
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights into cobalt(II/III)-catalyzed C–H oxidation: a combined theoretical and experimental study†
Xiao-Kang
Guo
,
Lin-Bao
Zhang
,
Donghui
Wei
* and
Jun-Long
Niu
*
The College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, Henan Province 450001, P. R. China. E-mail: donghuiwei@zzu.edu.cn; niujunlong@zzu.edu.cn
First published on 9th September 2015
Abstract
Cobalt-mediated C–H functionalization has been the subject of extensive interest in synthetic chemistry, but the mechanisms of many of these reactions (such as the cobalt-catalyzed C–H oxidation) are poorly understood. In this paper, possible mechanisms including single electron transfer (SET) and the concerted metalation–deprotonation (CMD) pathways of the CoII/CoIII-catalyzed alkoxylation of C(sp2)–H bonds have been investigated for the first time using the DFT method. CoII(OAc)2 has been employed as an efficient catalyst in our previous experimental study, but the calculated results unexpectedly indicated that the intermolecular SET pathway with CoIII as the actual catalyst might be the most favorable pathway. To support this theoretical prediction, we have explored a series of Cp*CoIII(CO)I2 catalyzed C(sp2)–H bond alkoxylations, extending the application of cobalt-catalyzed functionalization of C–H bonds. Furthermore, kinetic isotope effect (KIE) data, electron paramagnetic resonance (EPR) data, and TEMPO inhibition experiments also support the SET mechanism in both the Co-catalyzed alkoxylation reactions. Thus, this work should support an understanding of the possible mechanisms of the CoII/CoIII-catalyzed C(sp2)–H functionalization, and also provide an example of the rational design of novel catalytic reactions guided by theoretical calculations.
Introduction
Transition-metal-catalyzed C–H bond functionalization has attracted widespread attention due to its high efficiency and atom economy.1 However, most of these transformations have been accomplished with the aid of transition metals such as palladium,1l,2 rhodium,3 and ruthenium.4 Recent improvements in sustainable catalysis have focused on the development of cheaper and naturally abundant first-row transition-metal alternatives with comparable catalytic efficacies.5 Cobalt as a catalyst for C–H bond functionalization has been greatly developed with significant progress being achieved.6 For example, electron-rich CoI catalysts have been used in C–H activation/coupling reactions with alkynes, olefins, imines, alkyl/aryl halides, and phenol derivatives. Subsequently, high-valent Cp*CoIII complexes such as Cp*Co(CO)I2, [Cp*CoIII(arene)](PF6)2, and [Cp*Co(C6H6)][B(C6F5)4]2 were also discovered by several groups as effective catalysts with which to activate C–H bonds for addition to unsaturated compounds (imines, enones, and aldehydes) and amidation, cyanation, allylation, and halogenations.7 Additionally, Daugulis reported a new method for CoII-catalyzed C–H bond alkenylation, in which aminoquinoline is utilized as a directing group.8The formation of C–O bonds is more difficult than C–C bond generation by C–H activation, especially when alcohols are used as alkoxylation reagents9 and alkoxyl metal intermediates are prone to undergo β-hydride elimination to form the corresponding aldehydes, ketones, or carboxylic acids.10 So far, most of the cases that involve transition-metal-catalyzed C–H activation are focused on palladium catalysts11 but a few reports on copper catalyzed systems have also been disclosed.12 A Cu-catalyzed alkoxylation of arenes under basic and aerobic conditions (Scheme 1a) was also developed in this laboratory.13 Recently, we reported the first CoII-catalyzed alkoxylation of C(sp2)–H bonds under basic and oxidizing conditions (Scheme 1b).14 Due to the similarity between cobalt- and copper-catalyzed C–H functionalization, it might be expected that these reactions share a similar reaction mechanism, particularly in the early steps.
 | ||
| Scheme 1 Copper- and cobalt-catalyzed C(sp2)–H functionalization. | ||
For the Cu catalyzed reaction shown in Scheme 1a, a high kinetic isotope effect (KIE = 2.5) is observed, indicating that the C–H bond cleavage is the rate-determining step. It is generally accepted that the C–H bond is activated via a concerted metalation–deprotonation (CMD) mechanism.15 The similarity between the starting materials and reaction conditions depicted in Scheme 1a and b may imply a similar reaction mechanism for the two reactions but, unexpectedly, a preliminary mechanistic study of the CoII catalytic reaction depicted in Scheme 1b revealed that KIE ≈ 1, indicating that the C–H bond cleavage does not proceed by the same mechanism as the Cu catalyzed reaction. This phenomenon is similar to the CuII-catalyzed example reported by Yu's group (Scheme 1c),16 in which an intramolecular single electron transfer (SET) route, not the CMD route, was invoked. The similarity of the KIE results implies that our CoII-catalyzed system may also follow the SET mechanism. Stahl et al. observed a switch between SET-based and CMD-based pathways upon changing from acidic to basic reaction conditions in CuII-mediated aerobic C–H oxidation, in which a KIE is observed after the system is made alkaline.17 Consequently, a comparative study on both the SET-based and CMD-based routes is necessary to determine the detailed reaction mechanism of the CoII-catalyzed alkoxylation.
Despite the great progress that has been made in understanding cobalt catalysis, the detailed reaction mechanisms for these reactions remain poorly understood. In particular, CoII in the reaction system can be oxidized to CoIII by the excess oxidant additive (Scheme 1b), and it is unclear whether the exact oxidation state of the catalytic Co species is CoII or CoIII. In recent months, CoIII-catalyzed C(sp2)–H functionalization reactions have been reported (Scheme 2a and b),7e,h but the mechanisms of these reactions continue to be hotly debated. On the one hand, recent literature studies report that CoIII compounds are capable of catalyzing many chelate-directed C(sp2)–H functionalization reactions.7c,e,f,h,18 None of these works indicate the existence of a radical intermediate, all of them supporting organometallic mechanisms, even when the C–H activation is shown by KIE assessment to be not always rate-limiting. On the other hand, the oxidation of a π system by cobalt complexes is a well-known process which involves the intermediacy of carbon-centered radicals.19 For example, Kochi employed the intermolecular SET concept to elucidate the CoIII(TFA)3-mediated oxidation of aryl C–H bonds (Scheme 2c)20 and concluded that an aromatic cation-radical intermediate is formed after an arene electron is transferred to CoIII. Likewise, it is reasonable to speculate that the intermolecular SET mechanism may be appropriate in the CoIII-catalyzed C(sp2)–H functionalization described above.
 | ||
| Scheme 2 CoIII-catalyzed C(sp2)–H functionalization. | ||
No computational study on the mechanism of CoII/CoIII-catalyzed C(sp2)–H alkoxylation appears to have been performed, and experiments on the CoIII-catalyzed C(sp2)–H alkoxylation of arenes and olefins have not been reported. Several questions on the possible mechanisms of this kind of reaction need however to be resolved: (a) which mechanism (CMD or SET) is favored when CoII is the pre-catalyst? (b) In which state (CoII or CoIII) is the actual catalyst in high-valency Co-catalyzed C(sp2)–H alkoxylation? (c) Can the CoIII compound catalyze the C(sp2)–H alkoxylation? If so, which mechanism (CMD or SET) is favorable?
All of these questions led us to study the detailed mechanisms of Co-catalyzed C(sp2)–H alkoxylation not only in theory but also experimentally. In the present study, we first conducted a theoretical investigation of the detailed mechanism of CoII- and CoIII-catalyzed C(sp2)–H alkoxylation. This predicted that the CoIII compound might also catalyze the C–H alkoxylation. Subsequently, experimental results of the Cp*CoIII(CO)I2-catalyzed C(sp2)–H alkoxylation were found to confirm and support the theoretical prediction.
For practical assessment of the energetic viability of the possible routes, the CoII(OAc)2·4H2O catalyzed C(sp2)–H alkoxylation reaction between methanol and (E)-2-(2-methylbut-2-enamido)pyridine-1-oxide (denoted as R) was chosen for the theoretical investigation. Ag2O acts as the oxidant and NaOPiv as the base. It should be noted that Ag2O can react with methanol to generate AgOMe by the equation:
| Ag2O + 2MeOH = 2AgOMe + H2O |
Results and discussion
Several possible reaction pathways of CoII/CoIII-catalyzed C(sp2)–H alkoxylation have been studied by DFT, and further experimental studies have been performed to support the results predicted by the theoretical study.1. Possible pathways of CoII-catalyzed C(sp2)–H alkoxylation
In order to coordinate with Co, the nitrogen in the reactant R should initially be deprotonated. According to our calculated results shown in Scheme 3, the base OPiv− in the reaction system can abstract the proton attached to the nitrogen easily via transition state TS1, whose formation requires only 14.0 kcal mol−1. The carboxamide anion intermediate INT1 can coordinate with CoII forming INT2, as shown in Scheme 4. Two kinds of mechanisms, including SET and CMD, to activate the C–H bond are possible. We set the energy of reactant R at 0.0 kcal mol−1 in the energy profiles, and all the energies of the other minima discussed below are relative to that unless otherwise specified. | ||
| Scheme 3 Extraction of proton by the base additive. | ||
 | ||
| Scheme 4 Possible CMD mechanism (pathway 1) of CoII-catalyzed C(sp2)–H alkoxylation (energy: kcal mol−1, distance: Å). | ||
1.2.1 Intramolecular SET pathways. As described above, C–H bond activation may follow the SET mechanisms, and here two possible intramolecular SET pathways (including pathways 2 and 3) were explored. As shown in Scheme 5, pathway 2 consists of four steps: the ligand exchange between CoII and AgOMe via transition state TS4, the intramolecular single electron transfer of intermediate INT7, the OMe group transfer from CoI to the electron-deficient carbon via transition state TS5, and the solvent MeOH-assisted proton transfer from the olefinic carbon to the ligand OAc−via transition state TS6. The structure of intermediate INT7 still has only one unpaired electron, a doublet, after the intramolecular SET, and the two structures of intermediate INT7, before and after the SET, as in Scheme 5, resemble resonance structures of one another. We assumed that the SET may occur in this reaction pathway, and the details of this SET process are discussed in the next section. With the exception of the SET step, the Gibbs free energy barriers of the first and third steps in pathway 1 are only 3.6 and 19.8 kcal mol−1 (Scheme 5), respectively. However, the energy barrier of transition state TS6 for C–H bond breaking is extremely high (68.1 kcal mol−1), suggesting that pathway 2 is not energetically favorable.
 | ||
| Scheme 5 Possible intramolecular SET pathway (pathway 2) of CoII-catalyzed C(sp2)–H alkoxylation (energy: kcal mol−1, distance: Å). | ||
As shown in Scheme 6, pathway 3 consists of three reaction steps. The first is the intramolecular SET step for INT2. Here, we also assumed that the SET can occur in this reaction pathway. The OMe group can also be transferred directly from AgOMe to INT2via transition state TS7. The last step is a direct proton transfer process via transition state TS8. The energy barrier of the second step is only 14.9 kcal mol−1, but the energy barrier for the proton transfer is as high as 46.8 kcal mol−1, indicating that pathway 3 is unlikely to occur under the experimental conditions.
 | ||
| Scheme 6 Possible intramolecular SET pathway (pathway 3) of CoII-catalyzed C(sp2)–H alkoxylation (energy: kcal mol−1, distance: Å). | ||
According to the free energy profiles shown in Schemes 5 and 6, the two intramolecular SET pathways will not occur even if it is assumed that the SET process in INT2 and INT7 can take place. In an effort to find out whether the intramolecular SET can really happen, we performed time-dependent density functional theory (TD-DFT) calculations to evaluate the transition energy from the ground-state (GS) to the lowest excited state (ES). As shown in Fig. 1 and 2, the lowest-energy internal SET product arises from transfer of a α-spin electron from the olefin π electrons, the singly occupied orbital in INT2/INT7, to the lowest unoccupied orbital of the Co. The excitation energies for the transitions are 37.2 and 44.3 kcal mol−1 for INT2 and INT7 respectively, indicating that the intramolecular SET processes of CoII-catalyzed C(sp2)–H alkoxylation cannot occur.
 | ||
| Fig. 1 The lowest excited-state doublet derives from excitation of an α-electron from the singly occupied SOMO (GS(INT2)) into the nominally unoccupied LUMO (ES(INT2), dz2). The resulting excited state is 37.2 kcal mol−1 above the ground state, corresponding to a photon energy of 906.95 nm. | ||
 | ||
| Fig. 2 The lowest excited-state doublet derives from excitation of an alpha electron from the singly occupied SOMO (GS(INT7)) into the nominally unoccupied LUMO (ES(INT7), dz2). The resulting excited state is 44.3 kcal mol−1 above the ground state, corresponding to a photon energy of 761.09 nm. | ||
1.2.2 Intermolecular SET pathways. Inspired by the intermolecular SET mechanism of CuII-catalyzed C(sp2)–H functionalization reported by Stahl and Ertem,17 we considered the possible intermolecular SET transition processes for this reaction. As shown in Scheme 7, the Gibbs free energy differences for intermolecular SET processes range from 31.2 to 50.3 kcal mol−1, demonstrating that it would be difficult for the intermolecular SET to occur.
 | ||
| Scheme 7 Possible intermolecular SET processes of CoII-catalyzed C(sp2)–H alkoxylation. | ||
2. The possible pathways of CoIII-catalyzed C(sp2)–H alkoxylation
We have explored almost all the possible mechanisms for CoII-catalyzed C–H alkoxylation under basic conditions, but the above calculated results fail to explain the experimental results reasonably. As mentioned above, CoII might be oxidized to CoIII by AgOMe, and thus the CoIII-catalyzed C(sp2)–H functionalization reaction pathway would be feasible. As shown in Scheme 8, the energy barrier for oxidation of CoII to CoIII by AgOMe is only 2.5 kcal mol−1, showing that it would be very facile. Then, starting from CoIII(OAc)2(OMe), there are also two kinds of reaction mechanisms including SET and CMD starting with the CoIII catalyst and INT1. | ||
| Scheme 8 The oxidation process from CoII to CoIII by AgOMe. | ||
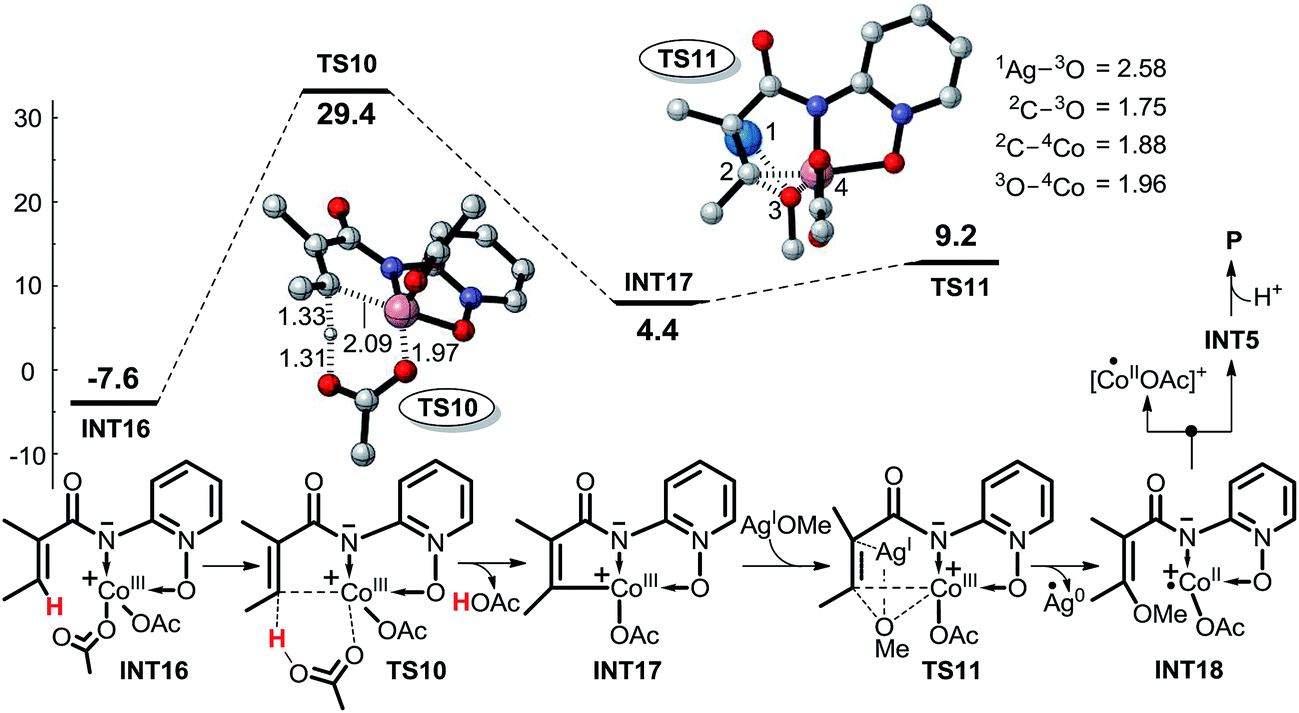 | ||
| Scheme 9 Possible CMD mechanism (pathway 4) of CoIII-catalyzed C(sp2)–H alkoxylation (energy: kcal mol−1, distance: Å). | ||
 | ||
| Scheme 10 Possible intermolecular SET pathways 5 (red, upper) and 6 (blue, lower) of CoIII-catalyzed C(sp2)–H alkoxylation (energy: kcal mol−1, distance: Å). | ||
As shown in Scheme 10, pathway 5 comprises three key reaction steps: coordination of intermediate INT11 with CoIII(OAc)2(OMe), the transfer of ligand OMe from CoIII to electron-deficient carbon via transition state TS12, and the proton transfer from the olefinic carbon to OAc−via transition state TS13. Furthermore, CoII is oxidized to CoIII by AgOMe and simultaneously INT18 dissociates, completing the cycle. Another OAc− coordinates with CoII to form INT21 before the proton transfer process takes place. The Gibbs free energy barriers for TS12 and TS13 are 4.6 and 12.6 kcal mol−1, respectively.
In pathway 6 depicted in Scheme 10 the coordination of INT11 with the CoII(OAc)(OMe) generated in the SET step affords intermediate INT13. There are still two transition states, TS14 and TS13 in pathway 6, which shares the same proton transfer mechanism with pathway 5. The energy barrier of the OMe transfer process via transition state TS14 (20.6 kcal mol−1) is higher than that (4.6 kcal mol−1) in pathway 5, indicating that pathway 5 should be more favorable.
In pathway 7, depicted in Scheme 11, intermediate INT23 is first formed by the weak interaction between intermediate INT13 and AgOMe, and then the OMe group is transferred directly from AgOMe to the olefinic carbon via transition state TS15. Subsequently, the OAc− anion abstracts the proton of the olefinic carbon via transition state TS16. The high energy barrier of 43.2 kcal mol−1 for transition state TS16 excludes this pathway. Furthermore, for pathway 8, depicted in Scheme 12, the other olefinic carbon forms a covalent bond automatically with the OMe group which renders it impossible for us to locate the structures of transition state TS17 and intermediate INT26, so we cannot explore this pathway in any more detail.
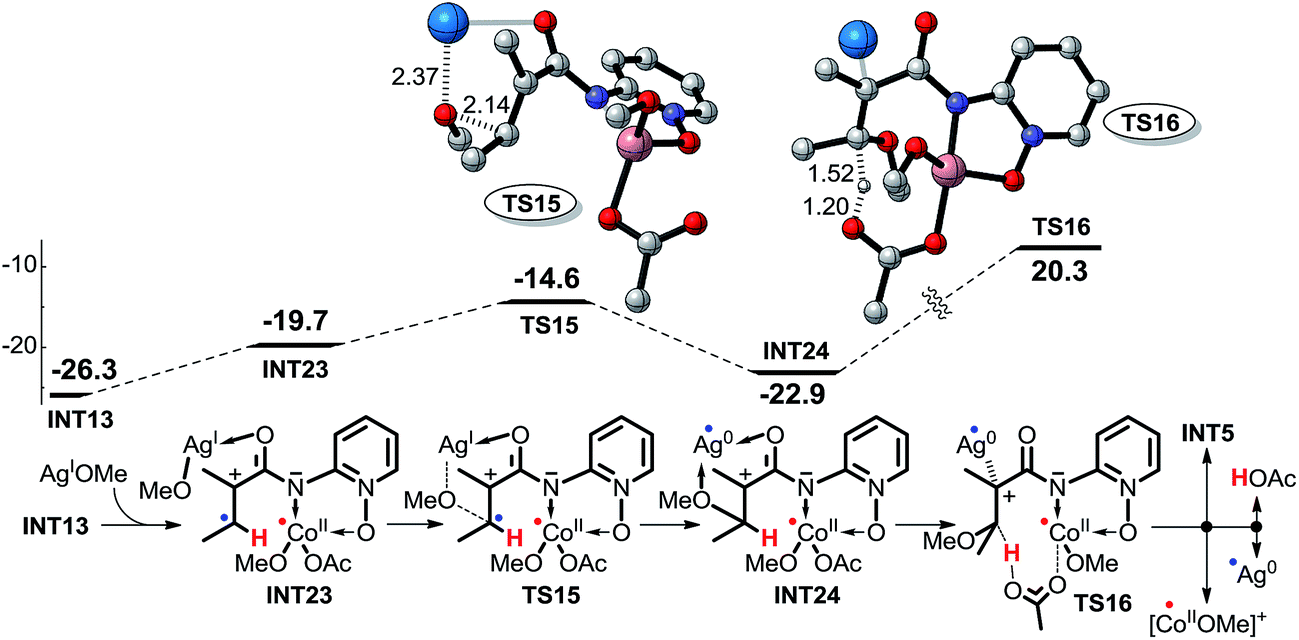 | ||
| Scheme 11 Possible intermolecular SET pathway 7 of CoIII-catalyzed C(sp2)–H alkoxylation (energy: kcal mol−1, distance: Å). | ||
 | ||
| Scheme 12 Possible intermolecular SET pathway 8 of CoIII-catalyzed C(sp2)–H alkoxylation (energy: kcal mol−1, distance: Å). | ||
If the pre-catalyst CoII(OAc)2 were to be oxidized to a CoIII intermediate, then the intermolecular SET pathway 5 becomes the most energetically favorable among all the eight possible pathways being considered, and we believe that the CoIII should be the actual catalyst for this kind of C(sp2)–H alkoxylation irrespective of the CoII or CoIII compound that was added to the reaction system. The INT11 has the highest energy (19.7 kcal mol−1) in the energy profile of pathway 5, so we can conclude that the intermolecular SET step rather than the C–H activation should be rate-determining in this reaction.
3. Combined experimental and theoretical exploration of CoIII-catalyzed C(sp2)–H alkoxylation
The above computational results prompt us to further explore whether the SET mechanism for the direct CoIII-catalyzed alkoxylation of C(sp2)–H bond is experimentally feasible. To confirm the plausibility of this prediction, experimental research was initiated with the reaction between ethanol and 2-benzamidopyridine 1-oxide (1a) catalyzed by Cp*Co(CO)I2. When a solution of benzamide (1a) in ethanol was treated at 70 °C with Cp*Co(CO)I2 (20 mol%), Ag2O (2 equiv.) and NaOAc (1 equiv.) under an air atmosphere (Scheme 13), the desired ethoxylated product 3aa was obtained in a good yield.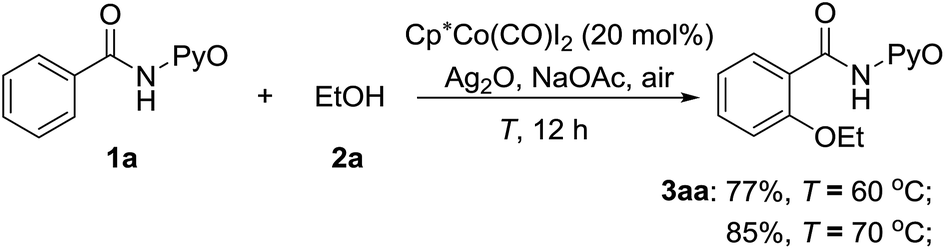 | ||
| Scheme 13 CoIII-catalyzed ethoxylation of 1a with ethanol. | ||
Diversely substituted amides were tolerated under the Cp*Co(CO)I2-catalyzed alkoxylation to furnish the desired products (3) in moderate to good yields (Scheme 14). The aryl substrates possessing electron-rich and electron-deficient functional groups underwent the transformation successfully (3aa–3ka). For meta-substituted amides (1g, 1h), the reaction tended to take place at the less hindered position and an iodo group on the aryl ring was also compatible. The heterocyclic substrate (1k) afforded the corresponding ethoxylated product in 63% yield. Several alcohols were tested and demonstrated to give the products in yields ranging from 48–85% (3ab–3ae). Olefinic carboxamides were also found to follow the protocol under the reaction conditions (3la–3oa).
 | ||
| Scheme 14 CoIII-catalyzed alkoxylation of benzamide derivatives with alcohols. | ||
In addition, control experiments revealed that the addition of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO, 1.5 equiv.) as a radical quencher completely inhibits the reaction (Scheme 15a). This result indicates that a radical pathway (SET mechanism) is involved. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1a and [D5]-1a was then treated with ethanol. No kinetic isotope effect (KIE = 1.0) was obtained (Scheme 15b), suggesting that C–H bond cleavage of arenes is not the rate-limiting step, in agreement with our calculated results. Additionally, the electron paramagnetic resonance (EPR) spectrum of the reaction system demonstrated the existence of the single electron (g = 2.23003, see the ESI†). The above experiments can be explained by our computational study, which they support.
:1 mixture of 1a and [D5]-1a was then treated with ethanol. No kinetic isotope effect (KIE = 1.0) was obtained (Scheme 15b), suggesting that C–H bond cleavage of arenes is not the rate-limiting step, in agreement with our calculated results. Additionally, the electron paramagnetic resonance (EPR) spectrum of the reaction system demonstrated the existence of the single electron (g = 2.23003, see the ESI†). The above experiments can be explained by our computational study, which they support.
 | ||
| Scheme 15 Controlled experiments for CoIII-catalyzed C(sp2)–H alkoxylation. | ||
We have performed a theoretical study on the mechanism of Cp*CoIII complex catalyzed C–H activation. As shown in Scheme 16, it is very similar to the favorable pathway 5 catalyzed by the CoIII complex, and the SET process is still the rate-determining step based on the energy profile, which is also in agreement with the KIE and EPR experiments. Based on the above computational and experimental results, we can propose the general mechanism for not only the CoII- but also the CoIII-catalyzed C(sp2)–H alkoxylation reaction. As shown in Scheme 17, AgOR (OR = alkoxy group) is generated and is the actual oxidant, and the reactant I is transformed to intermediate II by proton transfer to the base. First, it is an intermolecular SET process between intermediate II and CoIIIL3 (L = ligand) generating intermediate III, L−, and CoIIL2, which can be oxidized to CoIIIL2(OR) by AgOR. Second, intermediate IV is formed by the coordination of intermediate III with CoIIIL2(OR). Subsequently, the OR group is transferred from the Co to the substrate leading to the formation of intermediate V. Intermediate VI is formed by the coordination of base B−. As shown in Scheme 16, it should be noted that the coordination would not occur when the catalyst is Cp*CoIII. The next step is the proton transfer to base ligand and, finally, intermediate VII dissociates to VIII and CoII. CoII can be oxidized again to CoIII which catalyzes the next cycle, and intermediate VIII continues to be converted to the product IX. Although the tridentate chelate VII should be very stable, we still think it can be dissociated directly, because the structural transformation to VIII (i.e.INT18 in Scheme 10 or INT18′ in Scheme 16) is a highly exothermic process, which can compensate for the energy for the direct dissociation.
 | ||
| Scheme 16 The mechanism of Cp*CoIII complex catalyzed C(sp2)–H alkoxylation obtained at the M06-L(SMD, ethanol)/6-311++G(2df, 2pd)//SDD level (energy: kcal mol−1, distance: Å). | ||
 | ||
| Scheme 17 Fundamental reaction pathway of CoIII-catalyzed C(sp2)–H alkoxylation. | ||
Conclusions
We have explored multiple possible SET and CMD mechanisms of CoII/CoIII-catalyzed alkoxylation of C(sp2)–H bond in theory, and found that the intermolecular SET pathway in which the CoIII works as the actual catalyst (pathway 5) should be the most favorable pathway, even when the CoII(OAc)2 was added as a catalyst. Generally, there are three key steps in the favorable pathway of the high-valent Co-catalyzed C(sp2)–H alkoxylation, i.e. the intermolecular SET between the reactant and CoIII, the alkoxylation and, finally, the breaking of C(sp2)–H bond. The calculated results indicate that the SET step, rather than the C(sp2)–H activation, is rate-determining. Guided by the computed mechanism, the first CoIII-catalyzed C(sp2)–H alkoxylation is reported. A variety of amides with electron-rich and electron-poor functional groups were experimentally suitable for the Cp*Co(CO)I2-catalyzed alkoxylation, thus extending the application for cobalt-catalyzed functionalization of C–H bonds. All observations in the experiment can be explained by our computational results.This work provides another special SET-based example in the context of chelate-directed C(sp2)–H functionalization. The detailed CoII/CoIII-catalyzed SET and CMD models and pathways should be helpful for chemists to understand the general mechanism and the roles of the additives and catalysts in the CoII/CoIII-catalyzed C–H functionalization, and thus provide valuable insights into rational prediction and design of the more efficient catalysts in this kind of reaction.
Computational and experimental details
All of the calculations were performed using Gaussian 09.23 Computed structures are illustrated using CYLView.24 The density functional theory (DFT) method was applied since it has been successfully used in many studies of organocatalysis,25 organometallic catalysis,26 and biological reaction mechanisms.27 The density functional theory calculations were carried out with the M06-L functional in the presence of the SMD continuum solvation model with methanol (or ethanol) as the solvent. All the structures were completely optimized using a combined basis set: the LanL2DZ basis set28 was used for Co, I, and Ag along with the 6-31G(d, p) basis set for C, N, H, and O. The frequency calculations were performed at the same level at 298 K and 1 atm, and vibrational analysis was performed to confirm the optimized stationary points as true minima with no imaginary frequency, or transition states with one and only one imaginary frequency, on the potential energy surface and to obtain the thermodynamic data. On the basis of the optimized structures at the M06-L/6-31G(d, p)//LanL2DZ level, the energies were then refined by M06-L/6-311++G(2df, 2pd)//SDD29 single-point calculations with the same solvent effects. We checked the S2 values of all the stationary points obtained above, and found that all the differences between the obtained S2 value and the normal S2 value are less than 7.5%, so spin contamination can safely be ignored.We chose to conduct discussions based on Gibbs free energies rather than Born–Oppenheimer energies, which are the electronic (including nuclear-repulsion) energies plus ZPEs. Free energy contributions were added to single-point M06-L electronic energies computed with the SDD basis set on Co, I, and Ag and the 6-311++G(2df, 2pd) basis set on all other atoms to arrive at final, composite free energies.
TD-DFT calculations were performed to predict the UV/visible electronic excitations of postulated intermediates. The M06-L density functional, the LanL2DZ pseudopotential basis set on Co, I, and Ag, and the 6-311++G(2df, 2pd) basis set on all other atoms were used for the TD-DFT calculations in methanol with the SMD continuum solvation model.
Bakac et al. have performed TD-DFT calculations on several CoIII complexes, and confirmed that the computational results are close to the experimental results.30 In order to test the reliability of M06-L functional and the TD-DFT calculations in this paper, the structures of [CoIII(NH3)5Cl]2+,31TpPh2CoII(2,6-dibromophenolate),32 and (TF5PP)CoII depicted in Scheme 1833 have been chosen and completely optimized at the M06-L(SMD, water)/6-31G(d, p)//LanL2DZ, M06-L(SMD, chloroform)/6-31G(d, p)//LanL2DZ, and M06-L(SMD, dichloromethane)/6-31G(d, p)//LanL2DZ levels, respectively. It should be noted that the bond lengths of the optimized structures of TpPh2CoII(2,6-dibromophenolate) and (TF5PP)CoII have tiny differences (<0.05 Å) with those of the corresponding X-ray crystal structures reported in the experiments,32,33 indicating that the M06-L method is suitable for the Co complexes. Then, TD-DFT calculations based on the optimized structures have been carried out at the M06-L(SMD, water)/6-311++G(2df, 2pd)//LanL2DZ, M06-L(SMD, chloroform)/6-311++G(2df, 2pd)//LanL2DZ, and M06-L(SMD, dichloromethane)/6-311++G(2df, 2pd)//LanL2DZ levels, respectively. As summarized in Table 1, the computational λmax values for the three Co complexes are close to those reported in experiments,31–33 which demonstrates that the results of TD-DFT calculations should be reliable.
 | ||
| Scheme 18 The structures of representative Co complexes. | ||
| M06-L(SMD) | Experimental | |
|---|---|---|
| [CoIII(NH3)5Cl]2+ | 455.35 (2.72) | 474 (2.62) |
| 322.54 (3.84) | 338 (3.67) | |
| TpPh2CoII(2,6-dbp) | 1380.07 (0.90) | 1475 (0.84) |
| 810.68 (1.53) | 800 (1.55) | |
| 670.29 (1.85) | 660 (1.88) | |
| 586.77 (2.11) | 570 (2.18) | |
| (TF5PP)CoII | 542.92 (2.28) | 552 (2.25) |
| 527.16 (2.35) | 524 (2.37) |
We have calculated and compared the free energy ΔGtot and the ΔG50% (the entropy is cut by 50%) profiles of the favorable reaction pathway (pathway 5). As shown in Schemes 10 and 19, although a computational error does exist in the entropy calculation for a multimolecular reaction step (such as a SET process), the calculated results can still predict exactly that the SET process is the rate-determining step, which is in agreement with the KIE experiment. Therefore, we think that the calculated results can reliably explain the experimental results.
 | ||
| Scheme 19 Gibbs free energy ΔG50% for SET pathways 5 of CoIII-catalyzed C(sp2)–H alkoxylation (energy: kcal mol−1). | ||
Recently, Singleton et al. investigated the mechanism of alcohol-mediated Morita–Baylis–Hillman (MBH) reactions using the most popular DFT methods, i.e. B3LYP and M06-2X.34 They found that the calculated results by the two methods are remarkably different, and the rate-determining step cannot be predicted correctly by using the M06-2X method, so they concluded that the computations aid in interpreting observations but fail utterly as a replacement for experiment. In this work, similar computational tests to Singleton have been performed for the key C–H activation in the favorable reaction pathway (pathway 5) by using B3LYP and M06-L methods. As summarized in Table 2, the calculated results indicate that the free energy barriers ΔΔGtot, ΔΔG50%, and ΔΔGexplicit (calculated in the explicit solvents without implicit model) are close and the free energy barriers obtained by the different DFT methods are not significantly different, which is remarkably different from Singleton's work. As mentioned above, we believe that the computational errors in this system are not very significant and the calculated results using the M06-L method are consistent with experiment. In addition, multiple pathways involving explicit methanol for this kind of reaction35 are considered in both implicit and explicit models, but the H atom of HOMe returns automatically when we try to locate the possible structures of the transition states TS19, TS20, TS21, and intermediate INT27 (depicted in Scheme S1 of the ESI†), which is mainly because the basicity of OMe− is stronger than that of OAc−, so we did not further explore details of these pathways.
| Method | kcal mol−1 |
|---|---|
| a The transition state TS13explicit has firstly been located in the 10 Å box of the explicit solvents at the ONIOM(M06-L/6-31G(d, p)//LanL2DZ:UFF) level, it should be noted that additional five methanol molecules were putted into the high level, and all the other methanol molecules were putted into the low level. Then IRC calculation was performed to locate the corresponding intermediate INT21explicit. The single-point energies of the stationary points were refined at the higher ONIOM(M06-L/6-311++G(2df, 2pd)//SDD:UFF) level. | |
| M06-L(ΔΔGtot[TS13–INT21]) | 12.6 |
| M06-L(ΔΔG50%[TS13–INT21]) | 12.9 |
| M06-L(ΔΔG[TS13explicit–INT21explicit])a | 13.5 |
| B3LYP(ΔΔGtot[TS13B3LYP–INT21B3LYP]) | 8.0 |
| B3LYP(ΔΔG50%[TS13B3LYP–INT21B3LYP]) | 9.4 |
The M06-L method has been successfully used in the theoretical report on the mechanism of transition-metal catalyzed C–H oxidation,17 and the above computational tests also indicate that it should be suitable for investigating the systems including Co complexes.
Alcohol (1.2 mL) was added to a mixture of amide (0.15 mmol), Ag2O (69 mg, 0.3 mmol, 2 equiv.), NaOAc (12 mg, 0.15 mmol, 1 equiv.), Cp*Co(CO)I2 (14 mg, 20 mol%). Then, the reaction system was stirred at 70 °C for 12 h under an air atmosphere. After cooling to RT, the alcohol was evaporated under reduced pressure, and 2 N HCl (5 mL) was added to the residue. The mixture was extracted with CH2Cl2 and the organic layer was dried over anhydrous Na2SO4. Purification of the residue gave the product 3.
Acknowledgements
The work described in this paper was supported by the National Natural Science Foundation of China (No. 21303167), and the China Postdoctoral Science Foundation (No. 2013M530340).Notes and references
- For recent reviews, see: (a) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946 CrossRef CAS PubMed; (b) X. X. Guo, D. W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (c) S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (e) K. M. Engle and J. Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed; (f) K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597 CrossRef CAS PubMed; (g) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (h) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (i) B. J. Li and Z. J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (j) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (k) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (l) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (m) C. J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed.
- For selected reviews, see: X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed.
- For selected reviews, see: (a) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (b) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS; (c) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed.
- L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed.
- For selected reviews on the use of inexpensive first-row transition metal catalysts for C–H bond functionalization, see: (a) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412 CrossRef CAS PubMed; (b) J. Yamaguchi, K. Mito and K. Itami, Eur. J. Org. Chem., 2013, 19 CrossRef CAS PubMed; (c) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (d) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (e) Y. Nakao, Chem. Rec., 2011, 11, 242 CrossRef CAS PubMed; (f) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (g) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 CrossRef CAS PubMed; (h) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087 CAS.
- For reviews, see: (a) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (b) L. Ackermann, J. Org. Chem., 2014, 79, 8948 CrossRef CAS PubMed , For selected examples, see: ; (c) K. Gao, P. S. Lee, T. Fujita and N. Yoshikai, J. Am. Chem. Soc., 2010, 132, 12249 CrossRef CAS PubMed; (d) L. Ilies, Q. Chen, X. Zeng and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 5221 CrossRef CAS PubMed; (e) P. S. Lee, T. Fujita and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 17283 CrossRef CAS PubMed; (f) B. Li, Z.-H. Wu, Y.-F. Gu, C.-L. Sun, B.-Q. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 1109 CrossRef CAS PubMed; (g) Z. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 4698 CrossRef CAS PubMed; (h) W. Song and L. Ackermann, Angew. Chem., Int. Ed., 2012, 51, 8251 CrossRef CAS PubMed; (i) T. Andou, Y. Saga, H. Komai, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 3213 CrossRef CAS PubMed; (j) K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 9279 CrossRef CAS PubMed; (k) H. Lu, C. Li, H. Jiang, C. L. Lizardi and X. P. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7028 CrossRef CAS PubMed; (l) J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133 CrossRef CAS PubMed.
- For reviews, see: (a) N. Yoshikai, ChemCatChem, 2015, 7, 732 CrossRef CAS PubMed , For selected examples, see: ; (b) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207 CrossRef CAS PubMed; (c) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Chem.–Eur. J., 2013, 19, 9142 CrossRef CAS PubMed; (d) B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1381 CrossRef PubMed; (e) D. G. Yu, T. Gensch, F. de Azambuja, S. Vasquez-Cespedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS PubMed; (f) J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490 CrossRef CAS PubMed; (g) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3635 CrossRef CAS PubMed; (h) P. Patel and S. Chang, ACS Catal., 2015, 5, 853 CrossRef CAS; (i) D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508 CrossRef CAS PubMed.
- (a) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209 CrossRef CAS PubMed; (b) J. Y. Kim, S. H. Cho, J. Joseph and S. Chang, Angew. Chem., Int. Ed., 2010, 49, 9899 CrossRef CAS PubMed; (c) T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 775 CrossRef CAS PubMed; (d) T. M. Figg, S. Park, J. Park, S. Chang and D. G. Musaev, Organometallics, 2014, 33, 4076 CrossRef CAS.
- For selected reviews, see: (a) B. Liu and B.-F. Shi, Tetrahedron Lett., 2015, 56, 15 CrossRef CAS PubMed; (b) I. B. Krylov, V. A. Vil' and A. O. Terent'ev, Beilstein J. Org. Chem., 2015, 11, 92 CrossRef PubMed.
- (a) S. Gowrisankar, A. G. Sergeev, P. Anbarasan, A. Spannenberg, H. Neumann and M. Beller, J. Am. Chem. Soc., 2010, 132, 11592 CrossRef CAS PubMed; (b) P. Novak, A. Correa, J. Gallardo-Donaire and R. Martin, Angew. Chem., Int. Ed., 2011, 50, 12236 CrossRef CAS PubMed; (c) K. E. Torraca, X. Huang, C. A. Parrish and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 10770 CrossRef CAS.
- (a) L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141 CrossRef CAS PubMed; (b) W. Li and P. Sun, J. Org. Chem., 2012, 77, 8362 CrossRef CAS PubMed; (c) T. S. Jiang and G. W. Wang, J. Org. Chem., 2012, 77, 9504 CrossRef CAS PubMed.
- (a) S. Bhadra, C. Matheis, D. Katayev and L. J. Goossen, Angew. Chem., Int. Ed., 2013, 52, 9279 CrossRef CAS PubMed; (b) J. Roane and O. Daugulis, Org. Lett., 2013, 15, 5842 CrossRef CAS PubMed.
- L. B. Zhang, X. Q. Hao, S. K. Zhang, K. Liu, B. Ren, J. F. Gong, J. L. Niu and M. P. Song, J. Org. Chem., 2014, 79, 10399 CrossRef CAS PubMed.
- L. B. Zhang, X. Q. Hao, S. K. Zhang, Z. J. Liu, X. X. Zheng, J. F. Gong, J. L. Niu and M. P. Song, Angew. Chem., Int. Ed., 2015, 54, 272 CrossRef CAS PubMed.
- (a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (b) X. Li, Y. H. Liu, W. J. Gu, B. Li, F. J. Chen and B. F. Shi, Org. Lett., 2014, 16, 3904 CrossRef CAS PubMed; (c) B. Chen, X. L. Hou, Y. X. Li and Y. D. Wu, J. Am. Chem. Soc., 2011, 133, 7668 CrossRef CAS PubMed.
- X. Chen, X. S. Hao, C. E. Goodhue and J. Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed.
- A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed.
- (a) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3635 CrossRef CAS PubMed; (b) B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1491 CrossRef CAS PubMed; (c) T. Hyster, Catal. Lett., 2015, 145, 458 CrossRef CAS; (d) H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424 CrossRef CAS PubMed.
- J. Iqbal, B. Bhatia and N. K. Nayyar, Chem. Rev., 1994, 94, 519 CrossRef CAS.
- J. K. Kochi, R. T. Tang and T. Bernath, J. Am. Chem. Soc., 1973, 95, 7114 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- (a) Y. H. Lam, K. N. Houk, U. Scheffler and R. Mahrwald, J. Am. Chem. Soc., 2012, 134, 6286 CrossRef CAS PubMed; (b) Y. Qiao, K. Han and C. G. Zhan, Biochemistry, 2013, 52, 6467 CrossRef CAS PubMed; (c) Z. Li, D. Wei, Y. Wang, Y. Zhu and M. Tang, J. Org. Chem., 2014, 79, 3069 CrossRef CAS PubMed; (d) M. Zhang, D. Wei, Y. Wang, S. Li, J. Liu, Y. Zhu and M. Tang, Org. Biomol. Chem., 2014, 12, 6374 RSC; (e) Y. Li, Y. Zhu, W. Zhang, D. Wei, Y. Ran, Q. Zhao and M. Tang, Phys. Chem. Chem. Phys., 2014, 16, 20001 RSC; (f) Y. Qiao, K. Han and C. G. Zhan, Org. Biomol. Chem., 2014, 12, 2214 RSC; (g) Y. Wang, D. Wei, Z. Li, Y. Zhu and M. Tang, J. Phys. Chem. A, 2014, 118, 4288 CrossRef CAS PubMed.
- (a) T. Wang, Y. Liang and Z. X. Yu, J. Am. Chem. Soc., 2011, 133, 9343 CrossRef CAS PubMed; (b) E. Larionov, M. Nakanishi, D. Katayev, C. Besnard and E. P. Kundig, Chem. Sci., 2013, 4, 1995 RSC; (c) N. Quinones, A. Seoane, R. Garcia-Fandino, J. L. Mascarenas and M. Gulias, Chem. Sci., 2013, 4, 2874 RSC; (d) A. Varela-Alvarez and D. G. Musaev, Chem. Sci., 2013, 4, 3758 RSC; (e) S. Qu, Y. Dang, C. Song, M. Wen, K. W. Huang and Z. X. Wang, J. Am. Chem. Soc., 2014, 136, 4974 CrossRef CAS PubMed; (f) J. S. Cannon, L. F. Zou, P. Liu, Y. Lan, D. J. O'Leary, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 6733 CrossRef CAS PubMed; (g) Y. F. Yang, G. J. Cheng, P. Liu, D. Leow, T. Y. Sun, P. Chen, X. Zhang, J. Q. Yu, Y. D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344 CrossRef CAS PubMed; (h) G. J. Cheng, Y. F. Yang, P. Liu, P. Chen, T. Y. Sun, G. Li, X. H. Zhang, K. N. Houk, J. Q. Yu and Y. D. Wu, J. Am. Chem. Soc., 2014, 136, 894 CrossRef CAS PubMed; (i) Y. F. Dang, S. L. Qu, J. W. Nelson, H. D. Pham, Z. X. Wang and X. T. Wang, J. Am. Chem. Soc., 2015, 137, 2006 CrossRef CAS PubMed; (j) G. J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y. D. Wu, J. Am. Chem. Soc., 2015, 137, 1706 CrossRef CAS PubMed; (k) Y. Dang, S. Qu, Y. Tao, X. Deng and Z. X. Wang, J. Am. Chem. Soc., 2015, 137, 6279 CrossRef CAS PubMed; (l) F. Ye, S. Qu, L. Zhou, C. Peng, C. Wang, J. Cheng, M. L. Hossain, Y. Liu, Y. Zhang, Z. X. Wang and J. Wang, J. Am. Chem. Soc., 2015, 137, 4435 CrossRef CAS PubMed; (m) S. Z. Wu, R. Zeng, C. L. Fu, Y. H. Yu, X. Zhang and S. M. Ma, Chem. Sci., 2015, 6, 2275 RSC.
- (a) D. G. Xu, Y. S. Wei, J. B. Wu, D. Dunaway-Mariano, H. Guo, Q. Cui and J. L. Gao, J. Am. Chem. Soc., 2004, 126, 13649 CrossRef CAS PubMed; (b) S. S. Wu, D. G. Xu and H. Guo, J. Am. Chem. Soc., 2010, 132, 17986 CrossRef CAS PubMed; (c) Z. H. Ke, G. K. Smith, Y. K. Zhang and H. Guo, J. Am. Chem. Soc., 2011, 133, 11103 CrossRef CAS PubMed; (d) D. H. Wei, B. L. Lei, M. S. Tang and C. G. Zhan, J. Am. Chem. Soc., 2012, 134, 10436 CrossRef CAS PubMed; (e) D. M. Li, Y. Wang and K. L. Han, Coord. Chem. Rev., 2012, 256, 1137 CrossRef CAS PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS PubMed.
- P. Fuentealba, H. Preuss, H. Stoll and L. Vonszentpaly, Chem. Phys. Lett., 1982, 89, 418 CrossRef CAS.
- (a) A. Bakac, O. Pestovsky, B. L. Durfey and K. E. Kristian, Chem. Sci., 2013, 4, 2185 RSC; (b) E. A. Hull, A. C. West, O. Pestovsky, K. E. Kristian, A. Ellern, J. F. Dunne, J. M. Carraher, A. Bakac and T. L. Windus, Dalton Trans., 2015, 44, 3811 RSC.
- P. Kofod, Inorg. Chem., 1995, 34, 2768 CrossRef CAS.
- T. E. Machonkin, M. D. Boshart, J. A. Schofield, M. M. Rodriguez, K. Grubel, D. Rokhsana, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2014, 53, 9837 CrossRef CAS PubMed.
- K. M. Kadish, B. C. Han, M. M. Franzen and C. Araullo-McAdams, J. Am. Chem. Soc., 1990, 112, 8364 CrossRef CAS.
- R. E. Plata and D. A. Singleton, J. Am. Chem. Soc., 2015, 137, 3811 CrossRef CAS PubMed.
- A. Winter, Nat. Chem., 2015, 7, 473 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete Gaussian 09 reference, computational data, alternative methanol-assisted proton transfer pathways, the details of control experiments, EPR spectrum, characterization of products, NMR and mass spectra. See DOI: 10.1039/c5sc01807b |
| This journal is © The Royal Society of Chemistry 2015 |