
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilization of H3+ in the high pressure crystalline structure of HnCl (n = 2–7)
Ziwei
Wang
a,
Hui
Wang
*a,
John S.
Tse
*abc,
Toshiaki
Iitaka
d and
Yanming
Ma
ac
aState Key Laboratory of Superhard Materials, Jilin University, Changchun 130012, China. E-mail: huiwang@jlu.edu.cn
bDepartment of Physics and Engineering Physics, University of Saskatchewan, Saskatoon, S7N 5B2, Canada. E-mail: john.tse@usask.ca
cBeijing Computational Science Research Center, Beijing 10084, China
dComputational Astrophysics Laboratory, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 21st October 2014
Abstract
The particle-swarm optimization method has been used to predict the stable high pressure structures up to 300 GPa of hydrogen-rich group 17 chlorine (HnCl, n = 2–7) compounds. In comparison to the group 1 and 2 hydrides, the structural modification associated with increasing pressure and hydrogen concentration is much less dramatic. The polymeric HCl chains already present in the low temperature phase under ambient pressure persist in all the high pressure structures. No transfer of electrons from the chlorine atoms into the interstitial sites is found. This indicates the chemical bonding at high pressure in group 17 elements is fundamentally different from the alkali and alkaline elements. It is found that almost perfectly triangular H3+ ions can be stabilized in the crystalline structure of H5Cl.
Introduction
The suggestion that the hydrides of main group elements at high pressure may be superconductors with high critical temperature has stimulated recent interest in the search for the possible existence of hydrogen-rich alloys by theoretical and experimental means.1 The former has been greatly facilitated by the recent developments in practical strategies for the prediction of crystal structures. Although, the predicted hydrides have yet to be confirmed by experiment, the theoretical studies have revealed a myriad of novel structures for the high pressure hydrides and enriched the understanding of the nature of chemical bonding in the rarely explored high pressure regime.The hydrides of group 1 and 2 elements formed from the reaction of the metal and hydrogen molecules have been the most studied.2–6 Most of the structures and structural trends can be explained from the simple concept of electron transfer from the metal to the hydrogen due to the large electronegativity differences between the alkali and alkaline elements and hydrogen molecules. The predicted compounds display rich H species distinguished from the well-known H-ion in hydrides at traditional stoichiometric ratios. Perhaps one of the most exciting predictions is the emergence of symmetric and linear H3− at high pressures as observed in dense CsH3 and BaH6.7,8 Moreover, the formation pressures of these compounds of just a few tens of GPa are accessible by experiments. In comparison, the bonding pattern is quite different for group 14 and transition elements. For example, a Van der Waals solid with such molecular H2 units was found experimentally in SiH4 at low pressure.9 At higher pressure, the atoms of group 14 elements tend to aggregate to form a 2D layered structure decorated with molecular like H2 species as predicted for SiH4 and SnH4.10,11 In comparison, the high pressure chemistry of hydrogen with electron-rich group 17 halogens has not been investigated. In this paper, we present results on a study of the crystal structures and phase stabilities of hydrogen-rich HCl–H2 system. A major finding is the stabilization of cationic (H3)+ (H2) species in H5Cl. The geometry of H3+ becomes almost an equilateral triangle under very high pressure.
The observation of a triatomic hydrogen cation H3+ in the solid state is new and significant. The isolated molecule is important in various branches of science, such as physics, chemistry and astronomy. For example, it is known that the H3+ ion with a triangular configuration is stable in the interstellar medium thanks to the low temperature and low density of the interstellar space and the H3 molecule is commonly formed from the neutralization reaction of H3+ and an electron, and rather evanescent as a result of the repulsive nature of its ground state.12 Furthermore, the multicenter (H3)+ (H2) bond in group 17 Cl compounds signifies a deviation in the nature of chemical bonding from the charge transfer interactions in group 1 and 2 hydrides and covalent bonding in group 14 hydrides.
Computational details
The search for stable high pressure structures of the HnCl system was based on global minimization of free energy surfaces using ab initio total energy calculations and the particle–swarm-optimization scheme as implemented in the CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) code.13,14 The performance and reliability of this method has been demonstrated on many known systems. An example is the success on the prediction15–17 of an insulating orthorhombic (Aba2, Pearson symbol oC40) structure of Li and, more recently, two low-pressure monoclinic structures of Bi2 Te3. In both cases, the predicted structures were later confirmed by experiments.18,19 Structural searching was performed at 100, 200, and 300 GPa with a simulation cell consisting of 1–4 formula units. Ab initio electronic structure calculations and structural relaxations were carried out using density functional theory with the Perdew–Burke–Ernzerhof (PBE) exchange-correlation20 implemented in the Vienna ab initio Simulation Package (VASP) code.21 The predicted stable structures were carefully optimized at a high level of accuracy. A plane wave energy cutoff of 1000 eV was employed. Large Monkhorst-Pack k point sampling grids22 were used to ensure that all the enthalpy calculations were well converged to an accuracy of 1 meV per atom. The atomic charges were obtained from Bader topological analysis23–25 with very large grids to ensure sufficient accuracy. We also performed additional calculations employing the DF-2 van der Waals (vdW) functional26 to validate the results, in particular for the low pressure structures. Phonons were calculated with the supercell method27 implemented in the PHONOPY program.28 In essence, from finite displacements, the Hellmann–Feynman atomic forces computed at the optimized supercell by the VASP code were input to the PHONOPY code to construct the dynamical matrix. Diagonalization of the dynamical matrix gives the normal modes and their frequencies. Converged results were obtained with the use of a 2 × 2 × 1 supercell and 4 × 4 × 6 k-meshes for the Cc structure, and a 2 × 2 × 1 supercell and 4 × 4 × 6 k-meshes for the C2/c structure.Results and discussion
Before embarking on a detailed discussion of the structures of the predicted high pressure HnCl polymers, the relative energetics of the H–Cl system with H-rich stoichiometry from 100 to 300 GPa are summarized in the convex hull plot shown in Fig. 1. The enthalpies of formation were evaluated as the difference in the enthalpy of the predicted HnCl structure with solid HCl and H2 at the selected pressures. Since hydrogen has a small atomic mass, the zero point energy (ZPE) may be important. To investigate the vibrational effects on the phase stability, ZPEs for H2Cl and H5Cl were estimated at 100–300 GPa from the corresponding phonon spectra using the quasi-harmonic approximation.29 It is found that the ZPEs are quite small and the inclusion of ZPEs in the phase diagram only resulted in a slight shift in the formation pressures but the stability of both phases remains unaltered. Structures lying on the convex hull are thermodynamically stable or metastable and, in principle, can be synthesized. Fig. 1 reveals that only two HCl–H2 complexes, H2Cl and H5Cl are stable at 100 GPa. At this pressure H2Cl has the most negative enthalpy of formation. With increasing pressure, the stability of H5Cl relative to H2Cl increases and becomes the most stable phase at 300 GPa.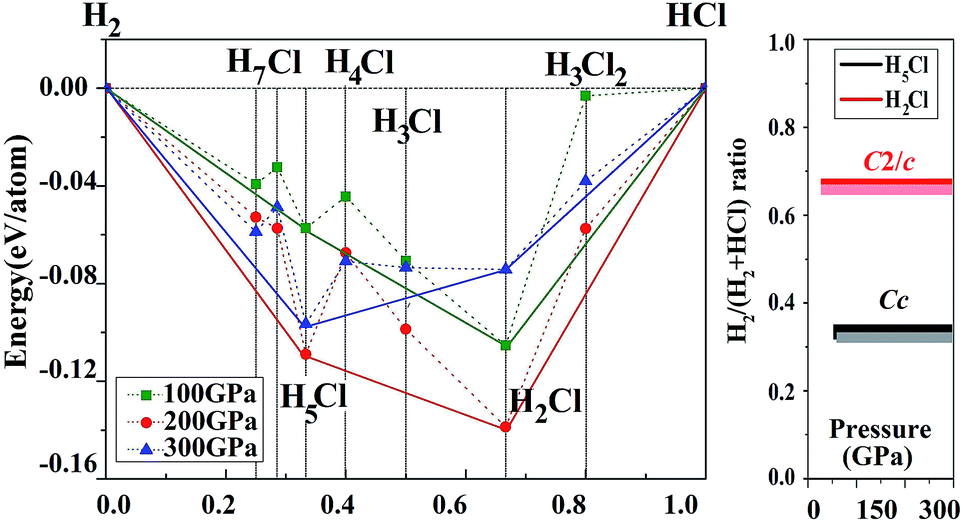 | ||
| Fig. 1 Stability of new hydrogen chlorides. (A) Enthalpies of formation (ΔH, with respect to HCl and H2 of their most stable phases at selected pressures) of HnCl (n = 1–7). The abscissa x is the fraction of H2 in the structures. Circles on the solid lines represent stable ground-state compounds under the corresponding pressure. (B) Pressure-composition phase diagram of the H–Cl system. The lighter colored lines represent the vdW corrections to corresponding structures. | ||
vdW effects may play an important role in the stabilization of a molecular solid. We have thus performed additional calculations on the H–Cl system with the vdW-DF2 method.26 The results show that the differences between calculations with and without vdW corrections on the formation enthalpies of the structures considered in Fig. 1 are small. The formation pressures were found to change slightly. For example, the stabilized pressure of H2Cl increased from 21.2 to 21.3 GPa, while for H5Cl it increased from 50 to 60 GPa. Otherwise, the energetic order remains the same.
Now we examine the development of the high pressure crystal structures in HnCl (n = 2–7). The starting point is the crystal structure of HCl under ambient pressure. At low temperature, X-ray and neutron diffraction show HCl crystallized in an orthorhombic structure (Bb21m).30 In the crystal, HCl molecules are linked via the H atoms forming zigzag chains running parallel to the crystallographic b axis. The nearest neighbour Cl–H and second nearest neighbour Cl⋯H distances are 1.25 Å and 2.44 Å, respectively. The Cl⋯Cl separation is 3.88 Å and the H–Cl⋯H valence angle is 93.6°. The predicted crystalline phase of H2Cl at 100 GPa has a C2/c space group and the structure is shown in Fig. 2. The crystal is formed from HCl chains interposed with H2 molecules. In this case, the H in the HCl chain is midway between the two Cl atoms with an H–Cl distance of 1.45 Å. The H–Cl–H angle has opened to 97.9° and the Cl⋯Cl separation is shortened to 2.90 Å. The H2 units in the structure all have a H–H distance of 0.74 Å, which is almost identical to that of the isolated molecule. The Bader charges for the H in the chain and Cl atoms are +0.44 and −0.35, respectively and 0.0 for the H atoms in the H2 units. The closest contact between a Cl atom and the H2 molecule is 1.98 Å. The crystal structure of H2Cl at 300 GPa differs little from that at 100 GPa. The H atoms in the H–Cl chains are still situated at the middle of the two neighbouring Cl atoms with a H–Cl distance of 1.35 Å. The H–Cl–H angle is 95.5° and the shortest separation between two Cl atoms has reduced further to 2.69 Å. The H–H bond length in the H2 unit is 0.73 Å. The closest H2⋯Cl distance is. 1.70 Å. Compression has a significant effect on the interatomic distances of the H–Cl chains but does not alter fundamentally the underlying bonding pattern. A longer H–Cl distance in the chain suggests increased ionicity of the Cl–H bonds.
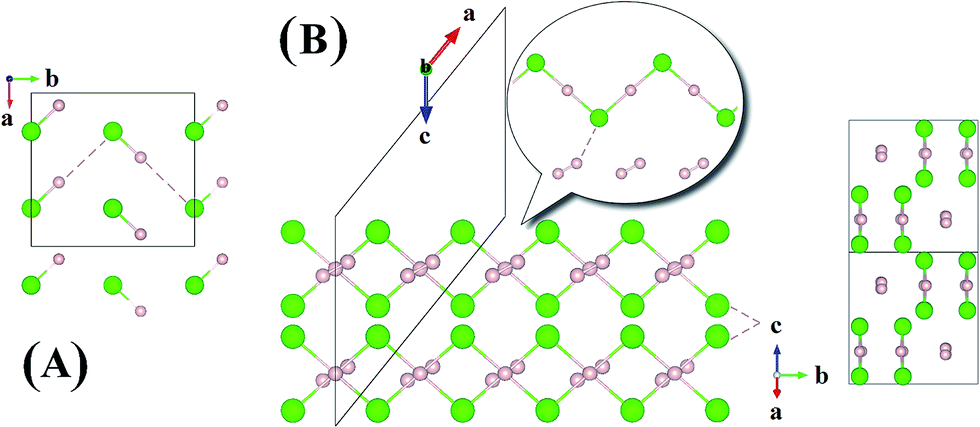 | ||
| Fig. 2 Crystal structures of hydrogen chlorides. (A) Experimental structure of HCl at ambient pressure and low temperature, (B) C2/c-H2Cl recovered at 100 GPa along a different angle. | ||
Although H3Cl and H4Cl are only metastable, it is instructive to examine the evolution of the crystal structure with increasing H2 concentration. The structures of H3Cl at 100 and 300 GPa are shown in Fig. 3. Both are composed of zigzag H–Cl chains. Like H2Cl, the H atom is equidistant from the two nearest Cl atom with H–Cl bond distances of 1.44 Å at 100 GPa and 1.43 Å at 300 GPa. The most significant difference between the low and high pressure structures is that the Cl–H–Cl angle is almost linear at 100 GPa but bends to 135° at 300 GPa. At 300 GPa, the closest contact between the H2 and the H in the chain is 1.27 Å. However, in both cases, the H–H distance of the H2 molecule remains 0.73 Å. The structure of H4Cl at 100 GPa differs dramatically from all the structures within this series of compounds. Instead of H–Cl chains, the structure is composed of isolated HCl and H2 molecules. The H–Cl distance is 1.38 Å and the H–H bond length is 0.74 Å. For comparison, the H–Cl bond of a free molecule is 1.276 Å. Therefore, the distance in the solid state at 100 GPa is slightly longer. The structure of H4Cl at 300 GPa again is different from that at 100 GPa. The basic building units are isolated Cl atoms, H2 molecules with H–H distance of 0.74 Å and a novel 2-D layer of slightly puckered fused hexagonal rings formed from 3 HCl units with additional H atoms attached to the Cl atoms. Each H atom in the ring is bonded to three Cl atoms. In addition, each Cl is bonded to an extra H atom which is not coordinated to other species in the crystal. The H–Cl distances in the fused ring are 1.59 Å and the terminal H–Cl is substantially shorter at 1.49 Å. Interestingly, the terminal H–Cl–H (ring) angles are 77° and the in-plane H–Cl–H and Cl–H–Cl angles are between 114–115°.
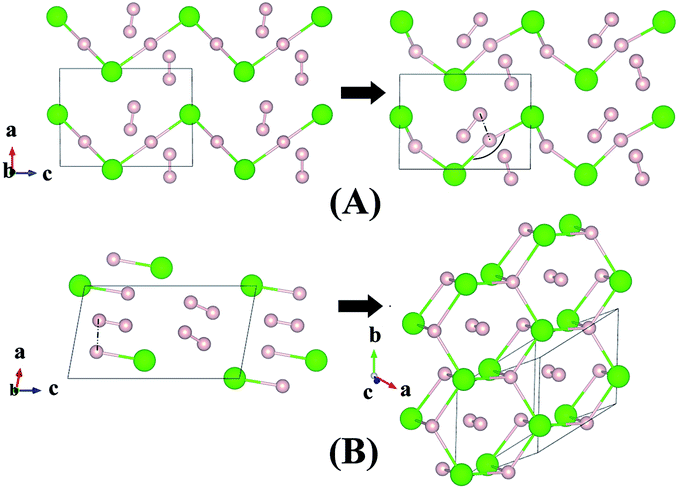 | ||
| Fig. 3 Crystal structures of hydrogen chlorides. Predicted metastable phase H3Cl (A) and H4Cl (B) at pressures of 100 to 300 GPa. | ||
An interesting structure was observed in H5Cl at 100 GPa. Although chains formed from Cl and H atoms are still clearly visible, the detailed construction of the chain is very different. In H5Cl, instead of placing one H atom midway between the two nearest neighbour Cl atoms, it is replaced by an H3 unit. The H3 is a distorted isosceles triangle and can be described as a loosely bound unit of an H atom and an elongated H2 with an apical angle of 63.8°. The apical H atom is linked to the two nearest Cl atoms in the chain with H–Cl distances of 1.47 Å. The distances from the apical H atom to the two H forming the H2 are 1.01 Å and 0.97 Å, respectively and the intermolecular H–H distance is 0.81 Å. The remaining H2 units in the structure have H–H distances of 0.74 Å. Moreover, the shortest distance from these H2 to the H3 is 1.36 Å and, therefore, may be considered as non-interacting molecules.
Compression of H5Cl to 300 GPa does not change the space group symmetry. The chain pattern with interpose H3 units is still maintained, but the local H⋯H interactions have changed dramatically. The H3 unit now approaches an equilateral triangle. The H–H lengths are 0.87, 0.87 and 0.88 Å with bond angles 59.7, 59.7 and 60.5°. The Cl–H distance has elongated from 1.47 Å at 100 GPa to 1.60 Å! The large lengthening of the Cl–H clearly suggests a substantial change in the Cl–H bonds. More significantly, the isolated H2 molecules are now pushed towards the H3 units and interact with one of the H atoms forming almost two H⋯H bonds at 1.15 Å. Concomitantly, the distance in H2 is lengthened to 0.76 Å. The Bader charges for the H atom in the H3 and H2 units and for the Cl atom are +0.16 and +0.014 and −0.48 respectively. In comparison to H2Cl the ionicity on both the H and Cl atoms have increased substantially. The plot of the electron localization function (ELF) shown in Fig. 4 shows localized spin paired electron density within the H3 ring and in the H2 molecule (ELF over 0.8). Weak pairing is also observed between one of the H in the H3 ring with the two H atoms of H2.
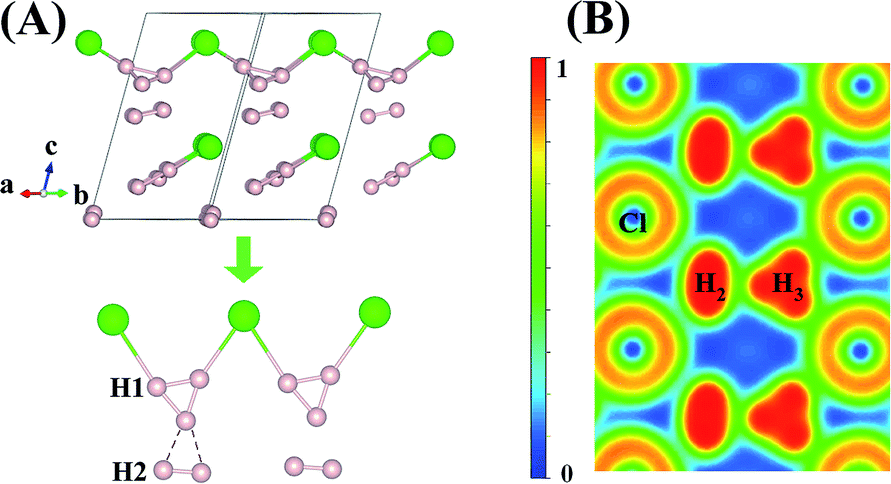 | ||
| Fig. 4 (A) crystal structure of Cc-H5Cl recovered at 100 GPa and its chain style when compressed to 300 GPa. (B) Electron localization function (ELF) maps of the planes where the hydrogen and chloride atoms lie for the Cc structure at 300 GPa. | ||
It is tempting to relate the positively charged H3 unit to an isolated trihydrogen cation H3+.
H3+ has a perfect triangular structure with H–H bond distance of 0.90 Å. For comparison, at 300 GPa the average intramolecular H–H distance in the H3 unit in H5Cl is 0.87 Å and a total charge of +0.48 (3 × 0.16). In addition, the H–H distance of 0.76 Å in the H2 unit is only slightly perturbed from the isolated molecule. Therefore, it is not unreasonable to suggest that the high pressure H5Cl structure is composed of H3+ stabilized in the solid state through primarily ionic interactions with the Cl atoms and secondary weak interactions with a pair of H2 molecules. To investigate further the properties of the H3+ unit in H5Cl, the phonon densities of states calculated at 300 GPa are shown in Fig. 5. The low-energy vibrations from 100–500 cm−1 are dominated by the Cl atomic motions. The phonon modes in the region from 1800 to 3400 cm−1 can be assigned to H3+ molecular vibrations and then near 4000 cm−1 to the H2 molecule. The H3+ bend vibrations are split into two bands centered at 1800 cm−1 and 2000 cm−1. The peak at 3350 cm−1 is attributed to the stretching vibration. In comparison, the fundamental frequencies for isolated H3+ are ν (stretch) = 3220 × 48 cm−1 and ν (bend) = 2545 × 99 cm−1, the latter is a degenerated mode.31 The vibration frequency is 4350 cm−1 for a free H2 molecule. The main mechanism for the synthesis of H3+ by experiment is via the chemical reaction:32 H+ + H2 → H3+. The concentration of H2+ is the limiting factor on the rate of this reaction. H2+ can only be produced in interstellar space by the ionization of H2 by a cosmic ray. In H5Cl, the electronegative chlorine atoms acquire electrons from the hydrogen. From previous studies, it is known that high pressure facilitates the transfer of electrons from the more electropositive element into the interstitials of a crystal, forming electrodes. In this case, these electrons, being transferred to the chlorine, are originating from the hydrogen. The formation of multicenter H3⋯H2 clusters helps to delocalize the positive charge and stabilizes the system. This explains the unexpected formation of H3+-like units in the crystal structure.
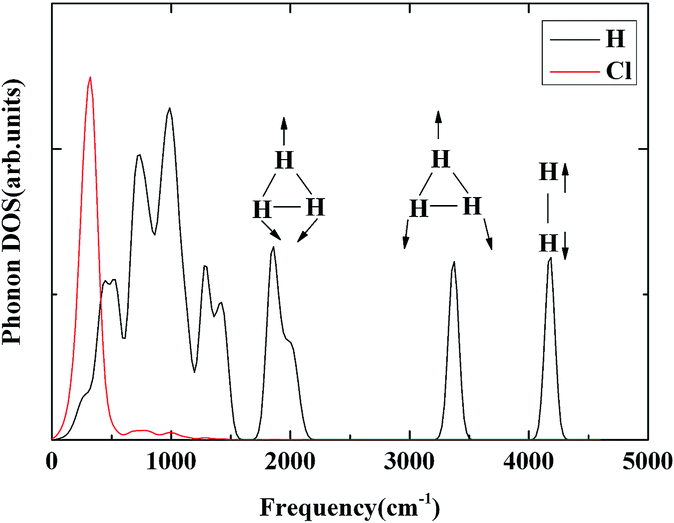 | ||
| Fig. 5 The phonon densities of states for Cc-H5Cl at 300 GPa are shown. The modes corresponding to the triatomic H3+ stretch, as well as the H2 vibron are denoted. | ||
The nature of the bonding in the group 17 hydrides at high pressure is different from group 1 and 2 and group 14 hydrides. One observed a gradual shift in the chemical interaction from electron transfer in electropositive group 1 and 3 compounds to covalent bonding group 14 and finally the ionic bonding in group 17 elements. Although this study was focused on Cl, we anticipate a similar bonding mechanism is applicable to other halogen hydrides.
Conclusions
We have investigated the phase stability of the HCl–H2 system at high pressure using the PSO algorithm in combination with ab initio density functional based electronic calculations. Between 100–300 GPa, two stable phases with the H5Cl and H2Cl stoichiometries were found. The basic structure of the high pressure phases is similar to the low temperature ambient pressure structure of HCl. H2Cl consists of zigzag H–Cl chains and non-interacting H2 molecules. The most usual and informative finding is that while the chain like structure is preserved in H5Cl, the H atoms connecting the Cl in the chains are replaced by units consisting of weakly interacting H2⋯H3. The H3+ is positively charged and stabilized from the formation of multicenter bonds. The similarity in the local structure, vibrational frequencies and electronic charge compel us to relate the unit to the isolated H3+ molecule. It is also found that the effect of pressure on the electronic structure of group 17 hydrides is very different from the more electropositive group 1 and 2 elements: the electron-rich Cl atom and anion do not transfer their electrons into interstitial space of the crystal under very high compression. In fact electrons are removed from the H atoms leading to the formation of cationic clusters that benefit from multicenter bonding.Acknowledgements
The authors appreciatively acknowledge the financial support by NSFC under Grant nos. 11104104, 11474126, 11474128, and 91022029, China 973 Program under Grant no. 2011CB808200, the 2013 Program for New Century Excellent Talents in University, 2012 Changjiang Scholar of Ministry of Education, Changjiang Scholar and Innovative Research Team in University (Grant no. IRT1132). T.I. was supported by MEXT of Japan (Grant nos. 20103001–20103005) and the RIKEN iTHES project. Part of the calculations were performed in the computing facilities at RIKEN Integrated Cluster of Clusters system (Japan) and the High Performance Computing Center of Jilin University. The work at the University of Saskatchewan was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant.Notes and references
- N. W. Ashcroft, Phys. Rev. Lett., 1968, 21, 1748 CrossRef CAS.
- J. Hooper and E. Zurek, Chem.–Eur. J., 2012, 18(16), 5013–5021 CrossRef CAS PubMed.
- E. Zurek, R. Hoffmann, N. W. Ashcroft, A. R. Oganov and A. O. Lyakhov, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17640 CrossRef CAS PubMed.
- J. Hooper and E. Zurek, J. Phys. Chem. C, 2012, 116(24), 13322–13328 CAS.
- Z. Wang, Y. Yao, L. Zhu, H. Liu, T. Iitaka, H. Wang and Y. Ma, J. Chem. Phys., 2014, 140, 124707 CrossRef PubMed.
- D. C. Lonie, J. Hooper, B. Altintas and E. Zurek, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 054107 CrossRef.
- A. Shamp, J. Hooper and E. Zurek, Inorg. Chem., 2012, 51(17), 9333–9342 CrossRef CAS PubMed.
- J. Hooper, B. Altintas, A. Shamp and E. Zurek, J. Phys. Chem. C, 2013, 117, 2982 CAS.
- T. A. Strobel, A. F. Goncharov, C. T. Seagle, Z. Liu, M. Somayazulu, V. V. Struzhkin and R. J. Hemley, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 144102 CrossRef.
- Y. Li, G. Gao, Q. Li, Y. Ma and G. Zou, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 064104 CrossRef.
- G. Gao, et al. , Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1317 CrossRef CAS PubMed.
- D. Martin, E. McDaniel and M. Meeks, Astrophys. J., 1961, 134, 1012 CrossRef CAS PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, CALYPSO: A method for crystal structure prediction, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS PubMed , CALYPSO code is free for academic use, please register at http://www.calypso.cn.
- P. Li, G. Gao, Y. Wang and Y. Ma, J. Phys. Chem. C, 2010, 114, 21745 CAS.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Phys. Rev. Lett., 2011, 106, 015503 CrossRef.
- L. Zhu, H. Wang, Y. Wang, J. Lv, Y. Ma, Q. Cui, Y. Ma and G. Zou, Phys. Rev. Lett., 2011, 106, 145501 CrossRef.
- C. L. Guillaume, E. Gregoryanz, O. Degtyareva, M. I. McMahon, M. Hanfland, S. Evans, M. Guthrie, S. V. Sinogeikin and H. K. Mao, Nat. Phys., 2011, 7, 211 CrossRef CAS.
- L. Zhu, H. Wang, Y. Wang, J. Lv, Y. Ma, Q. Cui, Y. Ma and G. Zou, Phys. Rev. Lett., 2011, 106, 145501 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354 CrossRef PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- K. Parlinski, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- Y. Ma and J. S. Tse, Solid State Commun., 2007, 143, 161 CrossRef CAS PubMed.
- E. Sándor and R. Farrow, Nature, 1967, 213, 171 CrossRef.
- G. D. Carney, Mol. Phys., 1980, 39(4), 923–933 CrossRef CAS.
- T. R. Hogness and E. G. Lunn, Phys. Rev., 1925, 26, 44 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |