
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
π-extended [12]cycloparaphenylenes: from a hexaphenylbenzene cyclohexamer to its unexpected C2-symmetric congener†
Florian E.
Golling
ab,
Silvio
Osella
c,
Martin
Quernheim
a,
Manfred
Wagner
a,
David
Beljonne
c and
Klaus
Müllen
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: muellen@mpip-mainz.mpg.de
bGraduate School Materials Science in Mainz, Staudinger Weg 9, 55128 Mainz, Germany
cChimie des Matériaux Nouveaux & Centre d'Innovation et de Recherche en Matériaux Polymères, Université de Mons-UMONS/Materia Nova, Place du Parc 20, 7000 Mons, Belgium
First published on 8th September 2015
Abstract
The synthesis of π-extended [12]cycloparaphenylene (CPP) derivatives from a kinked triangular macrocycle is presented. Depending on the reaction conditions for reductive aromatization, either a hexaphenylbenzene cyclohexamer or its C2-symmetric congener was obtained. Their structures were confirmed by NMR spectroscopy or X-ray crystallographic analysis. With the support of DFT calculations, a mechanistic explanation for the unexpected formation of the oval shaped bis(cyclohexadiene)-bridged C2-symmetric macrocycle is provided. The here employed congested hexaphenylbenzene mode of connectivity in conjunction with a non-strained precursor improves oxidative cyclodehydrogenation toward the formation of ultrashort carbon nanotubes (CNT)s. Thus, this strategy can pave the way for new conceptual approaches of a solution-based bottom-up synthesis of CNTs.
Introduction
Carbon nanotubes (CNT)s have gained a great deal of attention over the last decades due to their attractive physical and electronic properties.1 Special emphasis has been put on the controlled synthesis of CNTs, since their electronic features sensitively depend on size and shape (edge structure, chirality).2 Cycloparaphenylenes (CPP)s are envisioned as precursors for a concise bottom-up synthesis of carbon nanobelts, i.e., ultrashort CNTs.3,4 On surfaces, CPPs,5 besides other templates,6 have been successfully used for the growth of diameter-defined CNTs, whereas the advancement in solution-based approaches continues to be limited.7To reach this goal, several π-extended CPPs8 and structurally closely related derivatives9 have been synthesized over the last years. However, a necessary degree of axial π-extension of these structures, from a CPP to a nanobelt precursor (Fig. 1), was not accomplished to enable a solution-based bottom-up synthesis. In contrast, [9]cyclo-1,4-naphthylene10 and a [10]CPP dimer,11 are sufficiently π-extended and can thus be regarded as carbon nanobelt precursors, but dehydrogenation of the products was not reported.
 | ||
| Fig. 1 Schematic modes of π-extension: CPP (left), insufficiently π-extended CPP (second left), fully π-extended CPP to give a nanobelt precursor (second right), and CNT (right). | ||
Previously, we attempted a π-extension of CPPs using hexaphenylbenzene (HPB) as the general building block, which can be converted to hexa-peri-hexabenzocoronene (HBC) via oxidative cyclodehydrogenation. This concept has been widely employed for the bottom-up synthesis of 1D- and 2D-structures, such as graphene nanoribbons (GNR)s12 and nanographenes.13 To obtain π-lengthened CPPs (3D), HPB was embedded into a macrocycle to give a cyclic hexaphenylbenzene trimer ([3]CHPB) and its corresponding larger homologues.14 A further addition of phenyl rings afforded polyphenylene cylinders (PPC)s, such as 1, following the parallelogram motif (frame A, Fig. 2).15 A complete cyclodehydrogenation could not be achieved. Thus, the macrocycles have been redesigned with a different connectivity, which is entirely based on the congested HPB motif (frame B, Fig. 2), where one bridging-phenylene moiety is shared by two hexaphenylbenzenes. This structural motif leads to a lower degree of rotational freedom of the attached phenyl groups, which is favorable for the desired cyclodehydrogenation.
 | ||
| Fig. 2 Two different modes of connectivity. Previous work (left): CNT synthesis via the parallelogram motif A (left) using [9]PPC 1 and [15]PPC (not shown); in this work (right): CNTs were approached via the congested HPB motif B. | ||
Herein, we present the synthesis of PPCs that entirely consist of congested hexaphenylbenzene moieties. A congested cyclic hexaphenylbenzene hexamer ([6]CCHPB) 2 and its oval-shaped C2-symmetric congener 3 were synthesized via Suzuki coupling and subsequent reductive aromatization. The oxidative cyclodehydrogenation of both structures was investigated and a hitherto unprecedented degree of cyclodehydrogenation for PPC systems was achieved.
Results and discussion
For the synthesis of the new macrocycles [6]CCHPB 2 and 3 (Scheme 1), we applied a Suzuki-mediated cyclization introduced by Bertozzi and Jasti3a together with a strategy, developed in our group, to convert structurally related, linear polyphenylene (PP) precursors into GNRs,12b,16 using oxidative cyclodehydrogenation.17 Therefore, the para-HPB diboronate, employed for linear PPs, was exchanged with the kinked diboronate 4. Two different precursors, 6 and 7, were then foreseen to give the triangular macrocycle 8, as the kink angles of the three V-shaped units in 6 and 7 add up to ∼210°.14 Tetraphenyl and tetrakis(4-t-butylphenyl) were introduced in an alternating fashion to provide solubility and render cyclodehydrogenation possible.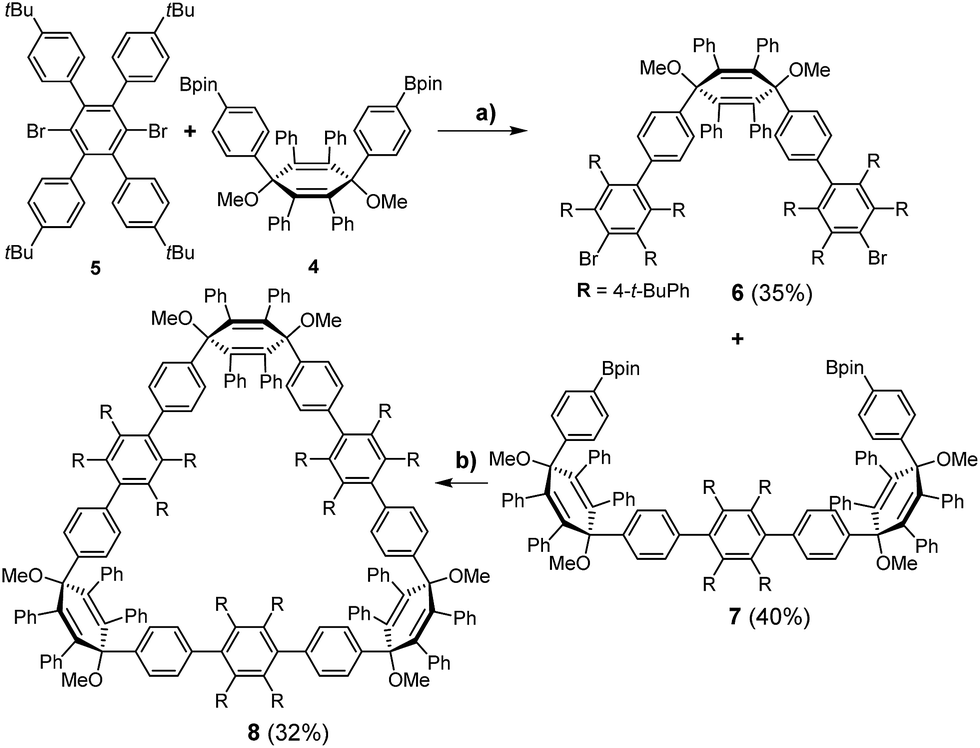 | ||
| Scheme 1 Synthesis of triangular macrocycle 8. (a) Pd(PPh3)4, Cs2CO3, 100 °C, 2 d, (toluene/H2O); (b) Pd(PPh3)4, Cs2CO3, 100 °C, 2 d, (toluene/H2O). | ||
As shown in Scheme 1, the unique “di-kinked” diboronate 7 and its “mono-kinked” counterpart 6 are synthesized via Suzuki coupling. For each approach, a fourfold excess of either diboronate 4 or dibromide 5 was used. Dibromide 6 and diboronate 7 could be obtained in yields of 35%, and 40% respectively. The macrocyclization of these two building blocks was achieved by Suzuki coupling in diluted media (0.1 mmol) to give 8 in 32% yield; side products such as higher linear homologues could be separated by recycling GPC.
An initial demethoxylation of 8 was achieved under reductive conditions that had previously been applied in our group: low-valent titanium was generated in situ and reacted with the triangular macrocycle 8 to give 2 in 46% (Scheme 2). The applied conditions are very effective at inducing reductive aromatization, but require elevated temperatures (80 °C). To unambiguously characterize cyclohexamer 2, 2D NMR experiments (Section 4.1, ESI†) were carried out. Single crystals of 2 were obtained after slow diffusion of MeCN into DCM. However, the obtained crystals did not yield sufficient reflections to resolve the structure.
 | ||
| Scheme 2 Synthesis of the [6]CCHPB 2. (a) TiCl4, LiAlH4, 80 °C, 3 d (THF). | ||
Therefore, we performed density functional theory (DFT) calculations of 2 with the B3LYP hybrid functional18 and the 6-31G(d) Pople basis set.19 The fully optimized geometry revealed a torsional angle between neighboring phenylene units of about 90° (Fig. 3, right), in an alternating fashion within the CPP ring. Interestingly, in comparison to PPC 1 (Fig. 2, left), which is based on an odd membered CPP ring with an ellipsoidal structure, cyclohexamer 2 exhibits a circular-shaped structure.
 | ||
| Fig. 3 Optimized geometry of 2 top (left) and side (right) view, as calculated at the B3LYP/6-31G(d) level of theory. Hydrogen atoms are omitted for clarity. | ||
In the second approach, a milder reduction of compound 8 with sodium naphthalenide (NaNp) (−78 °C vs. 80 °C) was attempted (Scheme 3). To our surprise, we did not obtain the expected [6]CCHPB 2, but rather a macrocycle containing four additional hydrogen atoms (Scheme 3). This was first indicated by the blue color of the reaction solution, hinting at the formation of anionic species, which can stem from an overreduction of the cyclohexadiene moieties, as reported in the literature.3q Therefore, the reaction was quenched with a proton source (MeOH) to trap the charged intermediate, while the use of iodine to quench the reaction or lowering the temperature did not afford any characterizable product. By mass spectrometry, we inferred that the obtained structure possesses four additional hydrogen atoms in comparison to [6]CCHPB 2. We expected that the reductive aromatization had not overcome ring strain at temperatures of −78 °C, but rather came to a halt after in situ formation of a tetraanionic species, which was then preserved by protonation. For this structure, only one of the three 3,6-dimethoxycyclohexadienes was reduced, when applying the above described conditions to give one phenylene moiety instead of the expected three, as shown in Scheme 3. Astonishingly, single crystal X-ray crystallography (Fig. 5) revealed the formation of 3 instead of the expected structure 9.
 | ||
| Scheme 3 Proposed reaction mechanism for the synthesis of 3. (a) NaNp, 1 h, −78 °C (THF); (b) −78 °C, H+. | ||
The formation of 3 can be explained by taking into account the mechanistic details of the aromatization: a stepwise two-electron reduction is proposed in the literature,3a forming a radical under loss of one methoxy group. After the second one-electron transfer, a monoanion is formed, which leads to an expulsion of the remaining methoxy group under concomitant aromatization, affording a phenylene moiety from the previous 3,6-dimethoxycyclohexadiene. Based on the obtained product 3, we propose a mechanism via a tetraanionic species with subsequent charge migration from the 3,6-bridging cyclohexadiene moiety to the tetrakis(t-butylphenyl)-substituted phenylene ring (Scheme 3), compound 10c. In detail, the reductive aromatization follows the above mechanism for one of the three cyclohexadiene moieties. Reduction of the second and third cyclohexadiene units does not yield the neutral hydrocarbon 2 but proceeds instead to tetraanion 10c (Scheme 3). Migration of the anionic charges in intermediate 10a and 10b, as shown by arrows in Scheme 3, gives the thermodynamically more favorable C2-symmetric tetraanion 10c.20 Protonation “freezes” the anionically charged structure, leading to the oval-shaped compound 3.
To support this mechanistic hypothesis, we performed DFT calculations for the different macrocycles shown in Fig. 4. Interestingly, the tetraanion 10 was not found as a stable intermediate, but rather spontaneously evolved into the C2-symmetric tetraanion 10c. On the other hand, the neutral counterpart of the anionic structures (namely 8a, 9, and 3; for all structures the methoxy groups are substituted by hydrogen atoms to reduce computational cost) were obtained as stable structures. To account for the observed charge migration, we compared the relative strain energies of 3, 8a, and 9 (Fig. 4). The calculations reveal an increase of strain energy by ∼24.3 kcal mol−1 when going from 8a to 9. This originates from the presence of one additional phenylene moiety obtained after reductive aromatization of 8a; hence a strain relief toward the starting material 8a is not feasible. On the other hand, a thermodynamically more favorable geometry with a reduced relative strain energy of ∼2.5 kcal mol−1 can be reached through a charge migration along one phenylene unit from 10a over 10b to its protonated form 3, as depicted in Scheme 3.
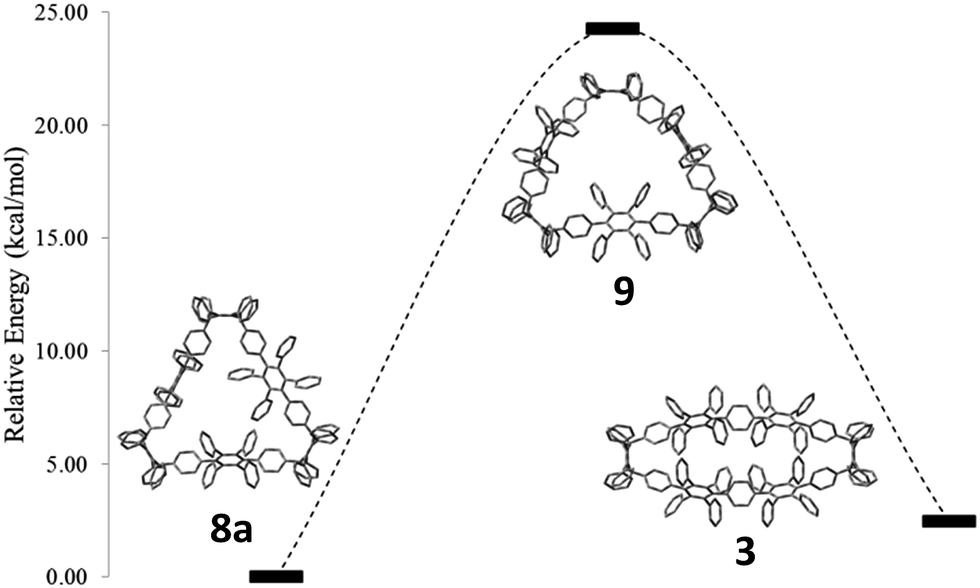 | ||
| Fig. 4 Comparison of strain energies of 8a, 9, and 3, using DFT calculations at the B3LYP/6-31G(d) level of theory. For all structures, methoxy groups are substituted by hydrogen atoms. Hydrogen atoms are omitted for clarity. | ||
To support the postulated resonance form of the tetraanionic intermediate 3a, the size of the holes of macrocycle 3 (neutral structure) and 10c (tetraanionic intermediate) were studied. The computed geometry optimization suggests an increase of the oval opening from 8.2 Å to 12.2 Å, respectively (opening of the neutral structure 9 is 13 Å), which is shown in Fig. 5. Thus, the equilibrium structure of the tetraanionic intermediate 10c is shifted toward 3 in comparison to 9. To understand whether the strain release is accompanied by a redistribution of charge over the entire aromatic system, a charge analysis for 11 (2e−) and 10c (4e−) was performed: the dianionic structure 11 is considered as a starting point and shows that the charges are mainly localized over the cyclohexadiene moieties where the hydrogen atoms have been removed. For 10c, the calculations reveal that the main part of the charges (∼2e−) is strongly localized over the cyclohexadiene moieties of the macrocycle, while the remaining partial charges (∼2e−) are delocalized over the substituted phenyl rings of the belt (Fig. S16, ESI†), leading to an oval shaped macrocycle. These findings additionally corroborate the postulated form of the tetraanionic intermediate 10c in opposition to 10a. The proposed charge migration based on DFT calculations is also supported for a dianionic species after formation of one phenylene moiety (Fig. S10 and S15, ESI†). An over-reduction of [6]CCHPB 2 to give the tetraanion 10c, which can be trapped by a proton source, has not been observed experimentally.
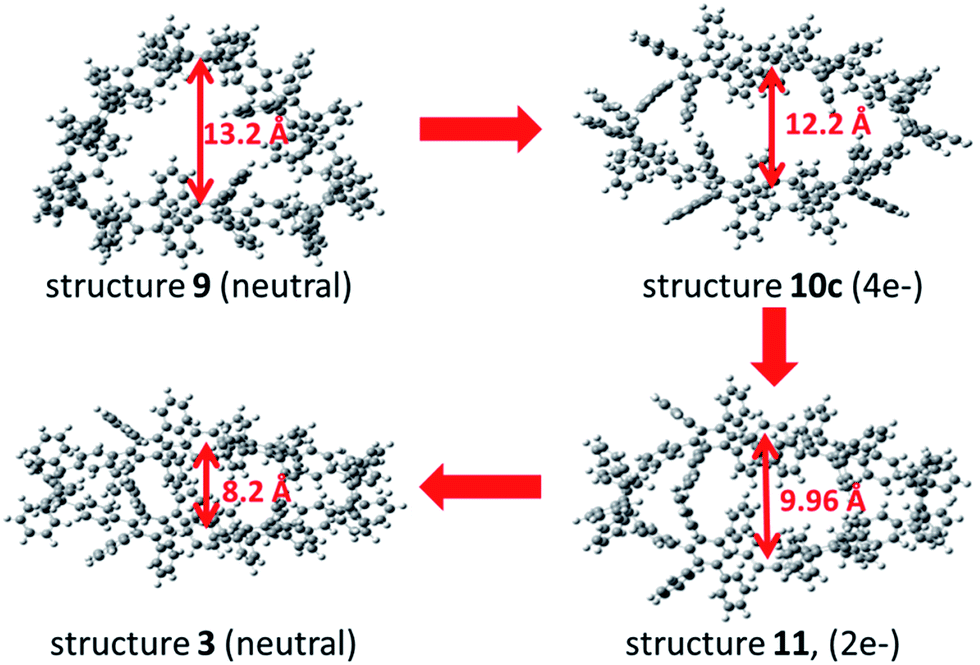 | ||
| Fig. 5 Geometric analysis of structures 9, 10c, 11 and 3 to understand the equilibrium structure of tetraanion 10c, using DFT calculations at the B3LYP/6-31G(d) level of theory. Structure 11 is twice negatively charged at one cyclohexadiene moiety, whereas the other contains two hydrogen atoms, analogously to the other structures. | ||
Single crystals of 3 were obtained after slow vapor diffusion of MeCN into a solution of DCM. The crystal structure confirmed the oval-shaped nature (Fig. 6) of 3. Pentaphenylene units (labeled in red) are bridged by two 3,6-substituted cyclohexa-1,4-dienes (labeled in blue). The pentaphenylene moieties are slightly bent (Fig. 6b) as the sp3-carbons cannot be fully torn out of plane due to angle constraints (Fig. 3, ESI†). Additionally, the crystal structure reveals the inherent design pattern: tetraphenyl and tetrakis-(4-t-butylphenyl) substituents are attached to the central ring in an alternating fashion.
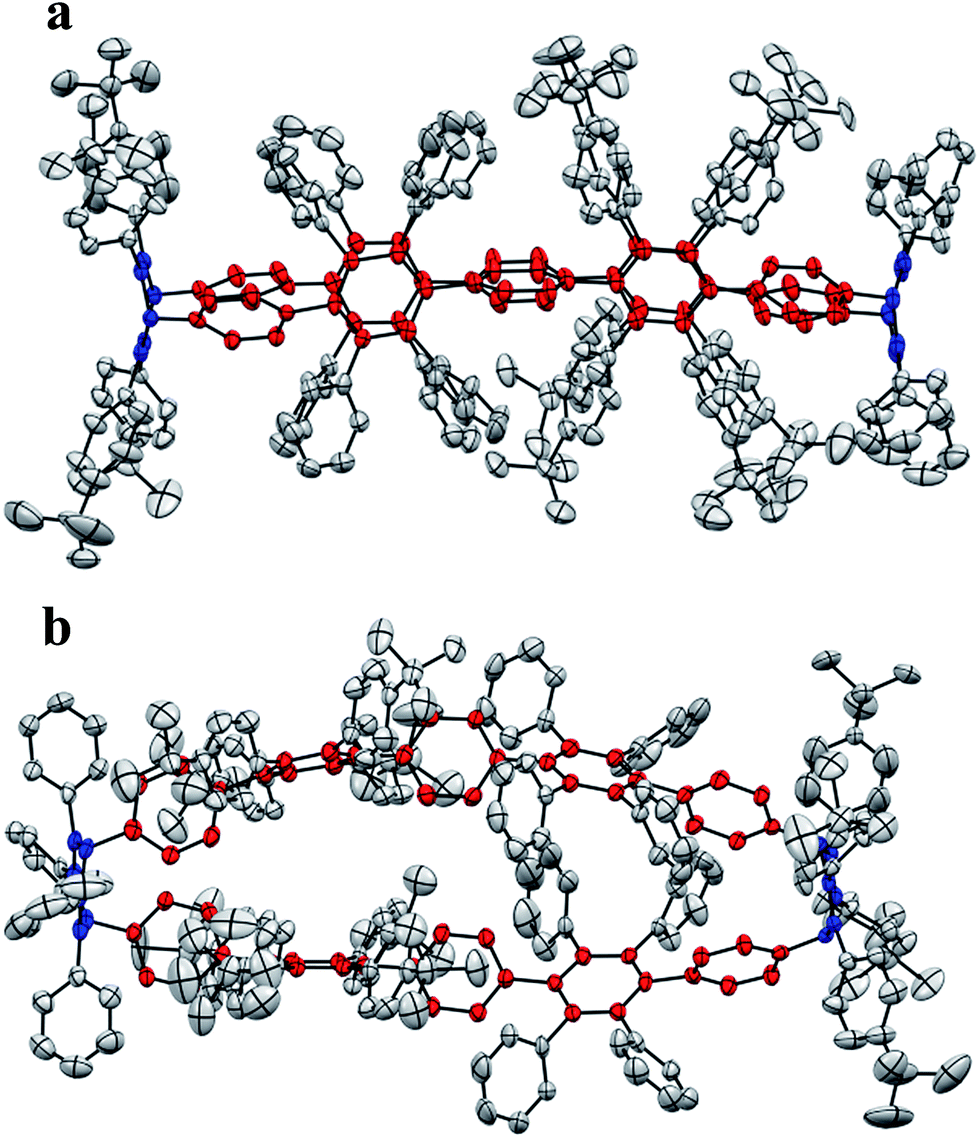 | ||
| Fig. 6 Crystal structure of 3. ORTEP drawing at 50% probability level. All hydrogen atoms and solvent molecules are omitted for clarity. Phenylene moieties are labelled in red. 3,6-Bridging cyclohexa-1,4-dienes are labelled in blue. Front view (a) and top view (b). | ||
As compounds 2 and 3 were designed in order to serve as direct precursors for a solution-based bottom-up synthesis of CNTs, we subjected both molecules to oxidative cyclodehydrogenation conditions using FeCl3 at room temperature.15 For 2, a loss of 20 hydrogen atoms of a possible 96 was observed by MALDI-MS, at which the cyclodehydrogenation came to a halt, despite prolonged reactions times. In contrast to our previous results, here we could, for the first time, determine the degree of cyclodehydrogenation due to isotopically resolved mass peaks. For the barely strained C2-symmetric congener 3, of the 96 hydrogen atoms to obtain a CNT a loss of 60 could be observed (see Scheme S3,† p. S92). The detailed analysis of the mass spectra hints at the formation of ribbon-like sidewall structures (Section 7.2, ESI†), rendering cyclodehydrogenation of the pentaphenylene sidewalls reasonable. However, since the structural analysis is based on mass spectrometry, well known side reactions for Scholl reactions, such as rearrangements, cannot be ruled out. Prolonged reaction times led to chloro-de-t-butylations; oxidative aromatization of the cyclohexadiene moieties was not observed, even with other reagents at elevated temperature (DDQ, 120 °C).
Conclusions
In summary, we presented the bottom-up synthesis of a [12]CPP based nanobelt precursor [6]CCHPB 2 and its unexpected weakly-strained C2-symmetric congener 3. Their structural features are based on congested hexaphenylbenzene units, i.e., two hexaphenylbenzenes are bridged by one shared phenylene unit. The surprising synthesis of 3 occurred due to the use of the milder reducing agent NaNp, whereas low valent titanium was applied for the synthesis of the congested HPB 2. We assume that two different products were obtained because of the high difference of thermal energy (ΔT 160 °C), giving rise to a kinetic control of the reaction. The mechanistic explanation for the astonishing charge migration during reductive aromatization of 3 was confirmed by DFT calculations. The inherent congested HPB structural feature leads to reduced rotational freedom for both macrocycles in comparison to the previously prepared PPCs such as 1. Finally, we attempted oxidative cyclodehydrogenation which resulted in a loss of 20 H and 60 H for 2 and 3, respectively. The higher loss for 3 may be assigned to the barely strained pentaphenylenes. The above discussed findings suggest that the oxidative cyclodehydrogenation runs more smoothly if (a) the congested HPB structural motif in comparison to the parallelogram approach is applied, as this mode of connectivity restricts rotation of the π-extended macrocycle, thus avoiding mismatched structural arrangements possible for 1 (parallelogram vs. trapezoid); and (b) a minimum of strain is present in the cyclic system, observed for 3 in comparison to 2. Thus, the synthesis of such non-strained precursors which can be pre-cyclodehydrogenated close to the target structure and are then being converted to strained geometries could open up new concepts for a concise bottom-up synthesis of carbon nanobelts.Acknowledgements
We would like to acknowledge funding from DFG project MU 334/36-1. We thank Dr Schollmeyer (University of Mainz) for X-ray crystal structure analysis. F. E. G. is a recipient of a fellowship through the Excellence Initiative (DFG/GSC 266). F. E. G. thanks A. N. Skabeev and J. W. Krumpfer for helpful discussions. This work was supported by the European Commission through the Marie Curie projects GENIUS (PITN-2010-GA-264694). The work in Mons is partly supported by the Belgian National Fund for Scientific Research (FNRS), the FNRS/FRFC ‘PHOTOGRAPH’ PDR project and the Belgian Science Policy Office (IAP 7/0505). D. B. is a FNRS Research Director.References
- (a) M. S. Dresselhaus, G. Dresselhaus, J. C. Charlier and E. Hernández, Philos. Trans. R. Soc. London, 2004, 362, 2065–2098 CrossRef CAS PubMed; (b) D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC.
- S. Louie, in Carbon Nanotubes, ed. M. Dresselhaus, G. Dresselhaus and P. Avouris, Springer Berlin Heidelberg, 2001, vol. 80, ch. 6, pp. 113–145 Search PubMed.
- (a) R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed; (b) H. Takaba, H. Omachi, Y. Yamamoto, J. Bouffard and K. Itami, Angew. Chem., Int. Ed., 2009, 48, 6112–6116 CrossRef CAS PubMed; (c) H. Omachi, S. Matsuura, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2010, 49, 10202–10205 CrossRef CAS PubMed; (d) S. Yamago, Y. Watanabe and T. Iwamoto, Angew. Chem., Int. Ed., 2010, 49, 757–759 CrossRef CAS PubMed; (e) T. Iwamoto, Y. Watanabe, Y. Sakamoto, T. Suzuki and S. Yamago, J. Am. Chem. Soc., 2011, 133, 8354–8361 CrossRef CAS PubMed; (f) T. J. Sisto, M. R. Golder, E. S. Hirst and R. Jasti, J. Am. Chem. Soc., 2011, 133, 15800–15802 CrossRef CAS PubMed; (g) Y. Segawa, P. Senel, S. Matsuura, H. Omachi and K. Itami, Chem. Lett., 2011, 40, 423–425 CrossRef CAS; (h) Y. Segawa, S. Miyamoto, H. Omachi, S. Matsuura, P. Šenel, T. Sasamori, N. Tokitoh and K. Itami, Angew. Chem., Int. Ed., 2011, 50, 3244–3248 CrossRef CAS PubMed; (i) E. Kayahara, Y. Sakamoto, T. Suzuki and S. Yamago, Org. Lett., 2012, 14, 3284–3287 CrossRef CAS PubMed; (j) J. Xia and R. Jasti, Angew. Chem., Int. Ed., 2012, 51, 2474–2476 CrossRef CAS PubMed; (k) J. Xia, J. W. Bacon and R. Jasti, Chem. Sci., 2012, 3, 3018–3021 RSC; (l) E. R. Darzi, T. J. Sisto and R. Jasti, J. Org. Chem., 2012, 77, 6624–6628 CrossRef CAS PubMed; (m) H. Isobe, S. Hitosugi, T. Yamasaki and R. Iizuka, Chem. Sci., 2013, 4, 1293–1297 RSC; (n) E. Kayahara, T. Iwamoto, T. Suzuki and S. Yamago, Chem. Lett., 2013, 42, 621–623 CrossRef CAS; (o) A.-F. Tran-Van, E. Huxol, J. M. Basler, M. Neuburger, J.-J. Adjizian, C. P. Ewels and H. A. Wegner, Org. Lett., 2014, 16, 1594–1597 CrossRef CAS PubMed; (p) E. Kayahara, V. K. Patel and S. Yamago, J. Am. Chem. Soc., 2014, 136, 2284–2287 CrossRef CAS PubMed; (q) P. J. Evans, E. R. Darzi and R. Jasti, Nat. Chem., 2014, 6, 404–408 CrossRef CAS PubMed; (r) N. Kubota, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 1356–1361 CrossRef CAS PubMed; (s) H. Ueno, T. Nishihara, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 3707–3711 CrossRef CAS PubMed; (t) H. Ito, Y. Mitamura, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 159–163 CrossRef CAS PubMed; (u) V. K. Patel, E. Kayahara and S. Yamago, Chem.–Eur. J., 2015, 21, 5742–5749 CrossRef CAS PubMed.
- (a) Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed; (b) V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS PubMed; (c) R. Jasti and C. R. Bertozzi, Chem. Phys. Lett., 2010, 494, 1–7 CrossRef CAS PubMed; (d) H. Omachi, Y. Segawa and K. Itami, Acc. Chem. Res., 2012, 45, 1378–1389 CrossRef CAS PubMed; (e) U. H. F. Bunz, S. Menning and N. Martín, Angew. Chem., Int. Ed., 2012, 51, 7094–7101 CrossRef CAS PubMed; (f) S. Yamago, E. Kayahara and T. Iwamoto, Chem. Rec., 2014, 14, 84–100 CrossRef CAS PubMed; (g) T. Nishiuchi and M. Iyoda, Chem. Rec., 2015, 15, 329–346 CrossRef CAS PubMed; (h) S. E. Lewis, Chem. Soc. Rev., 2015, 44, 2221–2304 RSC.
- H. Omachi, T. Nakayama, E. Takahashi, Y. Segawa and K. Itami, Nat. Chem., 2013, 5, 572–576 CrossRef CAS PubMed.
- (a) X. Yu, J. Zhang, W. Choi, J.-Y. Choi, J. M. Kim, L. Gan and Z. Liu, Nano Lett., 2010, 10, 3343–3349 CrossRef CAS PubMed; (b) J. R. Sanchez-Valencia, T. Dienel, O. Groning, I. Shorubalko, A. Mueller, M. Jansen, K. Amsharov, P. Ruffieux and R. Fasel, Nature, 2014, 512, 61–64 CrossRef CAS PubMed.
- (a) E. H. Fort, P. M. Donovan and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 16006–16007 CrossRef CAS PubMed; (b) M. A. Petrukhina and L. T. Scott, Fragments of Fullerenes and Carbon Nanotubes, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011 Search PubMed; (c) L. T. Scott, E. A. Jackson, Q. Zhang, B. D. Steinberg, M. Bancu and B. Li, J. Am. Chem. Soc., 2012, 134, 107–110 CrossRef CAS PubMed.
- (a) H. Omachi, Y. Segawa and K. Itami, Org. Lett., 2011, 13, 2480–2483 CrossRef CAS PubMed; (b) T. J. Sisto, X. Tian and R. Jasti, J. Org. Chem., 2012, 77, 5857–5860 CrossRef CAS PubMed; (c) J. Xia, M. R. Golder, M. E. Foster, B. M. Wong and R. Jasti, J. Am. Chem. Soc., 2012, 134, 19709–19715 CrossRef CAS PubMed; (d) T. Iwamoto, E. Kayahara, N. Yasuda, T. Suzuki and S. Yamago, Angew. Chem., Int. Ed., 2014, 53, 6430–6434 CrossRef CAS PubMed; (e) C. Huang, Y. Huang, N. G. Akhmedov, B. V. Popp, J. L. Petersen and K. K. Wang, Org. Lett., 2014, 16, 2672–2675 CrossRef CAS PubMed; (f) A. Yagi, G. Venkataramana, Y. Segawa and K. Itami, Chem. Commun., 2014, 50, 957–959 RSC.
- (a) Z. He, X. Xu, X. Zheng, T. Ming and Q. Miao, Chem. Sci., 2013, 4, 4525–4531 RSC; (b) S. Hitosugi, T. Yamasaki and H. Isobe, J. Am. Chem. Soc., 2012, 134, 12442–12445 CrossRef CAS PubMed; (c) S. Hitosugi, W. Nakanishi, T. Yamasaki and H. Isobe, Nat. Commun., 2011, 2, 492 CrossRef PubMed; (d) T. Matsuno, S. Kamata, S. Hitosugi and H. Isobe, Chem. Sci., 2013, 4, 3179–3183 RSC; (e) K. Matsui, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2014, 136, 16452–16458 CrossRef CAS PubMed; (f) K. Matsui, Y. Segawa, T. Namikawa, K. Kamada and K. Itami, Chem. Sci., 2013, 4, 84–88 RSC; (g) E. Kayahara, T. Iwamoto, H. Takaya, T. Suzuki, M. Fujitsuka, T. Majima, N. Yasuda, N. Matsuyama, S. Seki and S. Yamago, Nat. Commun., 2013, 4, 2694 Search PubMed.
- A. Yagi, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2012, 134, 2962–2965 CrossRef CAS PubMed.
- Y. Ishii, S. Matsuura, Y. Segawa and K. Itami, Org. Lett., 2014, 16, 2174–2176 CrossRef CAS PubMed.
- (a) V. S. Iyer, K. Yoshimura, V. Enkelmann, R. Epsch, J. P. Rabe and K. Müllen, Angew. Chem., Int. Ed., 1998, 37, 2696–2699 CrossRef CAS; (b) X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2008, 130, 4216–4217 CrossRef CAS PubMed; (c) Y. Fogel, L. Zhi, A. Rouhanipour, D. Andrienko, H. J. Räder and K. Müllen, Macromolecules, 2009, 42, 6878–6884 CrossRef CAS; (d) L. Dossel, L. Gherghel, X. L. Feng and K. Mullen, Angew. Chem., Int. Ed., 2011, 50, 2540–2543 CrossRef PubMed; (e) M. G. Schwab, A. Narita, Y. Hernandez, T. Balandina, K. S. Mali, S. De Feyter, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 18169–18172 CrossRef CAS PubMed; (f) T. H. Vo, M. Shekhirev, D. A. Kunkel, M. D. Morton, E. Berglund, L. Kong, P. M. Wilson, P. A. Dowben, A. Enders and A. Sinitskii, Nat. Commun., 2014, 5, 3189 Search PubMed; (g) A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunaga, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430–1437 CrossRef CAS PubMed; (h) A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. R. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. De Feyter and K. Müllen, Nat. Chem., 2014, 6, 126–132 CrossRef CAS PubMed; (i) K. Ozaki, K. Kawasumi, M. Shibata, H. Ito and K. Itami, Nat. Commun., 2015, 6, 6251 CrossRef CAS PubMed.
- (a) V. S. Iyer, M. Wehmeier, J. D. Brand, M. A. Keegstra and K. Müllen, Angew. Chem., Int. Ed., 1997, 36, 1604–1607 CrossRef CAS PubMed; (b) S. M. Draper, D. J. Gregg and R. Madathil, J. Am. Chem. Soc., 2002, 124, 3486–3487 CrossRef CAS PubMed; (c) M. Kastler, J. Schmidt, W. Pisula, D. Sebastiani and K. Müllen, J. Am. Chem. Soc., 2006, 128, 9526–9534 CrossRef CAS PubMed; (d) M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5524–5527 CrossRef CAS PubMed; (e) X. Feng, V. Marcon, W. Pisula, M. R. Hansen, J. Kirkpatrick, F. Grozema, D. Andrienko, K. Kremer and K. Mullen, Nat. Mater., 2009, 8, 421–426 CrossRef CAS PubMed; (f) A. C. Whalley, K. N. Plunkett, A. A. Gorodetsky, C. L. Schenck, C.-Y. Chiu, M. L. Steigerwald and C. Nuckolls, Chem. Sci., 2011, 2, 132–135 RSC; (g) K. N. Plunkett, K. Godula, C. Nuckolls, N. Tremblay, A. C. Whalley and S. Xiao, Org. Lett., 2009, 11, 2225–2228 CrossRef CAS PubMed; (h) C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206–12210 CrossRef CAS PubMed; (i) Y. Tokudome, T. Fukushima, A. Goto and H. Kaji, Org. Electron., 2011, 12, 1600–1605 CrossRef CAS PubMed; (j) M. Takase, T. Narita, W. Fujita, M. S. Asano, T. Nishinaga, H. Benten, K. Yoza and K. Müllen, J. Am. Chem. Soc., 2013, 135, 8031–8040 CrossRef CAS PubMed; (k) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed; (l) H. Arslan, F. J. Uribe-Romo, B. J. Smith and W. R. Dichtel, Chem. Sci., 2013, 4, 3973–3978 RSC.
- T. Nishiuchi, X. Feng, V. Enkelmann, M. Wagner and K. Müllen, Chem.–Eur. J., 2012, 18, 16621–16625 CrossRef CAS PubMed.
- (a) F. E. Golling, M. Quernheim, M. Wagner, T. Nishiuchi and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 1525–1528 CrossRef CAS PubMed; (b) M. Quernheim, F. E. Golling, W. Zhang, M. Wagner, H.-J. Räder, T. Nishiuchi and K. Müllen, Angew. Chem., Int. Ed., 2015, 54, 10341–10346 CrossRef CAS PubMed.
- (a) M. Müller, V. S. Iyer, C. Kübel, V. Enkelmann and K. Müllen, Angew. Chem., 1997, 109, 1679–1682 CrossRef PubMed; (b) M. Müller, C. Kübel, F. Morgenroth, V. S. Iyer and K. Müllen, Carbon, 1998, 36, 827–831 CrossRef; (c) M. Müller, V. S. Iyer, C. Kübel, V. Enkelmann and K. Müllen, Angew. Chem., Int. Ed., 1997, 36, 1607–1610 CrossRef PubMed.
- (a) A. A. O. Sarhan and C. Bolm, Chem. Soc. Rev., 2009, 38, 2730–2744 RSC; (b) M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed; (c) M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., 2013, 125, 10084–10115 CrossRef PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS PubMed.
- (a) A. Sekiguchi, K. Ebata, C. Kabuto and H. Sakurai, J. Am. Chem. Soc., 1991, 113, 1464–1465 CrossRef CAS; (b) A. Falceto, D. Casanova, P. Alemany and S. Alvarez, Chem.–Eur. J., 2014, 20, 14674–14689 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, crystallographic data for 3 and 5, 1H NMR, 13C NMR, 2D NMR, MALDI-TOF MS, as well as computational details. CCDC 10332671033268. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02547h |
| This journal is © The Royal Society of Chemistry 2015 |