor the fluorescence of bound ligand. FGF inhibition curves were generated by plotting the percentage of maximum FGF binding obtained in the absence of any antagonist versus the concentration of the molecule of interest (see Fig. 4 for a representative example). IC50 values were obtained by fitting the dose–response curves to a classic sigmoidal response (Table 1).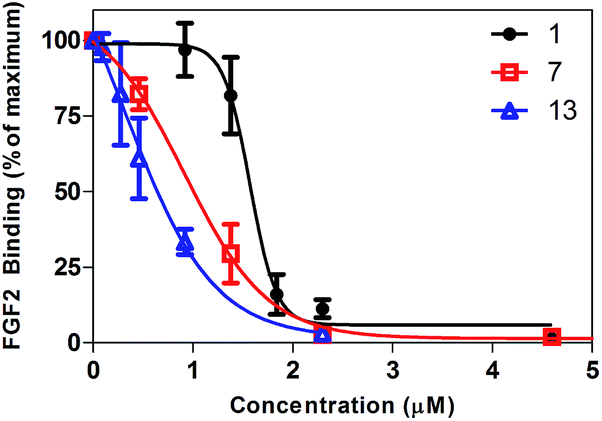 | ||
| Fig. 4 Representative inhibition curves. Surfen (1), oxalyl surfen (7), and diglycolyl surfen (13) were tested as inhibitors of FGF2 binding. | ||
| Compound | IC50 (μM) |
|---|---|
| a Values represent the mean ± SD of n = 3–4 experiments. | |
| Surfen (1) | 1.6 ± 0.04 |
| Hemisurfen (2) | >100 |
| Acetyl-hemisurfen (3) | >100 |
| Thio surfen (4) | 8.0 ± 0.20 |
| Methoxy surfen (5) | >100 |
| Deaminated surfen (6) | >100 |
| Oxalyl surfen (7) | 0.9 ± 0.08 |
| Malonyl surfen (8) | 1.2 ± 0.03 |
| Succinyl surfen (9) | 1.2 ± 0.11 |
| Glutaryl surfen (10) | 61.5 ± 0.70 |
| Adipoyl surfen (11) | 8.4 ± 1.2 |
| Pimeloyl surfen (12) | >100 |
| Diglycolyl surfen (13) | 0.7 ± 0.12 |
As shown in Table 1, analogs with oxalyl, malonyl, succinyl and diglycolyl amide linkers (7–9, 13) showed enhanced inhibition of FGF2 binding to HS compared to surfen (1), the parent compound. Interestingly, the glutaryl (10) and adipoyl (11) analogs showed slightly decreased activity compared to native surfen, while pimeloyl surfen (12) showed minimal inhibitory activity up to 100 μM. The thiocarbonyl analog (4) showed lower activity compared to surfen, while the diglycolyl derivative (13) showed the highest activity. Molecules with modification of the exocylic amine (5, 6) or that altered the dimeric structure of surfen (2, 3) were ineffective inhibitors up to 100 μM.
Surfen (1) and the most potent analogs (7, 9, 13) were tested for the biological relevance of their antagonistic properties by measuring inhibition of downstream FGF signaling in mouse embryonic fibroblasts (MEFs). In line with the binding studies, all four compounds showed a potent dose-dependent inhibition of FGF2-induced ERK phosphorylation relative to a control in the absence of antagonist (Fig. 5).
 | ||
| Fig. 5 Surfen and selected analogs inhibit FGF2-induced ERK phosphorylation. (a) Western blot analysis of the inhibition of downstream FGF-2 induced ERK phosphorylation (p-ERK), (b) band intensities were quantitated by densitometry and plotted as the ratio of p-ERK to ERK signal. | ||
To explore how analog variations might differentially alter binding of different proteins to HS we investigated the ability of surfen-type molecules to antagonize binding of the soluble ectodomain of RAGE (Receptor for Advanced Glycation Endproducts) to HS.29 Succinyl surfen (9) showed dose-dependent inhibition of binding (IC50 = 1.4 ± 0.2 μM) (Fig. 6a). Acetyl-hemisurfen (3) showed no inhibitory activity and was used as a negative control (IC50 > 100 μM).
 | ||
| Fig. 6 (a) Succinyl surfen (9) dose-dependently inhibits sRAGE binding to HS. (b) sRAGE binding inhibitory activity of selected compounds at 10 μM. The values represent the means ± SD. *P < 0.0001 compared with a control in the absence of antagonist. | ||
A full screen of surfen and its derivatives (10 μM) for sRAGE binding inhibition paralleled the observed trend for FGF2 binding (Fig. S2†). Specifically, the more potent FGF2 inhibitors (e.g.7–9, 13) also showed high potency for inhibition of sRAGE binding to cell surface HS, while certain surfen analogs (e.g.2, 3, 6) showed no inhibitory activity for either protein (Fig. 6b).
Surfen analogs neutralize the anticoagulant activity of heparin and low molecular weight heparins
Based on their capacity to inhibit HS–protein interactions in vitro, surfen (1) and the most active analogs (7, 9, and 13) were tested for their ability to neutralize the capacity of unfractionated heparin (UFH) to bind and activate antithrombin III (AT), a protease inhibitor that blocks the blood coagulation protease factor Xa. In this assay, heparin binds to AT, induces a conformation change, and triggers the inhibition of factor Xa. Thus, molecules that prevent heparin from interacting with AT prevent loss of factor Xa activity, which is readily assessed using a chromogenic substrate. IC50 values were determined by varying the concentration of surfen and its analogs. Surfen yielded an IC50 value of 2 ± 0.06 μM, whereas oxalyl surfen (7), succinyl surfen (9) and diglycolyl surfen (13) gave IC50 values of 1.2 ± 0.04, 4.6 ± 0.07, and 2 ± 0.03 μM, respectively (Fig. 7).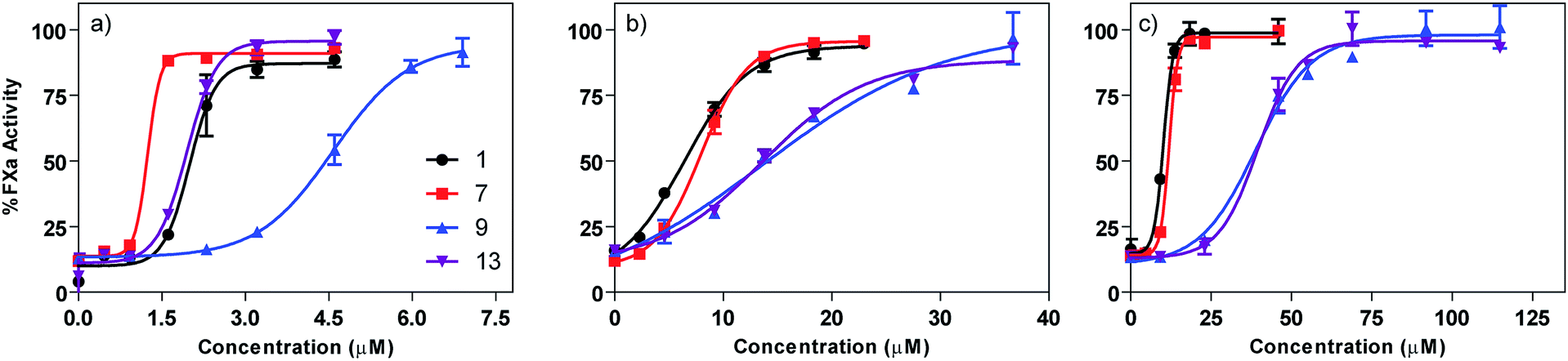 | ||
| Fig. 7 Dose-dependent neutralization of (a) unfractionated heparin (UFH), (b) enoxaparin (LMWH), (c) fondaparinux (pentasaccharide). Curves were fit using a classic sigmoidal-dose response. | ||
Selected surfen molecules were also tested as neutralizing agents of enoxaparin, a low molecular weight heparin. Using the factor Xa assay, we found that surfen, oxalyl surfen, succinyl surfen, and diglycolyl surfen neutralized enoxaparin in the low micromolar range (IC50 = 6.4 ± 0.4, 8 ± 0.2, 13.7 ± 2.7, 13.6 ± 0.6 μM, respectively). These compounds also neutralized fondaparinux, a synthetic pentasaccharide, albeit with slightly lower potency (IC50 = 10.2 ± 0.2, 11.9 ± 0.2, 38.9 ± 2.1, 39.5 ± 1.6 μM, respectively).
Oxalyl surfen neutralizes heparin and fondaparinux in mice
To test if the surfen analogs remained potent and had acceptable pharmacokinetic properties in vivo, the most potent analog (7) was evaluated as a reversal agent for heparin anticoagulant activity in mice treated with a high anticoagulant dose of either UFH or fondaparinux (Fig. 8). UFH at 1200 IU kg−1 (>5-fold the therapeutic dose) was injected subcutaneously and, after 1 hour, oxalyl surfen (6 mg kg−1) or vehicle control were injected intravenously. Protamine (16 mg kg−1) was used for comparison. After 5 minutes, blood was collected, centrifuged, and the plasma was isolated and analyzed for FXa activity. A calibration curve was established to determine the amount of active UFH or fondaparinux that remained. The concentration of UFH in the plasma was found to be significantly reduced in the oxalyl surfen-treated (0.08 ± 0.001 IU mL−1) and protamine-treated (0.09 ± 0.01 IU mL−1) groups compared to the control mice (1.46 ± 0.9 IU mL−1) (Fig. 8a). In a similar experiment, 0.5 mg kg−1 fondaparinux (ca. 5-fold the therapeutic dose) was injected subcutaneously, and after 10 minutes, protamine (16 mg kg−1), oxalyl surfen (16 mg kg−1) or vehicle control were injected intravenously. Fondaparinux in plasma was significantly reduced in oxalyl surfen-treated mice compared to the control-treated group (0.14 ± 0.06 mg L−1vs. 2.1 ± 0.33 mg L−1, respectively) and was equivalent to a 93 ± 2.8% decrease in anti-FXa activity (Fig. 8b). As previously reported,22 we found protamine ineffective in neutralizing fondaparinux in vivo. The results demonstrate that oxalyl surfen can reverse the anticoagulant activity of fondaparinux in vitro and in vivo and thus has the potential to be developed as an antidote in humans. | ||
| Fig. 8 In vivo neutralization of (a) UFH and (b) fondaparinux in mice (n = 3 for each group). The values represent the means ± SD. *P < 0.05 compared with the control group. | ||
Discussion
Surfen, a dimeric quinoline-containing molecule found in the NCI compound database, was recently described as a small molecule antagonist of certain HS–protein interactions.19 The nature of the interaction between surfen and HS is not, however, fully understood. To shed light on its interactions with HS and to identify more potent antagonists, several strategically modified surfen analogs were synthesized. Their ability to inhibit the binding of FGF2 to cell-surface HS on wild-type CHO cells was quantified to generate structure–activity relationships. FGF2, a potent mitogen and member of the heparin-binding growth factor family, has been shown to preferentially bind to N-sulfated, 2-O-sulfated sites of cell surface and extracellular matrix HS (Fig. 1).30,31 Due to the strong affinity of FGF2 for HS, this assay served as a useful tool to assess the ability of small molecules to antagonize these essential interactions. All derivatives were also tested as antagonists of soluble RAGE (sRAGE) protein binding to HS and as neutralizing agents of heparin and low molecular weight heparins.In general, surfen analogs with the dimeric aminoquinoline moiety intact showed a dose-dependent inhibition of FGF2 binding similar to that of surfen (Table 1). Analogs lacking the exocyclic amines on the quinoline ring systems (e.g., 5, 6) showed low inhibitory activity (IC50 > 100 μM). The lack of improvement in binding seen in these derivatives with modifications at the 4-position is not surprising. The protonated form of 4-aminoquinoline was previously shown to display lower acidity (pKa ∼ 8.5) compared with aminoquinoline derivatives substituted at other positions.32 Similarly, the basic 4-aminoquinoline moieties of surfen, upon protonation, can electrostatically interact with the negatively charged sulfate and carboxyl groups of HS via hydrogen bonding (Scheme S1†).
Hemisurfen (2) and acetyl-hemisurfen (3) did not inhibit FGF2 binding. These derivatives only contain one aminoquinoline moiety, while maintaining a urea or an amide unit mimicking the linker region. The lack of activity of these “monomeric” forms of surfen suggests a critical role for the dimeric structure of the parent molecule. A similar effect was observed recently with adhesamine, a small molecule that enhances cell adhesion through interactions with HS.33 It is possible that the three dimensional arrangement, length, or valency of surfen-type antagonists could affect their activity and interaction with HS. Investigation of multivalent analogs of HS-binding molecules reported cooperative and enhanced binding to HS upon increasing the number of cationic HS-binding moieties.34,35 These findings suggest that surfen analogs that display multiple aminoquinoline moieties might exhibit enhanced binding to HS as well.
Certain analogs with extended linkers (e.g., 7–9, 13), where the central urea moiety was replaced with a diamide linker, showed enhanced ability to inhibit FGF2 binding to HS compared to surfen. Similarly, these compounds also blocked binding of sRAGE to HS. Enhancing the H-bonding ability and spreading of the aminoquinoline recognition domains may account for enhanced potency, as engagement with additional sites along the long and heterogeneous HS chains might occur. The enhanced inhibitory activity of the oxalyl (7), malonyl (8), and succinyl (9) analogs compared to surfen (1) supports this idea. Interestingly, extending the linker region further (e.g., 10–12) hinders the interaction with HS and lowers the inhibitory activity, possibly due to their more hydrophobic nature. A similar loss of activity of derivatives with longer linker regions has been previously observed in a study of surfen-type compounds as inhibitors of anaphylatoxic C5a receptor.23 Due to the decreased activity of derivatives with more hydrophobic linkers, an analog with a more hydrophilic linker region, diglycolyl surfen (13), was synthesized. Diglycolyl surfen proved to be most potent in regards to inhibition of FGF2 binding to HS, suggesting that adding additional H-bonding sites to the linker region enhances the interaction with HS.
The observations discussed above correlating activity and linker lengths are not necessarily universal since the adipoyl surfen analog (11) showed better inhibitory activity compared to the glutaryl and pimeloyl analogs (10 and 12, respectively). Other reports have shown that the conformation of the binding domains of certain proteins is critical for their interaction with the anionic sugar residues of glycosaminoglycans, such as HS.36,37 In a similar way, it is possible that even-numbered linkers, due to preferred hydrocarbon conformation and impact on the projection of the heterocycles, display a more favorable molecular configuration for binding to HS compared to odd numbered linkers.
To assess the significance of the urea core, it was modified to a thiourea, while the rest of the structure was kept intact. Although a minor modification in regards to the entire molecular structure, the thio surfen analog (4) displayed lower FGF inhibitory potency compared to surfen, due to altered hydrogen bonding.38,39 Consequently, thio surfen is a less active HS antagonist compared to surfen, although it remains a micromolar inhibitor under the conditions of our assays.
Surfen (1) and the most potent compounds (7, 9, 13) were also able to dose-dependently antagonize downstream FGF signaling in MEFs. FGF2 signaling is dependent on the formation of a ternary complex between FGF, FGFR, and HS at the cell surface and initiates an intracellular signaling cascade leading to phosphorylation of ERK. These results are in line with previous observations using surfen (1) in endothelial cells19 and provide strong support for the biological relevance of surfen-type compounds as HS antagonists (Fig. 5). Variation in efficacy as compared to simple binding to FGF2 may be due to simultaneous inhibition of HS interaction with receptor as well as the ligand.
A screen of the entire library of surfen compounds for their ability to inhibit soluble RAGE (sRAGE) binding to cell surface HS revealed a similar potency trend to the FGF2 binding inhibition experiments (Fig. S2†). These results suggest that analog variations seem to similarly affect the binding properties of different HS ligands.
Heparin, a highly sulfated form of HS, binds to antithrombin III (AT), a serine protease inhibitor found in the coagulation cascade, and facilitates a conformational change that accelerates the inhibition of coagulation factors such as factor Xa and thrombin.40 Heparin has been used as an anticoagulant agent in surgery and treatment for certain thrombotic diseases for many years. However, adverse side effects can occur in some patients including excessive bleeding and thrombocytopenia.41 Numerous agents have been developed over the years to neutralize heparin.13,14,42–47 Currently, protamine, a mixture of arginine-rich proteins (MW ∼ 5000) is approved therapeutically for reversing the action of heparin in patients. However, protamine has displayed adverse side effects in some patients and demonstrates low efficacy towards reversing more recently developed low molecular weight heparins (LMWH), such as enoxaparin, and it is ineffective in neutralizing fondaparinux, a synthetic pentasaccharide analog of heparin.48–53
Interestingly, selected surfen analogs showed complete neutralization of unfractionated heparin (UFH), LMWH (enoxaparin) and the synthetic pentasaccharide (fondaparinux) both in vitro and in vivo (Fig. 7). Surfen (1) and oxalyl surfen (7) displayed the highest potency against each heparinoid (micromolar range), while succinyl surfen (9) had the lowest activity. Diglycolyl surfen (13) showed high activity against UFH compared to native surfen, while displaying inferior activity against LMWH. It appears that analogs with longer linker regions (9, 13) display lower activity against LMWH compared to surfen and oxalyl surfen (1, 7). All four surfen analogs tested show a potency trend of UFH > enoxaparin > fondaparinux, suggesting that neutralization may depend on the number of potential binding sites in the substrate. Although not studied here, we suspect that surfen molecules could align along each negatively-charged sugar chain in a cooperative manner and block the necessary AT binding sites. Regardless of their mechanism, these findings suggest that surfen analogs, such as oxalyl surfen, could be an alternative for protamine and can be further developed as a therapeutic agent.
Conclusions
In this study, a collection of surfen analogs was synthesized to illuminate their interaction with HS and to look for more potent HS antagonists. A cell culture-based structure–activity relationship, generated by monitoring the inhibition of FGF2 binding, suggests that the dimeric structure, exocyclic amines, and urea linker region play essential roles in the interaction of surfen with HS. Conserving the dimeric aminoquinoline moieties preserved the biological activity of the molecule, while certain modifications to the linker region either lowered or enhanced their antagonistic properties suggesting the hydrogen bonding capability of the linker as well as the separation and orientation of the heterocycles can impact the interaction with HS. Some extended analogs of surfen proved to be more potent antagonists of FGF2 binding to HS. Furthermore, these molecules were shown to dose-dependently antagonize the HS-binding of soluble RAGE protein and neutralize UFH and LMWH in a factor Xa chromogenic assay. Finally, a potent surfen analog was shown to neutralize heparin and fondaparinux in vivo in mice. Taken together, these findings illustrate the potential of small molecules as antagonists of HS and raise the possibility of using surfen-type compounds to serve as biochemical tools and as potential effectors in disorders that involve glycosaminoglycan–protein interactions.Experimental section
Mice
Eight-week old C57BL/6 mice were purchased from Charles Rivers Laboratories. All animals were housed in barrier conditions in the vivaria of the School of Medicine of the University of California San Diego that were approved by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures were approved by the local Animal Care Program. Mice were maintained on a 12 hour light–dark cycle, and were fed ad libitum water and PicoLab 5053 rodent chow.Materials
Unless otherwise specified, materials purchased from commercial suppliers were used without further purification. Surfen (bis-2-methyl-4-amino-quinolyl-6-carbamide) was obtained from the Open Chemical Repository in the Developmental Therapeutic Program of the National Cancer Institute (NSC 12155). All other anhydrous solvents and reagents were purchased from Sigma-Aldrich, with the exception of 4 M HCl in 1,4-dioxane (Alfa-Aesar) and 6-amino-2-methylquinoline (Acros Organics). NMR solvents were purchased from Cambridge Isotope Laboratories (Andover, MA). FGF2 (E. coli recombinant) was purchased from Peprotech, and the sulfo-NHS-LC-biotin linker was purchased from Thermo Scientific. PBS (Dulbecco's phosphate buffered saline), F12 Media, Cell Dissociation Buffer (PBS-based, Enzyme Free), and streptavidin-Cy5 were purchased from Life Technologies (Carlsbad, CA). FACS buffer (isotonic solution 0.85% w/v, phosphate buffered pH 7.1–7.3) and streptavidin-PE-Cy5 were purchased from BD Biosciences. Trypsin/EDTA was purchased from VWR (Mediatech). Tissue culture plates (20 mm) were from BD Falcon (BD Biosciences). Heparin (Scientific Protein Laboratories, 188.9 IU mg−1), enoxaparin sodium (Lovenox®, 100 mg mL−1) and fondaparinux sodium (Arixtra®, 5 mg mL−1) were obtained from their manufacturers. Salmon protamine (Grade IV) was purchased from Sigma-Aldrich.Instrumentation
NMR spectra were recorded on Varian Mercury 300 and 400 MHz, and Varian VX 500 MHz spectrometers. Mass spectra were recorded at the UCSD Chemistry and Biochemistry Mass Spectrometry Facility, utilizing an Agilent 6230 HR-ESI-TOF mass spectrometer. Flow cytometry studies were performed on a BD FACSCalibur. Absorbance measurements for the factor Xa assay were performed on a VersaMax microplate reader (Molecular Devices) using Softmax Pro software.Synthesis
Full synthetic procedures and characterization of all compounds can be found in the ESI.†FGF2 and sRAGE binding inhibition
Wild-type CHO cells (ATCC CCL-61) were grown in F12 growth medium supplemented with 10% (v/v) fetal bovine serum, 100 μg mL−1 of streptomycin sulfate, and 100 units per mL of penicillin G. Cells were grown to confluence, lifted with Cell Dissociation Buffer, washed with PBS buffer, and pre-incubated with specific concentrations of surfen or surfen analog in PBS/0.1% BSA on ice for 10 min. Next, biotinylated FGF2 (2.5 nM, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000) or biotinylated sRAGE (2 μg mL−1, 1:100) (ESI†) were added and incubated for 30 min or 1 hour on ice, respectively. Cells were washed, and bound biotinylated protein was detected by using streptavidin-PE-Cy5 or streptavidin-Cy5 (1:1000, PharMingen) and flow cytometry (FacsCalibur, BD Biosciences). Certain compounds (1, 4, 5, 7) required use of streptavidin-Cy5 due to interference. Raw data was interpreted using FlowJo Analytical Software (Tree Star Inc.). Protein binding was quantified by the geometric mean of the fluorescence intensity. Results are represented as the extent of binding compared with a sample incubated in the absence of surfen. These values were later plotted and further analyzed using GraphPad Prism v5.0.
:1000) or biotinylated sRAGE (2 μg mL−1, 1:100) (ESI†) were added and incubated for 30 min or 1 hour on ice, respectively. Cells were washed, and bound biotinylated protein was detected by using streptavidin-PE-Cy5 or streptavidin-Cy5 (1:1000, PharMingen) and flow cytometry (FacsCalibur, BD Biosciences). Certain compounds (1, 4, 5, 7) required use of streptavidin-Cy5 due to interference. Raw data was interpreted using FlowJo Analytical Software (Tree Star Inc.). Protein binding was quantified by the geometric mean of the fluorescence intensity. Results are represented as the extent of binding compared with a sample incubated in the absence of surfen. These values were later plotted and further analyzed using GraphPad Prism v5.0.
Stock solutions were made for each compound in DMSO (30 mM). For each experiment, the stock solutions were diluted first to 10 mM DMSO, then the appropriate serial dilutions were made in PBS buffer (with 0.1% BSA). Stock solutions for analogs 7 and 11 were made in 1:1 DMSO–H2O mixture to improve their solubility. Stock solutions were made and stored in glass containers because surfen has been previously found to bind to plastic. Compounds and DMSO stock solutions were stored under argon at −20 °C.
FGF2 signaling
For signaling assays, immortalized mouse embryonic fibroblast cells were cultured in 12-well plates. The growth medium was exchanged for serum-free DMEM (Invitrogen) for 5 hours before the addition of surfen or surfen analog. Cells were incubated with compounds for 10 min at 37 °C. Subsequently, FGF2 (20 ng mL−1) was added for 5 min then cells were put on ice, washed with ice-cold PBS, and lysed using ice-cold radioimmunoprecipitation assay buffer. Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Scientific) before Western blot analysis. Cells were resolved by SDS-PAGE and blotted with antiphospho-ERK and anti ERK antibodies (Cell Signaling Technology) at 1:1000 dilutions. Bands were visualized on an Odyssey Infrared imaging system (Li-Cor Biosciences) as described.54 Band intensities were quantitated by densitometry using Image J software.
Factor Xa inhibition assay
Human antithrombin III (66 μg mL−1; Enzyme Research Laboratories), heparin or heparinoid (0.2 IU mL−1), and various concentrations of surfen or surfen analog were prepared in a solution containing 25 mM HEPES (N-[2-hydroxyethyl] piperazine-N′-[2-ethanesulfonic acid]), 150 mM NaCl and 0.1% bovine serum albumin (pH 7.5) in a 96-well microtiter plate. Human factor Xa (0.4 μg mL−1, 50 μL; Enzyme Research Laboratories) was added to each well and the solutions were incubated for 10 min at room temperature with frequent mixing. Chromogenic substrate specific for factor Xa (0.5 mg mL−1, 50 μL; S-2765 Diapharma) was added, incubated for 5 min at room temperature, and absorbance was measured at 405 nm. Background absorption at 405 nm was subtracted prior to analysis of the results. A heparin/heparinoid concentration with approximately ∼85% factor Xa inhibition was selected for measurement of neutralization (0.2 IU per well). Results are represented as the extent of factor Xa activity compared with a sample incubated with heparin/heparinoid in the absence of surfen. Surfen and analogs had no direct effect on factor Xa activity measured in the absence of heparin.In vivo heparin neutralization
Wild-type mice (8 weeks old, weight ∼ 20–30 g) were divided into a control group (n = 3), an oxalyl surfen-treated group (n = 3), and a protamine-treated group (n = 3). Thirty min before heparin administration, a blood sample was collected by tail bleeding in 3.2% sodium citrate tubes (9:1 blood to citrate ratio). Each mouse received a subcutaneous injection of UFH (1200 IU kg−1, PBS) or fondaparinux (0.5 mg kg−1, PBS). Next, mice received an intravenous injection (via tail vein) of DMSO–H2O solution (control), protamine (16 mg kg−1, PBS), or oxalyl surfen (6 or 16 mg kg−1, DMSO–water) 60 min after UFH and 10 min after fondaparinux administration. Five min after administration of the reversal compound, blood was collected via submandibular bleeds into 3.2% sodium citrate tubes. Plasma was collected immediately (2200 g for 15 min) and stored at −80 °C. A heparin anti-FXa colorimetric assay was used to analyze the neutralization effect of protamine and oxalyl surfen (7). Their activity was compared to a heparin control without reversal agent as described above. The change in the absorption at 405 nm for each sample was compared with a standard calibration curve to determine the amount of heparin present. The purpose of this assay is to analyze the amount of UFH or fondaparinux remaining in plasma by indirectly measuring the residual activity of FXa. Heparinized plasma (3 μL) was mixed with antithrombin, forming an AT–heparin complex. The amount of UFH or fondaparinux remaining in plasma was plotted against the reversal agent dosage.
Acknowledgements
This work was supported by grants from the NIH (GM077471 to Y. T. and J. D. E. and HL107150 to J. D. E.). We are grateful to Drs Beth Wilson and Manuela Schuksz for their advice with the binding assays, Patrick Secrest for help with the mice injections, and Qiongyu Chen (UCSD Murine Hematology, Chemistry and Coagulation Core) for help with the factor Xa chromogenic assays. We also thank Drs Curtis Moore and Arnold Rheingold (UCSD Chemistry and Biochemistry X-ray Facility) for their help.Notes and references
- U. Hacker, K. Nybakken and N. Perrimon, Nat. Rev. Mol. Cell Biol., 2005, 6, 530–541 CrossRef PubMed.
- J. R. Bishop, M. Schuksz and J. D. Esko, Nature, 2007, 446, 1030–1037 CrossRef CAS PubMed.
- H. E. Bulow and O. Hobert, Annu. Rev. Cell Dev. Biol., 2006, 22, 375–407 CrossRef CAS PubMed.
- L. K. Hallak, D. Spillmann, P. L. Collins and M. E. Peeples, J. Virol., 2000, 74, 10508–10513 CrossRef CAS.
- A. N. Alexopoulou, H. A. B. Multhaupt and J. R. Couchman, Int. J. Biochem. Cell Biol., 2007, 39, 505–528 CrossRef CAS PubMed.
- F. Manetti, F. Corelli and M. Botta, Curr. Pharm. Des., 2000, 6, 1897–1924 CrossRef CAS.
- X. T. Zhu, B. T. Hsu and D. C. Rees, Structure, 1993, 1, 27–34 CrossRef CAS.
- P. A. Baeuerle and W. B. Huttner, Biochem. Biophys. Res. Commun., 1986, 141, 870–877 CrossRef CAS.
- T. A. Fritz, F. N. Lugemwa, A. K. Sarkar and J. D. Esko, J. Biol. Chem., 1994, 269, 300–307 CAS.
- A. Hekman, Biochim. Biophys. Acta, 1971, 251, 380–387 CrossRef CAS.
- S. M. Fuchs and R. T. Raines, Biochemistry, 2004, 43, 2438–2444 CrossRef CAS PubMed.
- A. K. Udit, C. Everett, A. J. Gale, J. R. Kyle, M. Ozkan and M. G. Finn, ChemBioChem, 2009, 10, 503–510 CrossRef CAS PubMed.
- G. L. Montalvo, Y. Zhang, T. M. Young, M. J. Costanzo, K. B. Freeman, J. Wang, D. J. Clements, E. Magavern, R. W. Kavash, R. W. Scott, D. Liu and W. F. DeGrado, ACS Chem. Biol., 2014, 9, 967–975 CrossRef CAS PubMed.
- S. Choi, D. J. Clements, V. Pophristic, I. Ivanov, S. Vemparala, J. S. Bennett, M. L. Klein, J. D. Winkler and W. E. DeGrado, Angew. Chem., Int. Ed., 2005, 44, 6685–6689 CrossRef CAS PubMed.
- L. D'Ilario, I. Francolini, A. Martinelli and A. Piozzi, Dyes Pigm., 2009, 80, 343–348 CrossRef PubMed.
- M. Schmidtke, A. Karger, A. Meerbach, R. Egerer, A. Stelzner and V. Makarov, Virology, 2003, 311, 134–143 CrossRef CAS.
- H.-C. Selinka, L. Florin, H. D. Patel, K. Freitag, M. Schmidtke, V. A. Makarov and M. Sapp, J. Virol., 2007, 81, 10970–10980 CrossRef CAS PubMed.
- N. Harris, F. Y. Kogan, G. Il'kova, S. Juhas, O. Lahmy, Y. I. Gregor, J. Koppel, R. Zhuk and P. Gregor, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 245–254 CrossRef CAS PubMed.
- M. Schuksz, M. M. Fuster, J. R. Brown, B. E. Crawford, D. P. Ditto, R. Lawrence, C. A. Glass, L. Wang, Y. Tor and J. D. Esko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13075–13080 CrossRef CAS PubMed.
- N. R. Roan, S. Sowinski, J. Muench, F. Kirchhoff and W. C. Greene, J. Biol. Chem., 2010, 285, 1861–1869 CrossRef CAS PubMed.
- L. M. Castellano and J. Shorter, Biology, 2012, 1, 58–80 CrossRef PubMed.
- J. Warford, C. D. Doucette, D. W. Hoskin and A. S. Easton, Biochem. Biophys. Res. Commun., 2014, 443, 524–530 CrossRef CAS PubMed.
- T. J. Lanza, P. L. Durette, T. Rollins, S. Siciliano, D. N. Cianciarulo, S. V. Kobayashi, C. G. Caldwell, M. S. Springer and W. K. Hagmann, J. Med. Chem., 1992, 35, 252–258 CrossRef CAS.
- F. C. Goble, J. Pharmacol. Exp. Ther., 1950, 98, 49–61 CAS.
- R. G. Panchal, A. R. Hermone, T. L. Nguyen, T. Y. Wong, R. Schwarzenbacher, J. Schmidt, D. Lane, C. McGrath, B. E. Turk, J. Burnett, M. J. Aman, S. Little, E. A. Sausville, D. W. Zaharevitz, L. C. Cantley, R. C. Liddington, R. Gussio and S. Bavari, Nat. Struct. Mol. Biol., 2004, 11, 67–72 CAS.
- T. E. Warkentin and M. A. Crowther, Can. J. Anaesth., 2002, 49, S11–S25 Search PubMed.
- H. Jensch, Angew. Chem., 1948, 60, 248 Search PubMed.
- C. T. Peng and T. C. Daniels, J. Am. Chem. Soc., 1956, 78, 3703–3708 CrossRef.
- D. Xu, J. H. Young, J. M. Krahn, D. Song, K. D. Corbett, W. J. Chazin, L. C. Pedersen and J. D. Esko, ACS Chem. Biol., 2013, 8, 1611–1620 CrossRef CAS PubMed.
- N. Turner and R. Grose, Nat. Rev. Cancer, 2010, 10, 116–129 CrossRef CAS PubMed.
- M. Maccarana, B. Casu and U. Lindahl, J. Biol. Chem., 1993, 268, 23898–23905 CAS.
- A. Albert and R. Goldacre, Nature, 1944, 153, 467–469 CrossRef CAS PubMed.
- N. Takemoto, T. Suehara, H. L. Frisco, S. Sato, T. Sezaki, K. Kusamori, Y. Kawazoe, S. M. Park, S. Yamazoe, Y. Mizuhata, R. Inoue, G. J. Miller, S. U. Hansen, G. C. Jayson, J. M. Gardiner, T. Kanaya, N. Tokitoh, K. Ueda, Y. Takakura, N. Kioka, M. Nishikawa and M. Uesugi, J. Am. Chem. Soc., 2013, 135, 11032–11039 CrossRef CAS PubMed.
- A. V. Dix, L. Fischer, S. Sarrazin, C. P. H. Redgate, J. D. Esko and Y. Tor, ChemBioChem, 2010, 11, 2302–2310 CrossRef CAS PubMed.
- K. S. Kawamura, M. Sung, E. Bolewska-Pedyczak and J. Gariépy, Biochemistry, 2006, 45, 1116–1127 CrossRef CAS PubMed.
- S. Gandrille, M. Aiach, D. A. Lane, D. Vidaud, P. Molho-Sabatier, R. Caso, P. de Moerloose, J. N. Fiessinger and E. Clauser, J. Biol. Chem., 1990, 265, 18997–19001 CAS.
- H. Margalit, N. Fischer and S. A. Ben-Sasson, J. Biol. Chem., 1993, 268, 19228–19231 CAS.
- S. M. Testa, M. D. Disney, D. H. Turner and R. Kierzek, Biochemistry, 1999, 38, 16655–16662 CrossRef CAS PubMed.
- T. Nishiguc and Y. Iwakura, J. Org. Chem., 1970, 35, 1591–1593 CrossRef.
- L. Jin, J. P. Abrahams, R. Skinner, M. Petitou, R. N. Pike and R. W. Carrell, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14683–14688 CrossRef CAS.
- T. E. Warkentin, M. N. Levine, J. Hirsh, P. Horsewood, R. S. Roberts, M. Gent and J. G. Kelton, N. Engl. J. Med., 1995, 332, 1330–1335 CrossRef CAS PubMed.
- L. C. Chang, J. F. Liang, H. F. Lee, L. M. Lee and V. C. Yang, AAPS PharmSci, 2001, 3, 18 CrossRef.
- L. M. Lee, L. C. Chang, S. Wrobleski, T. W. Wakefield and V. C. Yang, AAPS PharmSci, 2001, 3, 19 CrossRef PubMed.
- T. W. Wakefield, P. C. Andrews, S. K. Wrobleski, A. M. Kadell, S. Tejwani, M. S. Hulin and J. C. Stanley, J. Surg. Res., 1996, 63, 280–286 CrossRef CAS PubMed.
- S. Onoue, Y. Nemoto, S. Harada, T. Yajima and K. Kashimoto, Life Sci., 2003, 73, 2793–2806 CrossRef CAS.
- J. R. Fromm, R. E. Hileman, E. E. O. Caldwell, J. M. Weiler and R. J. Linhardt, Arch. Biochem. Biophys., 1997, 343, 92–100 CrossRef CAS PubMed.
- T. Mecca, G. M. L. Consoli, C. Geraci, R. La Spina and F. Cunsolo, Org. Biomol. Chem., 2006, 4, 3763–3768 CAS.
- W. A. Weiss, J. S. Gilman, A. J. Catenacci and A. E. Osterberg, JAMA, J. Am. Med. Assoc., 1958, 166, 603–607 CrossRef CAS PubMed.
- J. Choay, M. Petitou, J. C. Lormeau, P. Sinay, B. Casu and G. Gatti, Biochem. Biophys. Res. Commun., 1983, 116, 492–499 CrossRef CAS.
- L. S. Chawla, G. Moore and M. G. Seneff, Obes. Surg., 2004, 14, 695–698 CrossRef PubMed.
- J. J. van Veen, R. M. Maclean, K. K. Hampton, S. Laidlaw, S. Kitchen, P. Toth and M. Makris, Blood Coagulation Fibrinolysis, 2011, 22, 565–570 CrossRef CAS PubMed.
- D. Suryanarayan and S. Schulman, Thromb. Res., 2014, 133, S158–S166 CrossRef CAS.
- J. I. Weitz, J. Hirsh and M. M. Samama, Chest, 2008, 133, 234S–256S CrossRef CAS PubMed.
- J. C. Gonzales, P. L. S. M. Gordts, E. M. Foley and J. D. Esko, J. Clin. Invest., 2013, 123, 2742–2751 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new compounds, X-ray crystallographic analysis, binding curves, biotinylation of FGF2 and sRAGE, tabulated data. CCDC 1057474–1057477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01208b |
| This journal is © The Royal Society of Chemistry 2015 |