
In situ preparation of a MOF-derived magnetic carbonaceous catalyst for visible-light-driven hydrogen evolution†
Jing-Yin Xu‡
a,
Xin-Ping Zhai‡a,
Lin-Feng Gaoa,
Peng Chenb,
Min Zhaoa,
Hong-Bin Yanga,
Deng-Feng Caoa,
Qiang Wang*a and
Hao-Li Zhang*a
aState Key Laboratory of Applied Organic Chemistry (SKLAOC), Key Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, College of Chemistry and Chemical Engineering, Key Laboratory of Special Function Materials and Structure Design, Ministry of Education, Lanzhou University, Lanzhou, 730000, China. E-mail: qiangwang@lzu.edu.cn; haoli.zhang@lzu.edu.cn
bSchool of Pharmacy, Lanzhou University, Lanzhou, 730020, China
First published on 21st December 2015
Abstract
MOFs (Metal–Organic Frameworks) have emerged as novel photocatalysts for water reduction but are frequently plagued by their instability when exposed to moist and strongly acidic or alkaline reaction environments. Herein we employed a volatile Fe-based MOF in alkaline solution as precursor to evolve into a magnetic carbonaceous photocatalyst in situ, which demonstrated highly efficient visible-light-driven hydrogen evolution (∼125 μmol H2 produced within 6 h using 5 mg of MOF precursor) with a quantum efficiency of 1.8% even in the absence of noble metal cocatalyst, indicative of a possible photocatalytic system containing only earth-abundant elements for long-term conversion of solar light into hydrogen energy. The catalyst exhibited an apparent stoichiometric formula of FeO3.3C0.2H1.0 and was determined to be essentially a carbon–metal oxides/oxyhydroxides composite. Laser photolysis and electrochemical measurements were performed to visualize the fundamental multistep electron transfer processes during water reduction, which opens a strategy for the rational design of MOF-derived catalysts to dramatically increase H2 evolution efficiency.
Introduction
Metal–organic frameworks (MOFs), a class of hybrid materials built from the coordination of a limitless choice of metal ions or clusters with organic ligands,1–4 have exhibited intrinsically ordered three-dimensional (3D) structures that give rise to a myriad of potential applications such as gas adsorption and storage,5–8 sensing,9,10 catalysis,6,11–13 separation,14,15 and nonlinear optical devices.16,17 More recently, the employment of MOFs as precursors/templates for the fabrication of other functional materials has received mounting attention.18,19 Under appropriate experimental conditions such as thermolysis, MOFs are potentially capable of forming porous carbon materials, carbon–metal/metal oxide hybrids, micro/nanostructured metal oxides, etc., owing to their highly ordered porous structures constructed from a regular arrangement of metal nodes and abundant organic struts.18 The strategy expands the potential and functionality of MOFs, compensating their usual instability from exposure to moisture, high temperature, chemical reagents such as strong acid or alkali. As a result, a sizeable amount of MOF-derived functional materials have emerged as promising novel challengers in catalysis, lithium-ion batteries, capacitors, electrode, fuel cell applications and so on.18 However, to date MOF-derived functional materials for hydrogen evolution via water reduction is much less reported.Inspired by the natural hydrogenases mimics containing active site of Fe2S2 cluster that have been intensively investigated for photocatalytic H2 production,20,21 herein we developed MOF-derived carbon–metal oxides/oxyhydroxides composite in situ for visible-light-driven water reduction. MIL-101(Fe) is a MOF embedded with Fe-oxo metallic clusters, but is notoriously unstable in basic solution. However, through a mild in situ carbonization process, MIL-101(Fe) can serve as suitable precursors to be decomposed into micro/nano structured carbon–metal oxides/oxyhydroxides composite. This method bypassed thermolysis of the MOF precursors at high temperature and is rather facile and low-cost, indicative potential for large-scale fabrication. Moreover, the as-prepared carbonaceous catalyst is strongly magnetic and thus easily recoverable, one additional merit favored in photocatalytic application.
Indeed, the as-prepared carbon–metal oxides/oxyhydroxides composite acted as highly efficient catalyst for hydrogen evolution via water reduction, surpassing many of those previously reported H2 evolution performance based on MOF structures. It is noteworthy that even without the presence of noble metal cocatalyst, the catalysts are capable of attaining high H2 yield, indicative of a possible photocatalytic system containing only earth-abundant elements for long-term conversion of solar light into hydrogen energy. Laser photolysis and electrochemical measurements were employed to unravel their roles in H2 production, which pictured the fundamental multistep electron transfer processes during water reduction, open a strategy for the rational design of MOF-derived catalyst to highly raise H2 evolution efficiency, and the Fe-based, environmental-friendly catalysts with a facile and scaled-up synthesis may eventually enable sustainable artificial photosynthesis and tackle the urgent energy issues facing the world.
Experimental
Materials
1,4-Benzenedicarboxylic acid (BDC) was purchased from Tianjin GuangFu Technology Development Co. Ltd. Iron(III) chloride hexahydrate (FeCl3·6H2O) was obtained from Tianjin Kaitong Chemical Co. Ltd. Sodium sulfate (Na2SO4) was obtained from Tianjin Shengmiao Chemical Co. Ltd. Eosin Y alcohol soluble (EY) was from Aladdin Chemistry Co. Ltd. Triethylamine (TEA) and N,N-dimethylformamide (DMF, dried), ethanol (EtOH, dried), acetonitrile (MeCN), acetone was purchased from Rionlon Bohua (Tianjin) Pharmaceutical & Chemical Co. Ltd. Deuterium oxide (D2O, Qingdao Tenglong Weibo, 99.9% D). All solvents and chemicals were used without any further purification except for DMF and EtOH, which were dried with CaH2 and Na and distilled under reduced pressure. Deionized water (resistivity: 18.3 MΩ cm) was used to prepare aqueous solution throughout the experiment unless otherwise specified.Syntheses of MIL-101(Fe) precursor
The synthesis of size and morphology controlled crystals was accomplished following modified procedures as described in literatures.22–24 In a typical synthesis procedure, 0.206 g (1.24 mmol) of BDC and 0.675 g (2.45 mmol) of FeCl3·6H2O were dissolved in 15 mL of DMF. The mixture was then introduced into a 50 mL Teflon-lined microwave vessel and sealed. The reaction was then rapidly heated to 110 °C at 400 watt microwave irradiation, and was held at this temperature for 10 minutes (PreeKem WX-4000 microwave digestion system, Shanghai). After cooling to room temperature, the particles were isolated by centrifuging, and were washed with DMF and ethanol.MOF-derived catalyst in situ for hydrogen evolution
The as-synthesized MIL-101(Fe) of 5 mg was added to the acetonitrile/water solution in a 25 mL round-bottom flask, sealed with a silicone stopper, degassed, flushed with dry nitrogen, and sonicated in dark for ∼1–2 min to ensure a uniform mixture. Photocatalytic hydrogen evolution experiments were performed by irradiating the solution under continuous stirring in the presence of Eosin Y (EY) and triethylamine (TEA) using a white-light LED (light spectrum: 400–750 nm; energy density: 13.7 mW cm−2 at a distance of 6 cm) at room temperature. Thus the MOF-derived catalyst was synthesized in situ and the produced hydrogen was detected by a calibrated Varian GC-3380 Gas Chromatograph with a thermal conductivity detector and nitrogen as the carrier gas. For a repeated photocatalytic cycle measurement, the catalyst was first separated via centrifugation and thereafter the same procedure as the first H2 collection cycle was carried out with supplement of the same amount of reaction reagents, i.e., solvent, EY and TEA. The optimized reaction conditions are used for H2 production with suitable amount of catalyst in acetonitrile/water (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution in the presence of EY (8.0 × 10−4 M) and TEA (10% v/v), at a pH value of 10.0 adjusted by hydrochloric acid. To carry out the deuteration experiments, deuterium oxide was used to replace water, and the produced gas mixture was analyzed by gas chromatograph/mass spectrometry (GC/MS) on a SHIMADZU GCMS-QP2010 SE instrument. After hydrogen evolution measurement, the catalyst was separated via centrifugation for characterization.
:1) solution in the presence of EY (8.0 × 10−4 M) and TEA (10% v/v), at a pH value of 10.0 adjusted by hydrochloric acid. To carry out the deuteration experiments, deuterium oxide was used to replace water, and the produced gas mixture was analyzed by gas chromatograph/mass spectrometry (GC/MS) on a SHIMADZU GCMS-QP2010 SE instrument. After hydrogen evolution measurement, the catalyst was separated via centrifugation for characterization.
Characterization
:1) solution at a pH value of 10.Results and discussion
MIL-101(Fe) precursors were prepared following a modified route,22–24 where FeCl3·6H2O and terephthalic acid (BDC) were heated with the assistance of microwave in pure DMF. The precursors were then directly applied for photocatalytic reactions, which would be rapidly decomposed into the catalyst (Cat.) in situ subject to alkaline media and visible-light irradiation. As displayed by the scanning electron microscopy (SEM) images (Fig. 1a), MIL-101(Fe) particles exhibit an octahedron morphology and an average diameter of ∼500 nm, consistent with previous report.24 By contrast, the as-synthesized Cat. after separation showed amorphous and porous microstructures (Fig. 1b). The weight percentage of iron element was determined to be 49.6% by inductively coupled plasma atomic emission spectroscopy (ICP-AES). In combination with the weight ratio of 2.6%, 1.0%, 46.8% for C, H and O, respectively via elementary analysis, the stoichiometric formula of the carbon/metal or metal oxide composite was tentatively deduced to be FeO3.3C0.2H1.0. To disentangle the exact composition of the catalyst is challenging, as under the synthesis conditions, i.e., alkaline medium, light irradiation and absence of high temperature thermolysis, the Fe-based MOF precursors would experience complicated reactions and evolve into copious combinations between carbon and the extremely rich iron oxides/oxyhydroxides family, Fe2O3 of α, γ phases, Fe3O4, FeOOH of α, β, γ phases, to name a few. Therefore, in the following work, we tried various characterization techniques to provide information on the composition of the catalyst. | ||
| Fig. 1 SEM images of (a) MIL-101(Fe) (“MOFs”), (b) the MOF-derived catalyst (“Cat.”) and HRTEM image of the Cat. (c). (d) FTIR and (e) Raman spectra of MOFs, Cat. and the catalyst after digested by concentrated HCl to remove iron element (“Cat. digested by HCl”). | ||
One type of lattice fringes with the lattice spacings of 0.296 nm was shown in the high resolution transmission electron microscope (HRTEM) image of the Cat. (Fig. 1c), probably corresponding to the (220) plane of γ-Fe2O3 nanoparticles.25 FTIR and Raman spectroscopic measurements were further employed to confirm the generation of the Cat. via MIL-101(Fe) precursors (Fig. 1d and e, respectively). Clearly, the FTIR spectrum of MIL-101(Fe) exhibited typical vibrational bands in the region of 1000–1750 cm−1 for the carboxylic functional groups,26,27 which disappeared or transformed in the spectrum of the Cat. Specifically, 1380–1630 cm−1 were tentatively attributed to the deformation vibrations of H–O–H of iron oxyhydroxides (FeOOH) species contained in the Cat.28 The band at 547 cm−1 of MIL-101(Fe) was attributed to Fe–O–C bending vibrations,29 which however turned into the Fe–O vibration peak of 460 cm−1 for the Cat. and vanished after the Cat. was digested by concentrated HCl to remove iron element (“Cat. digested by HCl”). Both MOFs and the Cat. showed broad band centering around 3410 cm−1 due to stretching vibrations of hydroxyl groups. Raman spectra of the three materials were also distinctly different. For MIL-101(Fe), the peak at 466 cm−1 could be assigned to the Fe–O vibration.27 The bands at 631, 863 and 1144 cm−1 were attributed to the deformation vibrations of the aromatic –CH groups. The symmetric stretch of the carboxylate groups and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration could be found between 1370 and 1470 cm−1. The band with the highest intensity (1612 cm−1) was assigned to the asymmetric stretch of the carboxylate group. The Raman bands of the Cat., however implied a coexistence of iron oxides/oxyhydroxides in different phases. The band centering around 668 cm−1 was ascribed to either γ-Fe2O3 or Fe3O4.30,31 The bands at 216 and 394 cm−1 were considered characteristics of α-Fe2O3,31,32 whereas the peaks at 282, 588, and 668 cm−1 were typical bands of Fe3O4, and 282, 394, 1300 cm−1 could be also from α-FeOOH.30 The bands around 1300 and 1500 cm−1 were complicated, which could stem from γ-Fe2O3, α-FeOOH and carbon. Accordingly, by examining the Raman shifts of the Cat. digested by HCl, it was found that the typical bands for the metal oxides/oxyhydroxides vanished, and the residual broad bands at 1300 and 1540 cm−1 were expected to correspond to disordered amorphous carbon (D band) and graphite (G-band) respectively,31,33 indicative of the formation of amorphous carbon. Therefore based on the above discussions, γ-Fe2O3 and α-FeOOH could be the composites of the Cat. α-Fe2O3 is the most thermodynamically stable phase among the iron oxides family and the formation of it usually requires high thermolysis temperature,28 which contracts with our reaction condition. Hence the appearance of typical Raman shifts of α-Fe2O3 may be induced by the laser during measurement, which could transform γ-Fe2O3, Fe3O4 or FeOOH species into α-Fe2O3.30 Meanwhile, γ-Fe2O3 could be partially reduced to Fe3O4 during the in situ synthesis of the catalyst,31,33 which may account for certain characteristic Raman peaks of Fe3O4 in the spectra.
C vibration could be found between 1370 and 1470 cm−1. The band with the highest intensity (1612 cm−1) was assigned to the asymmetric stretch of the carboxylate group. The Raman bands of the Cat., however implied a coexistence of iron oxides/oxyhydroxides in different phases. The band centering around 668 cm−1 was ascribed to either γ-Fe2O3 or Fe3O4.30,31 The bands at 216 and 394 cm−1 were considered characteristics of α-Fe2O3,31,32 whereas the peaks at 282, 588, and 668 cm−1 were typical bands of Fe3O4, and 282, 394, 1300 cm−1 could be also from α-FeOOH.30 The bands around 1300 and 1500 cm−1 were complicated, which could stem from γ-Fe2O3, α-FeOOH and carbon. Accordingly, by examining the Raman shifts of the Cat. digested by HCl, it was found that the typical bands for the metal oxides/oxyhydroxides vanished, and the residual broad bands at 1300 and 1540 cm−1 were expected to correspond to disordered amorphous carbon (D band) and graphite (G-band) respectively,31,33 indicative of the formation of amorphous carbon. Therefore based on the above discussions, γ-Fe2O3 and α-FeOOH could be the composites of the Cat. α-Fe2O3 is the most thermodynamically stable phase among the iron oxides family and the formation of it usually requires high thermolysis temperature,28 which contracts with our reaction condition. Hence the appearance of typical Raman shifts of α-Fe2O3 may be induced by the laser during measurement, which could transform γ-Fe2O3, Fe3O4 or FeOOH species into α-Fe2O3.30 Meanwhile, γ-Fe2O3 could be partially reduced to Fe3O4 during the in situ synthesis of the catalyst,31,33 which may account for certain characteristic Raman peaks of Fe3O4 in the spectra.
Clearly, further evidence is required to ascertain the constitute of the Cat. XRD patterns alone are hard to distinguish the iron oxide phases as α-Fe2O3, γ-Fe2O3 and Fe3O4 possess resemble diffraction peaks.32–34 Moreover, the measured XRD patterns of the Cat. showed no obvious peaks corresponding to crystalline phases of carbon or Fe phase in any form (Fig. S1†), but similar to that of amorphous FeOOH. The XRD patterns of Cat. digested by HCl in the absence of iron element implied amorphous carbon. Therefore, X-ray photoemission spectroscopy (XPS) was utilized to further investigate the chemical compositions and the electronic structures of the composite in order to discern the varied phases thereof.
Fig. 2a indicate Fe, O and C peaks all exist in the survey XPS spectrum. Three characteristic peaks of Fe 2p3/2 at 711.0 eV, 717.4 eV and Fe 2p1/2 at 724.0 eV were shown in Fig. 2b of the high resolution curve-fitted XPS spectrum. It is known Fe 2p core-level spectra of some iron oxides/oxyhydroxides, such as α-Fe2O3, γ-Fe2O3 and α-FeOOH are very similar with each other although their crystal structures are distinct,35,36 and hence it is hard to identify them via XPS spectra. However, so called “shake-up” peaks sensitive to the electronic structure of iron oxides/oxyhydroxides are often employed as fingerprints to discern their different phases.35 Herein the satellite peak at 717.4 eV is regarded as the evidence of the presence of Fe3+ and absence of Fe2+,31,34,37 implying Fe3O4 in the Cat. is rare even if it does exist. On the other hand, the O 1s spectrum decomposed with the Gaussian distribution in Fig. 2c clearly showed the presence of Fe–O and O–H groups, with binding energies at 530.0 eV and 532.2 eV, respectively. According to previous literature,36 the result suggested the presence of α-FeOOH and the existence of water was also possible. Meanwhile, the decomposed C 1s spectrum implied the existence of different carbon-containing functional groups, such as CC, C–C, C–O, reflecting the electronic structures of carbon in the Cat. and was in agreement with aforementioned FTIR and Raman spectroscopic measurements.
 | ||
| Fig. 2 XPS spectra of the Cat.: (a) survey spectrum, and high-resolution (b) Fe 2p, (c) O 1s, (d) C 1s binding energy spectra. | ||
The composition of the catalyst is further analyzed by Thermogravimetric experiment (TGA). The TGA curve of the Cat. under N2 atmosphere is shown in Fig. 3a. The weight loss from 30 °C to 192 °C was attributed to the release of H2O, the existence of which had been suggested by the above FTIR and XPS measurement. There are two weight loss steps in the temperature. The next weight loss up to 637 °C is expected stem from the decomposition of different phases of iron oxides/oxyhydroxides, mainly α-FeOOH.38 The weight continues to lose at 637–698 °C, which is ascribed to the oxidation of carbon phase in the catalyst, and the weight loss of ∼1.5% is close to the carbon content of 2.6% of the catalyst via element analysis. The weigh-loss process ceased after 698 °C while the heating continues until to the eventual 800 °C. The weight of the stable residue was assigned as α-Fe2O3, accounting for a weight of ∼80.4%, consistent with the ∼49.6% of iron content measured by ICP-AES.
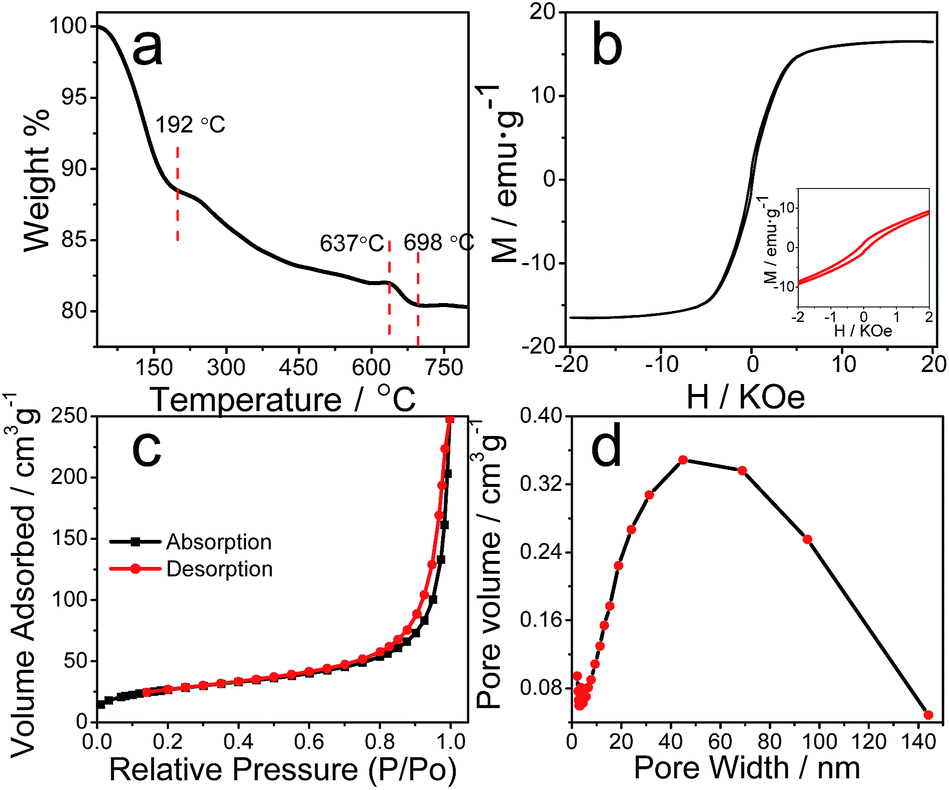 | ||
| Fig. 3 Thermogravimetric curve under nitrogen atmosphere (a), magnetization curve at ambient condition (the inset is the enlarged part) (b), N2 adsorption/desorption isotherms (c) and pore size distribution measured at 77 K (d) of the Cat. | ||
Magnetic feature is another way to effectively distinguish magnetic γ-Fe2O3 and Fe3O4 particles from weakly or none magnetic α-Fe2O3 and α-FeOOH.28,39 Fig. 3b displayed the field-dependent magnetization curve of the Cat., where a hysteresis loop (see the inset) indicated its ferromagnetic characteristic. A saturation magnetization value of 16.5 emu g−1 at 20 T is large enough for the Cat. to undergo efficient magnetic separation with external magnetic field.37 Combining with the above result, the observation further verified the γ-Fe2O3 constitute in the catalyst. In the end, nitrogen adsorption–desorption isotherm measurement were carried out to determine the pore properties of the Cat. Fig. 3c indicated a typical IV isotherm reflecting porous materials with BET surface areas of 93.7 m2 g−1, a value much less than that of its MOF precursor. Fig. 3d showed a broad range of pore diameter distribution from 2 to 140 nm, and average pore size of ∼16 nm, revealing the Cat. was a porous material mainly with mesopores. To sum up, the in situ generated magnetic carbonaceous catalyst from Fe-based MOFs exhibited an apparent stoichiometric formula of FeO3.3C0.2H1.0, but is essentially carbon–metal oxides/oxyhydroxides composite, with features partially characteristic of amorphous carbon, γ-Fe2O3 and α-FeOOH. Additionally, note another possibility was O-containing organic groups (–OH, –CHO, –COOH, etc.) tethered carbon with ferric oxide. The multiple-component system has potentials of executing synergetic effects in a photocatalytic reaction.
The utility of our catalysts was then demonstrated for visible-light-driven hydrogen evolution. MOF-based photocatalysts with integrated visible-light-responsive organic linkers would be ideal for hydrogen production under solar light irradiation, which however frequently involves multistep organic synthesis procedures.40,41 Alternatively, considering current dye-sensitization photocatalytic system usually comprising of commercial photosensitizer and sacrificial reagent is rather mature and cost-effective,42–44 we performed photochemical H2 evolution experiments by irradiating the catalysts with a white-light LED in the presence of Eosin Y (EY, photosensitizer) and triethylamine (TEA) as sacrificial reagent in acetonitrile/water solution (v/v = 1:1). Acetonitrile was used in the mixed solvent to improve the solubility of TEA and EY (ethanol soluble), which are not soluble well in pure water. This type of mixed solvent have been widely applied in previous photocatalytic water reduction system.45,46 The strategy could be a general way for photocatalytic water reduction via catalysts deficient in visible light response. Rates of photocatalytic H2 evolution with MOF-derived catalyst were shown in Fig. 4a. The visible-light-driven H2 evolution via the Cat. is highly efficient, even in the absence of the frequently indispensable noble metal cocatalyst as in previous reports.47,48 Within 6 hours, the amount of H2 produced was ∼125 μmol with just 5 mg MOFs precursors, and the calculated Quantum Efficiency (QE) was 1.8% (see the section on QE calculation procedures in ESI, and Fig. S2†). The Cat. also exhibited long durability and could be repeatedly used for several cycles without elemental content changes. The results emulated or surpassed those previously reported H2 evolution performance based on MOF structures in any form, either using MOFs as photosensitizers, semiconducting photocatalysts, or simply support for loading Pt particles.40,43,48–51 Control experiments indicated when neither the catalyst nor irradiation was present, H2 was not produced, implying that both the catalyst and the light are indispensable for the photocatalytic process. Commercial iron oxide nanoparticles like Fe2O3, Fe3O4 also did not produce H2. Likewise, individual components such as α-FeOOH also did not produce detectable hydrogen under the same reaction conditions. The result confirmed the multiple-component composite was not simply mixtures. It demonstrated synergetic effects in the photocatalytic reaction-the carbon content is low in the catalyst but may play crucial roles, and whether there is some special bonding between carbon and iron needs further exploration. Meanwhile, when deuterium oxide was used to replace water, the produced gas mixture analyzed by gas chromatograph/mass spectrometry (GC/MS) proved a dominant presence of D2 (Fig. S3†), strongly evidencing water was the origin for the hydrogen generated during the photocatalysis.52,53 In addition, although the catalyst exhibits considerable light response in the visible range as confirmed from diffuse reflection measurement in the following section, no detectable H2 production would be observed without the assistance of the photosensitizer.
 | ||
| Fig. 4 Rate of photocatalytic H2 evolution using the in situ prepared Cat., 4 repeated cycles (a). Transient absorption spectra of EY (b), EY + Cat. mixture (c), EY + TEA mixture (d), EY + Cat. + TEA mixture (e) and fitted decay curves for EY + Cat. mixture at 560 nm and 460 nm (f). The inset is the enlarged part for the very early time dynamics. The laser excitation wavelength is 525 nm. | ||
It is noteworthy that heterogeneous catalysts exhibited magnetic behaviors and can be conveniently recycled by an external magnet (Fig. S4†). Considering that isolation and reusability of one catalyst are crucial factors for industrial applications, the robustness and easy recyclability would endow the MOF-derived catalyst with great potential for practical use.
We also used other types of Fe-based MOFs as precursors to replace MIL-101(Fe) to investigate the impact of catalyst composition changes on the photoactivity. For instance, MIL-53(Fe) (Fig. S5†), which was prepared by using the same precursors of iron(III) chloride hexahydrate and 1,4-benzenedicarboxylic acid, gave a catalyst with a higher carbon content (weight ratio of 4.5%) than that of MIL-101(Fe)-derived catalyst. The H2 evolution efficiency of this as-prepared catalyst was ∼48 μmol gas produced within 5 h using 5 mg MOFs precursors, decreasing to less than half of that from MIL-101(Fe) derived catalyst under identical conditions. A systematic comparison of H2 evolution efficiency by catalysts derived from different precursors which therefore exhibited various elemental contents would be very interesting, and would be explored in separate work.
In order to disentangle the photocatalytic mechanism of the catalyst, we performed laser photolysis measurement to understand the dynamic processes during H2 formation. In Fig. 4b, the peak at 560 nm was attributed to the absorption of excited triplet state of EY (3EY*).42,54 Subsequently, 3EY* underwent one electron transfer to the Cat. and became a radical cation of EY+˙, as evidenced by its typical absorption at 460 nm (Fig. 4c),42,54 which is absent in the transient absorption spectra of sole EY. Fitting the absorption curve of 3EY* to a monoexponential decay equation gave one lifetime component of 31 μs (560 nm, Fig. 4f), which is remarkably reduced compared with the 132 μs from sole EY (see ESI for the Fitting methods, and Table S1†). Based on the expression of k = 1/τ(Cat.+EY) − 1/τEY, the electron transfer rate k from 3EY* to the Cat. was calculated to be 2.5 × 104 s−1 (Table S1†).42,55 While the absorption decay curves of EY+˙ (460 nm) followed a multiexponential decay and led to two lifetime components of 18 μs and 248 μs, respectively. The rapid rise of 18 μs corresponded to the formation time of EY+˙, which occurred concomitantly with the decay of 3EY* of 31 μs (Fig. 4f, inset), confirming the electron transfer from 3EY* to the Cat. The second 248-μs component is attributed to the lifetime of EY+˙, which thereafter will be regenerated if sacrificial donor exists in the system, as evidenced by the significantly quenched EY+˙ absorption in Fig. 4d in the presence of TEA. Control experiment had clearly showed that 3EY* was capable of acquiring electrons from TEA, indicated by the formation of EY−˙ with a typical absorption band at 403 nm with a long lifetime over 6 ms when the EY and TEA mixture was photo excited (Fig. 4e and Table S1†).42 Hence the entire photocatalytic system operate in the following cycle without intermittence: the catalyst attains electrons from EY (photosensitizer) for reduction of protons, and TEA (sacrificial reagent) on the other hand replenish electrons for EY to recover (Fig. 4e).
We further employed UV/VIS diffuse reflectance spectroscopy and electrochemical measurements to extract energy level information of the relevant parts in the photocatalytic system. From the diffuse-reflectance spectrum of the Cat. (Fig. 5a), the optical absorption of the sample can be approximated via the Kubelka–Munk function of F(R) = (1 − R)2/2R = α/S, where R is reflectance, α the absorption coefficient and S the scattering coefficient.56–58 The Tauc plot for the Cat. was then drawn with a standard relationship of (F(R) × hν)1/2 = A(hν − Eg), where h is Planck constant, ν the frequency, A the constant and Eg the sample band gap.49,56–58 Extrapolate a straight-line of the linear part of the curve within the lowest energy region to (F(R) × hν)1/2 = 0 (Fig. 5b), and then a band gap of 1.61 eV for the Cat. was obtained. In comparison, the band gap of MIL-101 (Fe) gave a value of 2.11 eV (Fig. S6†). We speculate that the narrower bandgap of the Cat. stemmed from its unique structure, which was in essence carbon–metal oxides/oxyhydroxides composite containing a considerable amount of amorphous carbon content.
 | ||
| Fig. 5 The UV/VIS diffuse reflectance spectra (a), corresponding Tauc plot (b), and Mott–Schottky plot in 0.5 M Na2SO4 aqueous solution (c) of the Cat. Proposed H2 production cycles photocatalyzed by the Cat. (d). | ||
Meanwhile, a positive slope of the Mott–Schottky (MS) plot indicated the Cat. was an n-type semiconductor (Fig. 5c).49 The Fermi level of the Cat. was determined to be −0.73 V vs. SCE (i.e., −0.49 V vs. NHE), equal to its flat band potential. Consequently, the conduction band (CB) edge of the Cat. was estimated to be −0.59 V, ca. 0.10 V more negative than the flat band potential.49 Compared with the LUMO level of EY (−1.05 V vs. NHE),59 CB edge of the Cat. is lower-lying, which would facilitate the electron transfer from EY to the Cat. and lead to an efficient charge separated state. The electron sequentially transfers to protons for a reduction to hydrogen as the CB edge of the Cat. is positioned above the reduction level for H2.
Hence the entire photocatalytic H2 evolution picture could be described by Fig. 5d. Upon photoexcitation, EY is prone to undergo intersystem crossing from singlet state to more active triplet state under deoxygenized environment. As its LUMO level is higher than the CB band of the catalyst, one electron transfers to the latter, which sequentially relays to even lower energy level for proton reduction. Meanwhile, EY can be supplemented with electrons from the sacrificial donor to extend the self-sustaining cycle incessantly. Once any link of the chain is broken, for instance, replacement of the catalyst by other materials with inappropriate CB bands, the electron transfer will be terminated and consequently no hydrogen will be produced. Overall, the transient dynamic process and the energy level determined from the electrochemical measurement substantiate the proposed scenario.
Conclusions
In this study, we reported a magnetic carbonaceous catalyst decomposed from earth abundant Fe-based MOFs in situ for highly efficient visible-light-driven hydrogen evolution for the first time. With this general strategy, instable MOF structures when exposed to moisture, thermal treatments, or chemical agents etc. however can be utilized as precursors to evolve into stable micro/nano materials. One striking result is high H2 yield of the MOF-derived catalyst even in the absence of noble metal co-catalyst. The employment of earth-abundant elements as cost-effective substitute for Pt, Pd, Ru, Ir and Rh in photocatalytic water reduction renders the production of hydrogen energy more pragmatic. Additionally, various methods such as laser photolysis and electrochemical measurements were applied to thoroughly investigate the roles of the catalyst in H2 production. Time-resolved spectra specifically provided unambiguous spectroscopic evidence on the transient species and the electron transfer dynamics during catalysis, providing new insights into the design of highly efficient catalyst for H2 evolution from the perspective of photocatalytic dynamics.Acknowledgements
This work is supported by National Basic Research Program of China (973 Program, No. 2012CB933102), Gansu Province Science Foundation for Distinguished Young Scholars (Grant No. 1308RJDA014), Longyuan Support Project for Young Creative Talents (Grant No. GANZUTONGZI [2014] no. 4), the Fundamental Research Funds for the Central Universities (lzujbky-2013-66), SRF for ROCS, SEM.Notes and references
- J. Kim, B. L. Chen, T. M. Reineke, H. L. Li, M. Eddaoudi, D. B. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239–8247 CrossRef CAS PubMed.
- O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS PubMed.
- W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC.
- Q. L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- H. J. Lee, W. Cho, S. Jung and M. Oh, Adv. Mater., 2009, 21, 674–677 CrossRef CAS.
- Y. Y. Liu, Y. M. Yang, Q. L. Sun, Z. Y. Wang, B. B. Huang, Y. Dai, X. Y. Qin and X. Y. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 7654–7658 CAS.
- M. Savage, S. Yang, M. Suyetin, E. Bichoutskaia, W. Lewis, A. J. Blake, S. A. Barnett and M. Schröder, Chem.–Eur. J., 2014, 20, 8024–8029 CrossRef CAS PubMed.
- B. Panella, M. Hirscher, H. Putter and U. Muller, Adv. Funct. Mater., 2006, 16, 520–524 CrossRef CAS.
- X. Lin, G. Gao, L. Zheng, Y. Chi and G. Chen, Anal. Chem., 2013, 86, 1223–1228 CrossRef PubMed.
- M. Zhang, G. Feng, Z. Song, Y.-P. Zhou, H.-Y. Chao, D. Yuan, T. T. Y. Tan, Z. Guo, Z. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241–7244 CrossRef CAS PubMed.
- X. Zhang, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2009, 265, 155–160 CrossRef CAS.
- J.-L. Wang, C. Wang and W. Lin, ACS Catal., 2012, 2, 2630–2640 CrossRef CAS.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS.
- D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637–20640 CrossRef CAS PubMed.
- W. Wu, A. M. Kirillov, X. Yan, P. Zhou, W. Liu and Y. Tang, Angew. Chem., Int. Ed., 2014, 126, 10825–10829 CrossRef.
- J. Yu, Y. Cui, C. Wu, Y. Yang, Z. Wang, M. O'Keeffe, B. Chen and G. Qian, Angew. Chem., Int. Ed., 2012, 51, 10542–10545 CrossRef CAS PubMed.
- J. Zou, Q. Peng, Z. Wen, G. Zeng, Q. Xing and G. Guo, Cryst. Growth Des., 2010, 10, 2613–2619 CAS.
- J.-K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071 CAS.
- M. Y. Masoomi and A. Morsali, Coord. Chem. Rev., 2012, 256, 2921–2943 CrossRef CAS.
- C.-B. Li, Z.-J. Li, S. Yu, G.-X. Wang, F. Wang, Q.-Y. Meng, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2013, 6, 2597–2602 CAS.
- F. Wang, W. J. Liang, J. X. Jian, C. B. Li, B. Chen, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2013, 52, 8134–8138 CrossRef CAS PubMed.
- X. Cheng, A. Zhang, K. Hou, M. Liu, Y. Wang, C. Song, G. Zhang and X. Guo, Dalton Trans., 2013, 42, 13698–13705 RSC.
- N. V. Maksimchuk, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, Chem. Commun., 2012, 48, 6812–6814 RSC.
- K. M. L. Taylor-Pashow, J. D. Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef CAS PubMed.
- P. Wu, N. Du, H. Zhang, L. Jin and D. Yang, Mater. Chem. Phys., 2010, 124, 908–911 CrossRef CAS.
- M.-T. H. Nguyen and Q.-T. Nguyen, J. Photochem. Photobiol., A, 2014, 288, 55–59 CrossRef CAS.
- C. Scherb, Controlling the Surface Growth of Metal–Organic Frameworks, Dissertation, LMU München, 2009.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, Wiley-VCH, 2nd edn, 2003 Search PubMed.
- G. Tong, F. Du, L. Xiang, F. Liu, L. Mao and J. Guan, Nanoscale, 2014, 6, 778–787 RSC.
- D. L. A. d. Faria, S. V. Silva and M. T. d. Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef.
- F. Zhang, H. Hu, H. Zhong, N. Yan and Q. Chen, Dalton Trans., 2014, 43, 6041 RSC.
- T. Yu, Y. Zhu, X. Xu, K. S. Yeong, Z. Shen, P. Chen, C. T. Lim, J. T. Thong and C. H. Sow, Small, 2006, 2, 80–84 CrossRef CAS PubMed.
- G. Gao, H. Wu, Y. Zhang, K. Wang, P. Huang, X. Zhang, S. Guo and D. Cui, J. Mater. Chem., 2011, 21, 12224 RSC.
- Y. Liu, L. Yu, Y. Hu, C. Guo, F. Zhang and X. Wen Lou, Nanoscale, 2012, 4, 183–187 RSC.
- T. Fujii, F. M. F. d. Groot and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 3195–3202 CrossRef CAS.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529 CrossRef CAS.
- J.-D. Xiao, L.-G. Qiu, X. Jiang, Y.-J. Zhu, S. Ye and X. Jiang, Carbon, 2013, 59, 372–382 CrossRef CAS.
- X. Wang, X. Chen, L. Gao, H. Zheng, M. Ji, C. Tang, T. Shen and Z. Zhang, J. Mater. Chem., 2004, 14, 905 RSC.
- D. Li, X. Hu, Y. Sun, S. Su, A. Xia and H. Ge, RSC Adv., 2015, 5, 27091–27096 RSC.
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- X.-J. Yang, B. Chen, L.-Q. Zheng, L.-Z. Wu and C.-H. Tung, Green Chem., 2014, 16, 1082 RSC.
- J. He, J. Wang, Y. Chen, J. Zhang, D. Duan, Y. Wang and Z. Yan, Chem. Commun., 2014, 50, 7063–7066 RSC.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- R. P. Sabatini, W. T. Eckenhoff, A. Orchard, K. R. Liwosz, M. R. Detty, D. F. Watson, D. W. McCamant and R. Eisenberg, J. Am. Chem. Soc., 2014, 136, 7740–7750 CrossRef CAS PubMed.
- M. P. McLaughlin, T. M. McCormick, R. Eisenberg and P. L. Holland, Chem. Commun., 2011, 47, 7989–7991 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- S. Saha, G. Das, J. Thote and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 14845–14851 CrossRef CAS PubMed.
- Y. Kataoka, K. Sato, Y. Miyazaki, K. Masuda, H. Tanaka, S. Naito and W. Mori, Energy Environ. Sci., 2009, 2, 397–400 CAS.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CAS.
- G.-G. Luo, K. Fang, J.-H. Wu and J. Mo, Chem. Commun., 2015, 51, 12361–12364 RSC.
- F. Wang, Y. Jiang, A. Gautam, Y. Li and R. Amal, ACS Catal., 2014, 4, 1451–1457 CrossRef CAS.
- E. Joselevich and I. Willner, J. Phys. Chem., 1995, 99, 6903–6912 CrossRef CAS.
- S. Yu, Y. H. Kim, S. Y. Lee, H. D. Song and J. Yi, Angew. Chem., Int. Ed., 2014, 53, 11203–11207 CrossRef CAS PubMed.
- R. R. Yeredla and H. Xu, Nanotechnology, 2008, 19, 055706 CrossRef PubMed.
- A. Murphy, Sol. Energy Mater. Sol. Cells, 2007, 91, 1326–1337 CrossRef CAS.
- R. López and R. Gómez, J. Sol-Gel Sci. Technol., 2011, 61, 1–7 CrossRef.
- S. Min and G. Lu, J. Phys. Chem. C, 2011, 115, 13938–13945 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S6, eqn (1–3), Table S1. See DOI: 10.1039/c5ra23838b |
| ‡ The authors equally contributed to the manuscript. |
| This journal is © The Royal Society of Chemistry 2016 |