
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclopentadithiophene–benzothiadiazole copolymers with permutations of repeating unit length and ratios; synthesis, optical and electrochemical properties and photovoltaic characteristics†
Shu-Wei
Chang
a,
Jeff
Kettle
b,
Huw
Waters
b and
Masaki
Horie
*a
aDepartment of Chemical Engineering, National Tsing-Hua University, 101, Section 2, Kuang-Fu Road, Hsinchu, 30013, Taiwan. E-mail: mhorie@mx.nthu.edu.tw
bSchool of Electronics, Bangor University, Dean st., Bangor, Gwynedd, LL57 1UT, Wales, UK
First published on 11th December 2015
Abstract
Conjugated copolymers with varying ratios and lengths of cyclopentadithiophene (CPDT) to benzothiadiazole (BT) repeating units, –[(CPDT)x–(BT)y]n–, have been synthesized by palladium complex catalysed Suzuki coupling polymerisation, direct arylation polymerisation or oxidative polymerisation using iron(III) chloride. The different permutations of the co-polymers allow for tuning of the optical and electrical properties. To our knowledge, this is one of the first studies of its kind for conjugated polymers for use in OPVs. The optical band gaps were measured between 1.7 and 2.0 eV in film, in which the polymer with ratios of CPDT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BT (x:y = 1:1) units showed the lowest bandgap followed by x:y = 1:2, 2:1, 2:2 and 3:3. Hole mobility and solar cell performance of these polymers with PC61BM as the electron acceptor were measured. A relatively small variation in hole mobility was observed between polymers. However, the best reported power conversion efficiency (PCE) was measured at 2.5% when processed in the absence of any additives using the polymer with ratios of CPDT:BT (x:y = 2:2), which was followed by the ratios of x:y = 2:1, 1:1, 1:2 and 3:3, indicating that variation of the ratios and length of donor and acceptor units affect OPV performance and better performance is achievable by careful consideration of the donor-to-acceptor ratio.
:BT (x:y = 1:1) units showed the lowest bandgap followed by x:y = 1:2, 2:1, 2:2 and 3:3. Hole mobility and solar cell performance of these polymers with PC61BM as the electron acceptor were measured. A relatively small variation in hole mobility was observed between polymers. However, the best reported power conversion efficiency (PCE) was measured at 2.5% when processed in the absence of any additives using the polymer with ratios of CPDT:BT (x:y = 2:2), which was followed by the ratios of x:y = 2:1, 1:1, 1:2 and 3:3, indicating that variation of the ratios and length of donor and acceptor units affect OPV performance and better performance is achievable by careful consideration of the donor-to-acceptor ratio.
1. Introduction
Organic photovoltaics (OPVs) based on solution processable polymers and fullerenes have attracted remarkable interest in energy generation as they provide the potential for lower cost manufacture, using technique such as roll-to-roll (R2R) printing. Recent research in this area has led to a reported power conversion efficiency (PCE) of over 11%.1 In addition, the modules can be made entirely flexible, which enables a wider range of applications and finally, they can provide benefits in terms of aesthetics, as the appearance can be tuned by altering the active layer.1–7 The latter is particularly significant to architects and to achieve this, molecular design has been vigorously studied. The main strategy pursued by researchers to vary the optical properties is to fuse ring building block and to use donor–acceptor (D–A) alternating structure in the polymer main chain, which enables a reduction of the bandgap of the polymer, leading to effective conversion of sunlight into electricity by maximizing the polymer absorption.2–4 One of most studied donor type conjugated polymers is poly[4,4-bis(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene-2,6-diyl-alt-2,1,3-benzothiadiazole-4,7-diyl] (PCPDTBT) (Fig. 1) which is categorized in a third generation of conjugated polymers for OPV application.5,6 Whilst the D–A chemical structure is the most commonly reported polymer structure, D–A–D structures are also investigated, but the improvement on molecular design by further combinations of D- and A-units are possible but have so far not been well reported.7–10 | ||
| Fig. 1 PCPDTBT with permutations of repeating unit length and ratios. | ||
Previously we reported PCPDTBT analogues containing alternating structure of two 4,4-bis(2-ethylhexyl)cyclopentadithiophene (CPDT) units with one 2,1,3-benzothiadiazole (BT) unit, which showed significant improvement in organic field effect transistors (OFETs) hole mobility and moderate improvement in a PCE of OPV devices.8
The superior performance was perceived to be due to a higher molecular weight (number average molecular weight, Mn = 100k) of the polymer synthesized via iron(III) chloride oxidative polymerisation of (CPDT)–(BT)–(CPDT) oligomer.
Despite the importance of the fundamental studies on altering of D–A ratio on the polymer properties such as tailoring HOMO–LUMO levels and superior OFET and OPV device performances, PCPDTBT analogues with alternative D–A ratios have not been systematically studied, primarily due to the difficulties in the synthetic procedure. Here we report the synthesis and characterisation of new PCPDTBT analogues comprising of varying ratios of CPDT to BT repeating units, –[(CPDT)x–(BT)y]n– (Fig. 1). This is the first such report achieved in this area. The polymers have been synthesized by palladium complex catalysed Suzuki coupling, direct arylation or oxidative polymerisation using iron(III) chloride. UV-vis absorption spectrometry and cyclic voltammetry were used to compare the optical and electrochemical properties of the polymers. The novel polymer structures show differing HOMO–LUMO levels, indicating that the optical and electrochemical properties can be ‘tuned’ and show an increase in solar cell performance when compared to a standard –CPDT–BT–polymer material system. Whilst this work focused on the CPDT–BT material system, the results could provide researchers insight into the influence D–A ratios in other material systems.
2. Results and discussion
2.1. Synthesis of monomers and polymers
Scheme 1 shows synthesis of monomers. Compound 1 was synthesized by palladium catalysed direct arylation of CPDT with 4,7-dibromo-2,1,3-benzothiadiazole (BT-Br2) as the first coupling product in a cosolvent of DMF:toluene = 1:1 in 26% yield. Compound 2 could be obtained by increasing the proportion of CPDT also using direct arylation in the solvent of DMF; in this case a 40% yield was achieved.9 Compound 1 was used as the base-material for the subsequent synthesis of the other three monomers: 3, 4 and 5. Compound 3 was synthesized via Yamamoto coupling in the presence of Ni(cod)2 (cod = 1,5-cyclooctadiene), bipyridine and 1,5-cyclooctadiene in DMF at 60 °C for a day, whereas 4 was synthesized via Suzuki coupling of two equivalents of 1 with an equivalent of 2,1,3-benzothiadiazole-4,7-bis(boronic acid pinacol ester). These monomers were isolated by column chromatography and preparative size exclusion chromatography (SEC), and then characterized by 1H, 13C and 2D NMR spectroscopies in deuterated chloroform, mass spectroscopy and elemental analysis (see the experimental section and the ESI†).
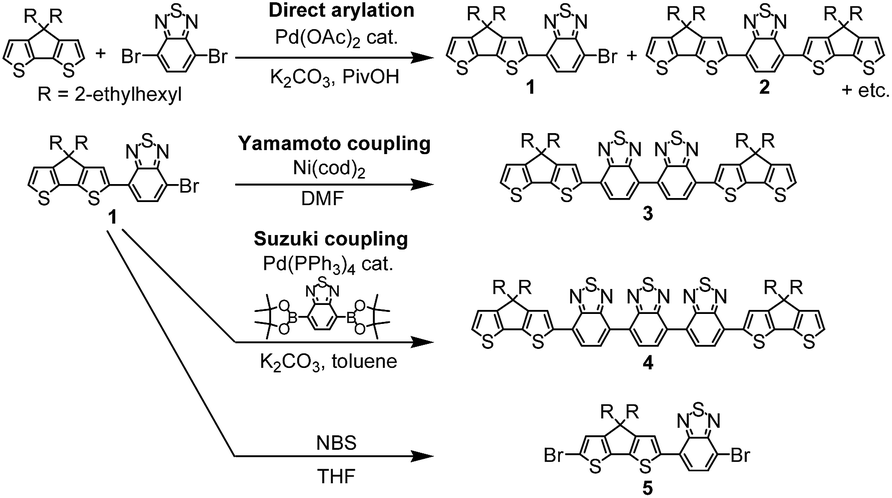 | ||
| Scheme 1 Synthesis of monomers. | ||
Scheme 2 and Table 1 show the synthetic scheme and polymerisation results for polymers P1–P5. PCPDTBT (P1) possesses a 1:1 ratio of CPDT and BT in its alternating sequence and was obtained via palladium catalysed Suzuki coupling of dibromo-CPDT and 2,1,3-benzothiadiazole-4,7-bis(boronic acid pinacol ester). In this reaction, trace amount of palladium catalyst (5.9 ppm, analysed by inductively coupled plasma mass spectrometry (ICP-MS)) provided the high reactivity to produce P1 with Mn of 53200 and polydispersity index (PDI) of 3.7. The polymer, P2, comprising CPDT:BT ratio = 2:2, was synthesized by an oxidative polymerisation of 3 using iron(III) chloride in chlorobenzene. Here, the reaction condition was maintained at a relatively low temperature of 60 °C, but with a long reaction time for 48 hours, which was critical in order to avoid formation of branched structures that could be produced due to reaction on 3- and 5-positions of CPDT. Using similar conditions from previous reports,8 such as an 80 °C reaction temperature, led to an insoluble polymer cluster, or a gelation in the formed products (see Table S2 in ESI†). After polymerisation, P2 was firstly de-doped using hydrazine and then purified, in order to remove any insoluble by-products and residues of iron, which was then followed by precipitation in methanol, Soxhlet extractions, and flash silica gel column chromatography. The molecular weight (Mn = 20300) was not as high as expected from using oxidative polymerisation, presumably due to the lower reaction temperature (60 °C).
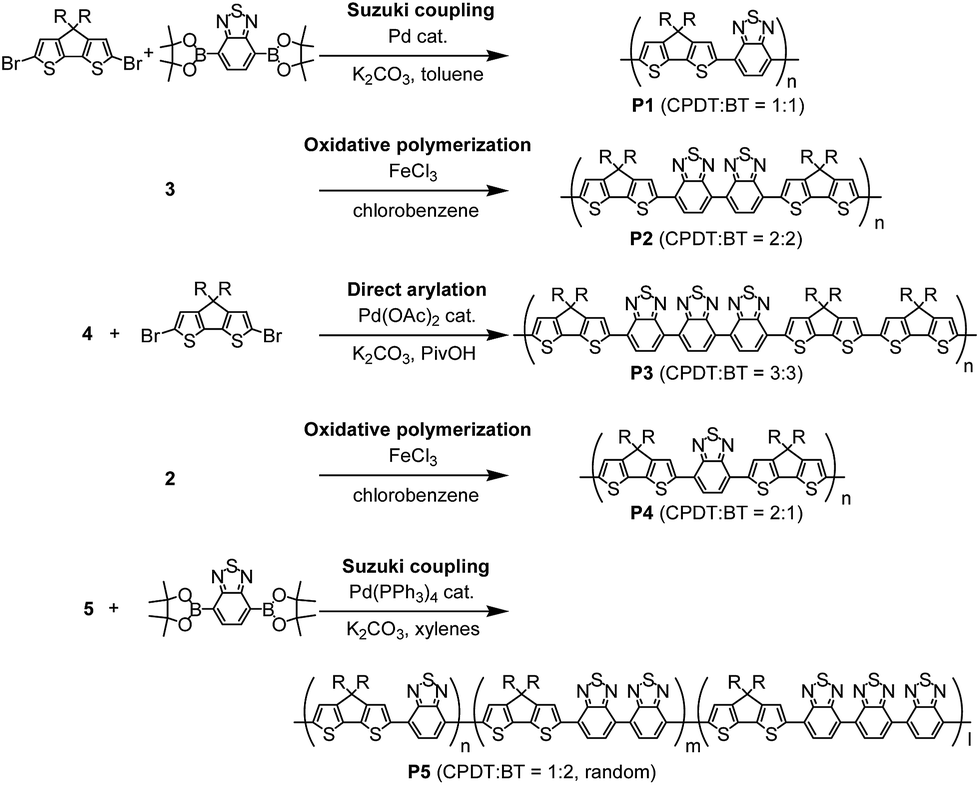 | ||
| Scheme 2 Synthesis of polymers. | ||
| Polymer (CPDT:BT) |
Reaction | M n a | M w/Mna | Yieldb (%) |
|---|---|---|---|---|
| a Calculated from gel-permeation chromatography (GPC) measurements carried out using THF as the solvent and calibrated by polystyrene standards. b After Soxhlet extractions. | ||||
|
P1 (1:1) |
Suzuki coupling | 53200 |
3.7 | 88 |
|
P2 (2:2) |
Oxidative | 20300 |
5.2 | 55 |
|
P3 (3:3) |
Direct arylation | 9000 | 1.7 | 50 |
|
P4 (2:1) |
Oxidative | 51300 |
3.7 | 76 |
|
P5 (1:2) |
Suzuki coupling | 30300 |
9.1 | 81 |
Monomer 4, comprising of three BT capped with two CPDT units, is joined with dibromo-CPDT by direct-arylation, in order to synthesize P3, which possesses of a CPDT:BT ratio = 3:3. The reaction conditions were systematically optimised, with the final synthesis undertaken with pivalic acid in N-methyl-2-pyrrolidone (NMP) at 80 °C for 12 hours (see Table S3 in ESI†). The optimised reaction time was much shorter than our previous report for synthesis of P1via direct arylation between CPDT and BT-Br2 (20 hours),11 because it was discovered that longer reaction time for 20 hours produced a high proportion of insoluble gels, thus lowering the yield of soluble part (<10%). However, by using a shorter reaction for 3 hours, lower molecular weight P3 (Mn = 6500) was obtained. Under the same reaction condition (3 hours reaction time), higher molecular weight of P1 (Mn = 9000) could be produced via direct arylation of CPDT with BT-Br2. The lower reactivity of monomer 4 for synthesis of P3 than CPDT for P1 in direct arylation is due to the presence of electron-deficient BT units in monomer 4. Kuwabara et al. reported that palladium catalysed direct arylation of electron-deficient monomers was unreactive in a polar solvent such as dimethylacetamide.12
The polymer, P4, comprising of two CPDT units and one BT unit, was synthesized and purified by an analogous method to P2. P4 was shown to have a higher molecular weight of Mn = 51300 and lower PDI of 3.7, because of enhanced solubility of the CPDT–BT–CPDT unit over the CPDT–BT–BT–CPDT unit, as well as higher optimal reaction temperature (80 °C).
The random copolymer, P5, comprising CPDT:BT = 1:2 was synthesized via palladium catalysed Suzuki coupling polymerisation of 1 and 2,1,3-benzothiadiazole-4,7-bis(boronic acid pinacol ester) at 90 °C in xylenes. Due to the symmetric structure of both monomers, the resulting polymer should have random sequence of CPDT:BT = 1:1, 1:2 and 1:3 in the backbone, which shows a broad peak around 7.5–8.8 ppm for aromatic region in the 1H NMR spectrum. Nevertheless, an average ratio of CPDT:BT is 1:2 was confirmed, according to the number of protons between aromatic units and 2-ethylhexyl groups. Janssen et al. reported an alternating copolymer with –CPDT–BT–BT-sequence,5h which possessed the same CPDT:BT ratio (1:2) with P5. However, the molecular weight of their polymer was low (Mn = 4300) due to the limited solubility of 7,7′-diiodo-4,4′-bis(2,1,3-benzothiadiazole) during Suzuki coupling polymerisation. Therefore, the synthesis of the random copolymer P5 using the approach outlined in this paper is advantageous for the production of the high molecular weight polymer (Mn = 30300), which should lead to higher performances in organic electronic devices.
2.2. Optical and electrochemical properties
The optical properties of polymers were examined by UV-vis absorption spectroscopy in tetrahydrofuran (THF) solution and in thin film, as shown in Fig. 2. The optical band gap (Eg-opt) were estimated from the onset of the absorption spectra in thin film, and results are summarized in Table 2. | ||
| Fig. 2 UV-vis spectra of polymers in (a) THF and (b) thin films. A photo of the polymers in THF is inserted. | ||
| Polymer (CPDT:BT) |
λ max in THF (nm) | λ max in film (nm) | λ onset in film (nm) | E g-opt a (eV) | HOMOb (eV) | LUMOc (eV) | E g-ec d (eV) |
|---|---|---|---|---|---|---|---|
| a E g-opt = optical band gap calculated from absorption onset of films. b HOMO = −(4.8 + Epa-onset − EFc). Half wave potential of ferrocene, EFc (=0.55 V vs. AgCl/Ag), was measured in MeCN solution. c LUMO = −(4.8 + Epc-onset − EFc). d Electrochemical band gap, Eg-ec = LUMO − HOMO. | |||||||
|
P1 (1:1) |
704 | 732 | 861 | 1.44 | −5.00 | −3.19 | 1.81 |
|
P2 (2:2) |
646 | 663 | 785 | 1.58 | −5.21 | −3.36 | 1.85 |
|
P3 (3:3) |
607 | 613 | 761 | 1.63 | −5.20 | −3.61 | 1.59 |
|
P4 (2:1) |
696 | 697 | 816 | 1.52 | −4.98 | −3.11 | 1.87 |
|
P5 (1:2) |
690 | 685 | 816 | 1.52 | −5.14 | −3.37 | 1.77 |
The maximum absorption wavelength (λmax) for P1 (CPDT:BT = 1:1) is observed at 704 nm in a THF solution and this was seen to decrease with the increasing CPDT–BT ratio length; a peak absorption of 646 nm was observed for P2 (2:2) and 607 nm for P3 (3:3). These results can be considered as due to weaker charge transfer in P2 and P3 derived from increased spacing between CPDTs and BTs sequences. The suppressed charge transfer ability observed from the successive structures provides insight into the influence of donors and acceptors ratios on the conjugated length in alternating copolymer. The absorption maximum of P4 (2:1) and P5 (1:2, random) was observed at 696 nm and 690 nm, respectively, which represents a slightly blue wavelength shift of ca. 10 nm from P1. These results show that efficient charge transfer structure can be obtained when the donor and the acceptor units contain at least one D–A connection with neighbouring units, however a higher proportion of CPDT or BT in the polymer backbone does not lead to higher wavelength absorption.
In thin films, the trend of λmax variations for P1–P3 is similar to that in the solutions; λmax at 732 nm for P1 > 663 nm for P2 > 613 nm for P3. The absorption spectra of all polymers, except for the random copolymer P5, showed a red-shift in absorption and thus they show the smaller optical bandgap than solution, due to increased intermolecular association in the solid state (Fig. 2b). Because P5 possesses a random combination of CPDT and BT across the backbone, the stacking or organization ability may be poor in the solid state when compared to other polymers. Among the polymers P1–P3 which have the same equivalent of CPDT:BT ratio, P3 (3:3) exhibits the smallest red-shift by 6 nm, while P1 (1:1) shows highest red-shift by 68 nm. Previously we observed X-ray single crystallographic structure of the CPDT–BT–CPDT oligomer having ethyl groups instead of 2-ethylhexyl group on CPDT, which shows π–π stacking at the central BT unit of the molecule but no intermolecular interaction between CPDT units.9 Therefore, the continuous CPDT sequence probably has large intermolecular steric hindrance due to the proximity of neighbouring 2-ethylhexyl side chains on 4-position of CPDT, which suppress the stacking of polymer chains.
The electrochemical properties of polymers were investigated by cyclic voltammetry (Fig. 3), and these results are summarized in Table 2. Thin film of the polymers were deposited on a platinum plate and cyclic voltammograms (CVs) were recorded in an acetonitrile solution containing [(n-C4H9)4N]PF6 (0.10 M) using a AgCl/Ag reference electrode. Interestingly, P2 and P3 show multi-step oxidation and reduction peaks, whereas P1, P4 and P5 show single-step oxidation and reduction peaks. These results suggest that polymers with more than two continuous sequence of BT units tend to stabilize radical dications and dianions in –(CPDT)x– and –(BT)y– sequences during electrochemical oxidation and reduction, respectively. The energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) for these polymers were estimated form the onset potential of oxidation and reduction peaks of the CV curves (Table 2). The electrochemical band gap (Eg-ec) of the polymers was estimated from the onset of these redox peaks. Fig. 4 shows a summary of the calculated energy diagrams of the polymers. When comparing P1–P3, it appears that the BT unit more profoundly determines the HOMO and LUMO levels; increasing the BT units leads to lower HOMO and lower LUMO levels. The HOMO level of P1, P2 and P3 were estimated to be −5.00 eV, 5.21 eV and −5.20 eV, respectively, suggesting the presence of BT can reduce the HOMO level, although saturation in HOMO level is observed around −5.2 eV. Conversely, a stepwise decrease of LUMO level is observed (−3.19 eV for P1, −3.36 eV for P2, −3.61 eV for P3). Similarly, when comparing P1, P4 and P5, P5 with higher number of BT unit (CPDT:BT = 1:2) shows the lowest HOMO and LUMO levels, compared to P1 (1:1) and P4 (2:1), which possess fewer BT units. These results indicate that the HOMO and LUMO energy levels are more strongly affected by the number of BT units in the polymer backbone, than the CPDT units.
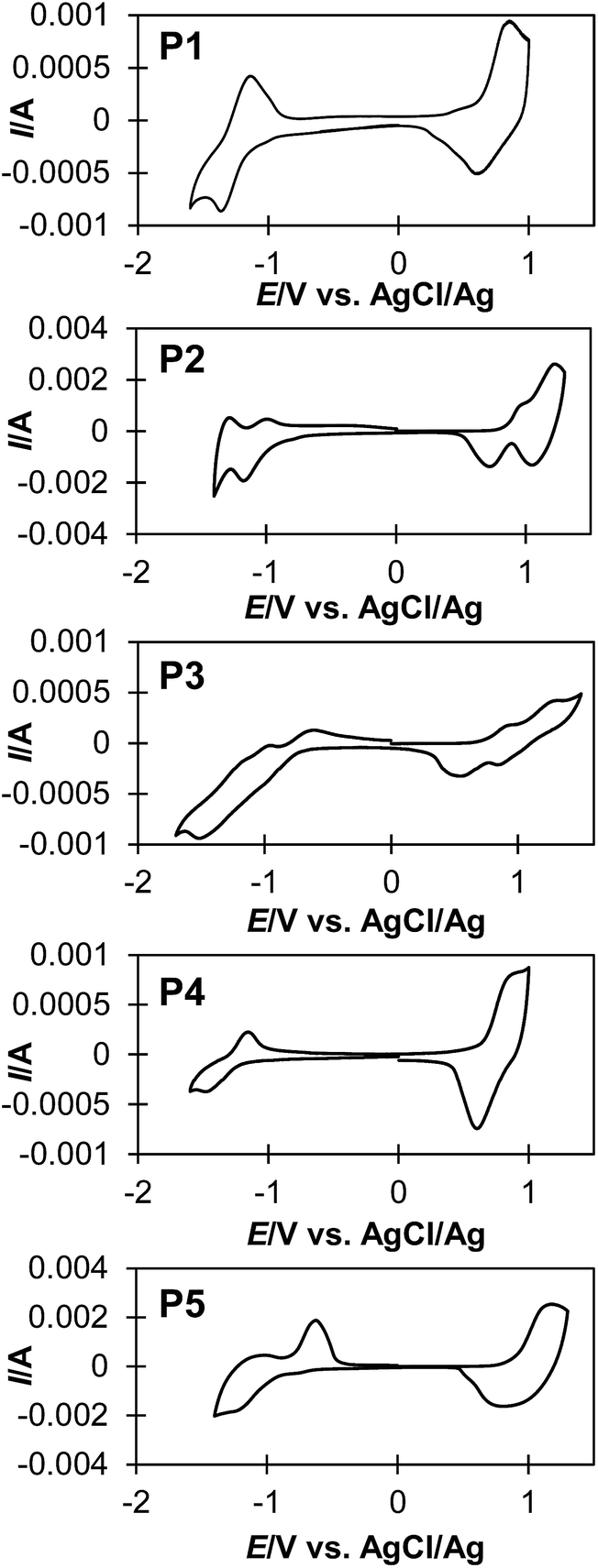 | ||
| Fig. 3 Cyclic voltammograms of polymers. | ||
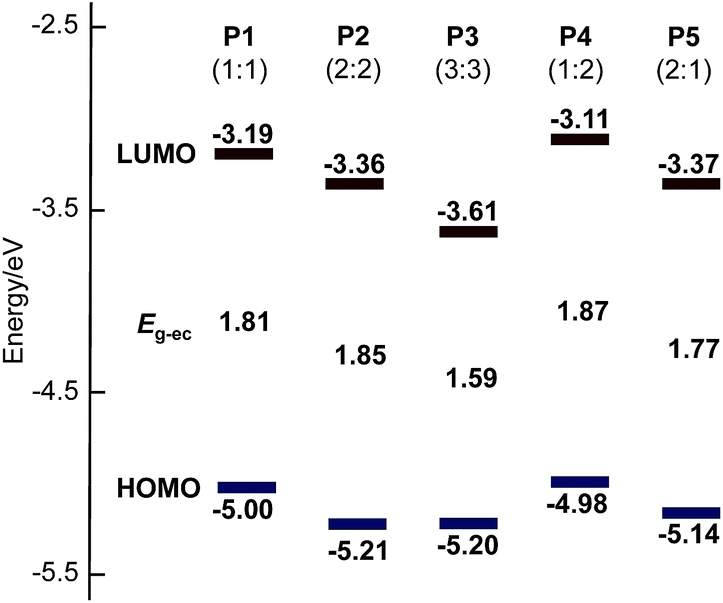 | ||
| Fig. 4 HOMO and LUMO energy diagrams of P1 to P5. The number of BT units have the greatest influence on the measured energy levels. | ||
2.3. SCLC and OPV characteristics
Hole-only devices with the structure ITO/PEDOT:PSS/polymer (P1–P5)/Au were made by spin coating a 100 nm thick layer of pristine polymer on to PEDOT:PSS coated ITO substrates followed by evaporation of a gold top electrode through a shadow mask. Steady state current density–voltage measurements were taken at room temperature in the dark using a Keithley 237 Source Measure Unit, with the device mounted in a partially evacuated sample chamber. Fitting the data to the Mott–Gurney law13 and assuming no difference in injection efficiency upon purification, the best mobility's are summarized in Table 3. The mobility of P2 was calculated at 1.5 × 10−4 cm2 V−1 s−1, which was similar to P1. However, the mobility of P3 was about 5 times lower than that of P2. These results indicate that the polymer with D–A ratio of 2:2 possesses the optimal charge transport properties and D–A ratio of 3:3 lead to reduced charged transfer. P4 and P5 show mobility of 2.0 × 10−4 cm2 V−1 s−1 and 1.5 × 10−4 cm2 V−1 s−1, respectively, which values are similar to P2, suggesting that the polymers with D–A ratio of 2:1 and 1:2 also possess efficient charge transport. The variations in the mobility observed between polymers P1, P2, P4 and P5 are relatively minor. P3 shows a significant drop in mobility and is likely to be due to the larger coalescing of donor or acceptor units, which inhibit and suppress charge transfer across the respective polymer.
| Polymer (CPDT:BT) |
Optimum annealing temperature (°C) | μ h a (cm2 V−1 s−1) | V oc b (V) | J sc c (mA cm−2) | FFd (%) | PCEe (%) |
|---|---|---|---|---|---|---|
| a Average hole mobility is shown across 6 devices. In brackets, the highest recorded hole mobility is reported. b Open circuit voltage. c Short current density. d Fill factor. e Power conversion efficiency. f Hole mobility of P1 is shown in our previous report.14 | ||||||
|
P1 (1:1)f |
60 | 2.0 × 10−4 | 0.611 | −6.92 | 43.8 | 1.95 |
|
P2 (2:2) |
60 | 1.5 × 10−4 (2.5 × 10−4) | 0.760 | −8.22 | 39.3 | 2.45 |
|
P3 (3:3) |
90 | 3.2 × 10−5 (1.0 × 10−4) | 0.550 | −1.73 | 35.3 | 0.33 |
|
P4 (2:1) |
120 | 2.0 × 10−4 (2.0 × 10−4) | 0.550 | −9.14 | 48.5 | 2.44 |
|
P5 (1:2) |
90 | 1.5 × 10−4 (3.5 × 10−4) | 0.747 | −6.51 | 39.5 | 1.92 |
Photovoltaic devices of the polymers were fabricated on glass substrates with an indium tin oxide (ITO)/poly(3,4-ethyl-enedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) anode. The active layer was spin cast from chlorobenzene solutions of the polymer mixed with PC61BM at a concentration of 3 wt%. Spin speeds were varied between 1.5k and 3k rpm for each polymer in order to deduce the optimum layer thickness. Prior to coating, the polymer:fullerene blends were allowed to dissolve for one week on a hotplate stirrer and filtered using a 0.45 μm PTFE filter. The preliminary OPV tests for these polymers are shown in Fig. 5 and Table 3. All polymers show PCE around 2% (except P3), which was obtained without the use of processing additives, as these are known to degrade lifetime, which could impact the reproducibility of OPV results between material systems.15 Among them, P2 with CPDT:BT ratio of 2:2 exhibited the best PCE of 2.45% and P4 exhibits a PCE of 2.44%, both higher than the more commonly reported polymer PCPDTBT (P1).
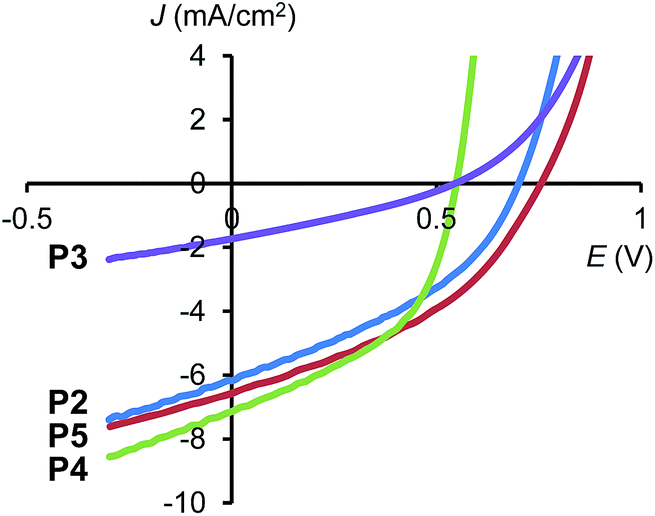 | ||
| Fig. 5 Current density–voltage characteristics of P2–P5 blended with PC61BM, measured under AM1.5G illumination. | ||
This data indicates that by careful selection of the donor-to-acceptor ratio in polymers, not only is ‘tuning’ of the optical and electrochemical properties possible, but an enhancement in performance is achievable. By altering the polymer structure to P2 and P4, a relative enhancement of ∼25% is demonstrated over the original polymer. Compared to many literature reports, the efficiencies are low, but this is achieved without the use of processing additives, which are known to increase the efficiency by a factor 2–3 times in the PCPDTBT material system.13 Based upon this expected factor enhancement, the use of processing additives could boost the efficiency of OPVs made with P2 and P4 to ∼6%.
With respect to P2 (with CPDT:BT ratio 2:2), the primary reason for this is due to higher Voc (0.76 V) derived from deeper HOMO level (−5.21 eV). However, the high PCE associated with P4 (with CPDT:BT ratio 2:1) appears to be due to the higher Jsc and FF than the other polymers, as it possesses the lowest Voc (0.55 V). The major difference in P4 is that it possesses lower molecular weight (Mn = 51300), compared to the same polymer previously reported (Mn = 107000);8 therefore P4 has better solubility which enables a better film morphology with PC61BM to be established, leading to higher Jsc and fill factor.
P3 showed the lowest performance, which is not surprising as the mobility was very poor and likely to lead to high charge recombination, which is supported by the low value for FF (0.35). Finally, P5 showed no improvement in PCE, when compared to the material with a 1:1 ratio and was measured at 1.92%. OPVs made with P5 have a high Voc (0.75 V) but lower Jsc (−6.5 mA cm−2). According to the absorption spectrum in film, the random conformation of P5 weakened the interchain π–π stacking, which may attribute to the lower Jsc. Solubility of the polymers decrease with increase of BT unit, resulting in rough morphology and low fill factor. When the polymers possess more than one BT unit in their repeating unit, the fill factor reduces to <0.4, which can be equated to poor active layer morphology derived from the lower solubility of the polymers, leading to increased shunts in the devices.
The morphology of the polymer:PC61BM films were investigated using atomic force microscopy (AFM). The AFM images are shown in the ESI.† All films show similar homogeneous morphology with an average roughness (Ra) in the range between 0.30 nm and 0.39 nm. These results suggest that differences in PCE are due to different electronic state derived from fundamental energy levels obtained from the polymer main chain.
3. Conclusions
The ratio of CPDT to BT repeating units in polymers has been systematically varied to understand the influence of repeating units on polymer properties and optoelectronic device performance. This is particularly important given the recent interest in OPVs for use in building integrated PVs.16,17 Here the ability to ‘tune’ the colour to suit architectural demands is imperative. The approach set out in this paper enables this without adjusting the fundamental polymer material and without compromising the efficiency of the solar cell. To our knowledge, this is the first time such a wide range of polymers with diverse backbone structures has been employed in OPVs. The polymers of –[(CPDT)x–(BT)y]n– have been studied with x:y = 1:1, 2:2, 3:3, 2:1, 1:2, which were synthesised by Suzuki coupling, direct arylation and oxidative polymerisation. The optical and electrochemical properties of these polymers have been compared in terms of UV-vis spectra and CVs. With increase of both CPDT and BT units, optical band gap is increased due to weaker charge transfer between donor and acceptor units, and HOMO–LUMO levels are decreased, in which the energy levels are mainly affected by number of BT units rather than CPDT units. These results provide insight of maximum conjugated length for the building block of conjugated polymers. All polymers showed similar hole mobility up to 2 × 10−4 cm2 V−1 s−1, except x:y = 3:3, according to SCLC. The copolymer with 2:2 ratio showed the best OPV performance of 2.5% with PC61BM because of high open-circuit voltage derived from appropriate energy levels and relatively high Jsc derived from suitable photoabsorption range among these polymers. The results indicate a clear benefit is achieved by using a more complex structure as evidenced by the increase in solar cell performance and ability to ‘tune’ the optical properties of the polymer.
4. Experimental
4.1. General methods
4,7-Dibromo-2,1,3-benzoxadiazole, 2,1,3-benzoxadiazole-4,7-bis(boronic acid pinacol ester), Pd(OAc)2, Pd(PPh3)4 and pivalic acid were purchased from Sigma-Aldrich and used without further purification. Compounds 1 and 2 and 4,4-bis(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene were synthesized according to our previous report.8,9,11 Synthesis was performed under nitrogen atmosphere using standard Schlenk techniques. Compounds were purified by preparative size exclusion chromatography (SEC) using JAI LC-9204 with a column GAIGEL-1H-40 eluted with chloroform (flow rate = 14 mL min−1). Polymer molecular weight was determined by GPC in tetrahydrofuran (THF) solution using a JASCO 880-PU and a JASCO 870-UV detector (referenced to polystyrene standards). 1H NMR spectra were obtained using a Bruker 500 MHz spectrometer. UV-vis absorption spectra were recorded on a Hitachi U-3300 UV-vis spectrophotometer. Cyclic voltammetry was performed at 0.10 V s−1 in a DY2300 electrochemical analyzer with a three-electrode cell, AgCl/Ag as reference electrode, platinum wire as counter electrode and polymer film on a platinum plate as the working electrode in nitrogen-purged anhydrous 0.10 M tetrabutylammonium hexafluorophosphate acetonitrile solution at room temperature.4.2. Synthetic procedures
:DCM = 5:2 as eluent. Furthermore, the product was purified by preparative SEC using chloroform as eluent to give the purple solid (0.484 g, 26%). 1H NMR (500 MHz, CDCl3): δ 8.42 (m, 2H, BT, each peak was split into three singlets with the ratio of 1:2:1 due to racemic 2-ethylhexyl groups), 8.12 (s, 2H, 3-CPDT, the ratio was 1:2:1 due to racemic 2-ethylhexyl groups), 8.01 (m, 2H, BT, each peak was split into three singlets with the ratio of 1:2:1 due to racemic 2-ethylhexyl groups), 7.20 (d, J = 5.0 Hz, 2H, 6-CPDT), 6.97 (d, J = 5.0 Hz, 2H, 5-CPDT, each peak was split into three singlets with the ratio of 1:2:1 due to racemic 2-ethylhexyl groups), 1.98 (m, 8H, CH2), 1.00–0.88 (m, 28H, CH & CH2), 0.74 (t, J = 7.5 Hz, 12H, CH3), 0.60 (t, J = 7.5 Hz, 12H, CH3). FABMS: m/z = 1071 [M]+. Anal. calcd for (C62H78N4S6): C 69.48, H 7.34, N 5.23; found: C 67.90, H 6.90, N 5.13.
:DCM = 2:1 as the eluent. Furthermore, the product was purified by preparative SEC using chloroform as eluent to give the purple solid (0.264 g, 80%). 1H NMR (500 MHz, CDCl3): δ 8.56 (m, 2H, middle BT), 8.42 (d, J = 7.5 Hz, 2H, 5-BT), 8.16 (s, 2H, 3-CPDT, the ratio was 1:2:1 due to racemic 2-ethylhexyl groups), 8.04 (d, J = 7.5 Hz, 2H, 6-BT, each peak was split into three singlets with the ratio of 1:2:1 due to racemic 2-ethylhexyl groups), 7.21 (d, J = 5.0 Hz, 2H, 6-CPDT), 6.98 (d, J = 5.0 Hz, 2H, 5-CPDT, each peak was split into three singlets with the ratio of 1:2:1 due to racemic 2-ethylhexyl groups), 1.97 (m, 8H, CH2), 1.01–0.90 (m, 28H, CH & CH2), 0.75 (t, J = 7.5 Hz, 12H, CH3), 0.61 (t, J = 7.5 Hz, 12H, CH3). 13C{1H} NMR (125 MHz, CDCl3): δ 158.6, 154.3, 152.5 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N on BT), 139.6, 138.6, 136.9 (Cquart.), 131.6 (5-BT), 130.9 (middle BT), 129.6, 128.7, 128.5, 127.0 (Cquart.), 125.6 (6-CPDT), 123.7 (6-BT), 123.6 (3-CPDT), 123.4 (Cquart.), 122.4 (5-CPDT), 53.8 (4-CPDT), 43.2 (CH2), 35.2 (CH), 34.2, 28.6, 27.4, 22.8 (CH2), 14.1, 10.7 (CH3). FABMS: m/z = 1205 [M]+. Anal. calcd for (C68H80N6S7): C 67.73, H 6.69, N 6.97; found: C 67.71, H 6.90, N 6.84.
:2:1 due to racemic 2-ethylhexyl groups), 7.80 (d, 1H, J = 8.5 Hz, 5-BT), 7.64 (d, 1H, J = 8.0 Hz, 6-BT, each peak was split into three singlets with the ratio of 1:2:1 due to racemic 2-ethylhexyl groups), 6.97 (s, 1H, 5-CPDT, the ratio was 1:2:1 due to racemic 2-ethylhexyl groups), 1.98–1.82 (m, 4H, CH2), 1.03–0.88 (m, 14H, CH & CH2), 0.76 (t, 6H, J = 7.0 Hz, CH3), 0.60 (t, 6H, J = 7.0 Hz, CH3). 13C{1H} NMR (125 MHz, CDCl3): δ 157.7, 157.3 (CN on BT), 153.9, 151.6, 138.7, 138.2, 137.0 (Cquart.), 132.3 (5-BT), 127.9 (Cquart.), 125.4 (5-CPDT), 124.1 (6-BT), 123.4 (3-CPDT), 111.8, 111.2 (C–Br), 54.6 (4-CPDT), 43.1 (CH2), 35.2 (CH), 34.1, 28.5, 27.4, 22.7 (CH2), 14.0, 10.7 (CH3). FABMS: m/z = 694 [M]+. Anal. calcd for (C31H38Br2N2S3): C 53.60, H 5.51, N 4.03; found: C 59.81, H 7.32, N 3.36.
200, Mw/Mn = 3.7. UV-vis: λmax = 704 nm in THF, and λmax = 732 nm in film.
300, Mw/Mn = 5.2. UV-vis: λmax = 646 nm in THF, and λmax = 663 nm in film.
300, Mw/Mn = 3.7. UV-vis: λmax = 696 nm in THF, and λmax = 697 nm in film.
300, Mw/Mn = 9.1. UV-vis: λmax = 690 nm in THF, and λmax = 685 nm in film.
N on BT), 139.6, 138.6, 136.9 (Cquart.), 131.6 (5-BT), 130.9 (middle BT), 129.6, 128.7, 128.5, 127.0 (Cquart.), 125.6 (6-CPDT), 123.7 (6-BT), 123.6 (3-CPDT), 123.4 (Cquart.), 122.4 (5-CPDT), 53.8 (4-CPDT), 43.2 (CH2), 35.2 (CH), 34.2, 28.6, 27.4, 22.8 (CH2), 14.1, 10.7 (CH3). FABMS: m/z = 1205 [M]+. Anal. calcd for (C68H80N6S7): C 67.73, H 6.69, N 6.97; found: C 67.71, H 6.90, N 6.84.
:2:1 due to racemic 2-ethylhexyl groups), 7.80 (d, 1H, J = 8.5 Hz, 5-BT), 7.64 (d, 1H, J = 8.0 Hz, 6-BT, each peak was split into three singlets with the ratio of 1:2:1 due to racemic 2-ethylhexyl groups), 6.97 (s, 1H, 5-CPDT, the ratio was 1:2:1 due to racemic 2-ethylhexyl groups), 1.98–1.82 (m, 4H, CH2), 1.03–0.88 (m, 14H, CH & CH2), 0.76 (t, 6H, J = 7.0 Hz, CH3), 0.60 (t, 6H, J = 7.0 Hz, CH3). 13C{1H} NMR (125 MHz, CDCl3): δ 157.7, 157.3 (CN on BT), 153.9, 151.6, 138.7, 138.2, 137.0 (Cquart.), 132.3 (5-BT), 127.9 (Cquart.), 125.4 (5-CPDT), 124.1 (6-BT), 123.4 (3-CPDT), 111.8, 111.2 (C–Br), 54.6 (4-CPDT), 43.1 (CH2), 35.2 (CH), 34.1, 28.5, 27.4, 22.7 (CH2), 14.0, 10.7 (CH3). FABMS: m/z = 694 [M]+. Anal. calcd for (C31H38Br2N2S3): C 53.60, H 5.51, N 4.03; found: C 59.81, H 7.32, N 3.36.
200, Mw/Mn = 3.7. UV-vis: λmax = 704 nm in THF, and λmax = 732 nm in film.
300, Mw/Mn = 5.2. UV-vis: λmax = 646 nm in THF, and λmax = 663 nm in film.
300, Mw/Mn = 3.7. UV-vis: λmax = 696 nm in THF, and λmax = 697 nm in film.
300, Mw/Mn = 9.1. UV-vis: λmax = 690 nm in THF, and λmax = 685 nm in film.
4.3. Device fabrication
Acknowledgements
The authors thank Yanhua Hong and Dinesh Kumar from the Bangor University for AFM. The authors thank the National Science Council of Taiwan for financial support. HW would like to thank Bangor University for his 125 Scholarship.Notes and references
- H. Zhou, Y. Zhang, C. K. Mai, S. D. Collins, G. C. Bazan, T. Q. Nguyen and A. J. Heeger, Adv. Mater., 2015, 27, 1767 CrossRef PubMed.
- (a) H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef; (b) H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed; (c) M.-C. Yuan, M.-Y. Chiu, S.-P. Liu, C.-M. Chen and K.-H. Wei, Macromolecules, 2010, 43, 6936–6938 CrossRef CAS; (d) G. Y. Chen, Y. H. Cheng, Y. J. Chou, M. S. Su, C. M. Chen and K. H. Wei, Chem. Commun., 2011, 47, 5064–5066 RSC; (e) T. Y. Chu, J. Lu, S. Beaupre, Y. Zhang, J. R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed; (f) S. K. Lee, J. M. Cho, Y. Goo, W. S. Shin, J. C. Lee, W. H. Lee, I. N. Kang, H. K. Shim and S. J. Moon, Chem. Commun., 2011, 47, 1791–1793 RSC; (g) Z. Li, S.-W. Tsang, X. Du, L. Scoles, G. Robertson, Y. Zhang, F. Toll, Y. Tao, J. Lu and J. Ding, Adv. Funct. Mater., 2011, 21, 3331–3336 CrossRef CAS.
- (a) L. Dou, J. Gao, E. Richard, J. You, C. C. Chen, K. C. Cha, Y. He, G. Li and Y. Yang, J. Am. Chem. Soc., 2012, 134, 10071–10079 CrossRef CAS PubMed; (b) L. Huo, Y. Huang, B. Fan, X. Guo, Y. Jing, M. Zhang, Y. Li and J. Hou, Chem. Commun., 2012, 48, 3318–3320 RSC; (c) I. Osaka, M. Shimawaki, H. Mori, I. Doi, E. Miyazaki, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2012, 134, 3498–3507 CrossRef CAS PubMed; (d) S. Beaupré and M. Leclerc, J. Mater. Chem. A, 2013, 1, 11097 RSC; (e) C. Cabanetos, A. El Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. Frechet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656–4659 CrossRef CAS PubMed; (f) X. Guo, N. Zhou, S. J. Lou, J. Smith, D. B. Tice, J. W. Hennek, R. P. Ortiz, J. T. L. Navarrete, S. Li, J. Strzalka, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Nat. Photonics, 2013, 7, 825–833 CrossRef CAS.
- (a) J. Huang, Y. Ie, M. Karakawa, M. Saito, I. Osaka and Y. Aso, Chem. Mater., 2014, 26, 6971–6978 CrossRef CAS; (b) J.-H. Kim, M. Lee, H. Yang and D.-H. Hwang, J. Mater. Chem. A, 2014, 2, 6348 RSC; (c) Z. R. Owczarczyk, W. A. Braunecker, S. D. Oosterhout, N. Kopidakis, R. E. Larsen, D. S. Ginley and D. C. Olson, Adv. Energy Mater., 2014, 4, 1301821 Search PubMed; (d) Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed; (e) A. M. Schneider, L. Lu, E. F. Manley, T. Zheng, V. Sharapov, T. Xu, T. J. Marks, L. X. Chen and L. Yu, Chem. Sci., 2015, 6, 4860–4866 RSC; (f) Z. Zhang, F. Lin, H.-C. Chen, H.-C. Wu, C.-L. Chung, C. Lu, S.-H. Liu, S.-H. Tung, W.-C. Chen, K.-T. Wong and P.-T. Chou, Energy Environ. Sci., 2015, 8, 552–557 RSC.
- (a) D. Mühlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. Brabec, Adv. Mater., 2006, 18, 2884–2889 CrossRef; (b) I. W. Hwang, C. Soci, D. Moses, Z. Zhu, D. Waller, R. Gaudiana, C. J. Brabec and A. J. Heeger, Adv. Mater., 2007, 19, 2307–2312 CrossRef CAS; (c) J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed; (d) C. Soci, I. W. Hwang, D. Moses, Z. Zhu, D. Waller, R. Gaudiana, C. J. Brabec and A. J. Heeger, Adv. Funct. Mater., 2007, 17, 632–636 CrossRef CAS; (e) Z. Zhu, D. Waller, R. Gaudiana, M. Morana, D. Mühlbacher, M. Scharber and C. Brabec, Macromolecules, 2007, 40, 1981–1986 CrossRef CAS; (f) J. Hou, H. Y. Chen, S. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144–16145 CrossRef CAS PubMed; (g) A. J. Moulé, A. Tsami, T. W. Bünnagel, M. Forster, N. M. Kronenberg, M. Scharber, M. Koppe, M. Morana, C. J. Brabec, K. Meerholz and U. Scherf, Chem. Mater., 2008, 20, 4045–4050 CrossRef; (h) J. C. Bijleveld, M. Shahid, J. Gilot, M. M. Wienk and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 3262–3270 CrossRef CAS.
- (a) H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, Y. Yoon do, R. Graf, W. Pisula, H. W. Spiess and K. Mullen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed; (b) G. C. Welch and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 4632–4644 CrossRef CAS PubMed; (c) S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, J. Kniepert, S. Inal, P. Pingel, K. Fostiropoulos, N. Koch and D. Neher, J. Am. Chem. Soc., 2012, 134, 14932–14944 CrossRef CAS PubMed; (d) J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C. C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed; (e) P. C. Chow, S. Gelinas, A. Rao and R. H. Friend, J. Am. Chem. Soc., 2014, 136, 3424–3429 CrossRef CAS PubMed; (f) C. Gu, M. Xiao, X. Bao, L. Han, D. Zhu, N. Wang, S. Wen, W. Zhu and R. Yang, Polym. Chem., 2014, 5, 6551–6557 RSC; (g) J. S. Kim, Z. Fei, S. Wood, D. T. James, M. Sim, K. Cho, M. J. Heeney and J.-S. Kim, Adv. Energy Mater., 2014, 4, 1400527 Search PubMed; (h) J. Lee, T. Marszalek, K. C. Lee, J. Kim, W. Pisula and C. Yang, Macromol. Chem. Phys., 2015, 216, 1244–1250 CrossRef CAS; (i) J. Kesters, P. Verstappen, J. Raymakers, W. Vanormelingen, J. Drijkoningen, J. D'Haen, J. Manca, L. Lutsen, D. Vanderzande and W. Maes, Chem. Mater., 2015, 27, 1332–1341 CrossRef CAS.
- (a) L. E. Polander, L. Pandey, S. Barlow, S. P. Tiwari, C. Risko, B. Kippelen, J.-L. Brédas and S. R. Marder, J. Phys. Chem. C, 2011, 115, 23149–23163 CrossRef CAS; (b) P. M. Beaujuge, H. N. Tsao, M. R. Hansen, C. M. Amb, C. Risko, J. Subbiah, K. R. Choudhury, A. Mavrinskiy, W. Pisula, J. L. Bredas, F. So, K. Mullen and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 8944–8957 CrossRef CAS PubMed; (c) L. Biniek, S. Fall, C. L. Chochos, N. Leclerc, P. Lévêque and T. Heiser, Org. Electron., 2012, 13, 114–120 CrossRef CAS; (d) J.-H. Kim, J. B. Park, F. Xu, D. Kim, J. Kwak, A. C. Grimsdale and D.-H. Hwang, Energy Environ. Sci., 2014, 7, 4118–4131 RSC.
- M. Horie, J. Kettle, C.-Y. Yu, L. A. Majewski, S.-W. Chang, J. Kirkpatrick, S. M. Tuladhar, J. Nelson, B. R. Saunders and M. L. Turner, J. Mater. Chem., 2012, 22, 381–389 RSC.
- S.-W. Chang, H. Waters, J. Kettle and M. Horie, Org. Electron., 2012, 13, 2967–2974 CrossRef CAS.
- C.-H. Li, J. Kettle and M. Horie, Mater. Chem. Phys., 2014, 144, 519–528 CrossRef CAS.
- S.-W. Chang, H. Waters, J. Kettle, Z.-R. Kuo, C.-H. Li, C.-Y. Yu and M. Horie, Macromol. Rapid Commun., 2012, 33, 1927–1932 CrossRef CAS PubMed.
- J. Kuwabara, K. Yamazaki, T. Yamagata, W. Tsuchida and T. Kanbara, Polym. Chem., 2015, 6, 891–895 RSC.
- J. Kettle, M. Horie, L. A. Majewski, B. R. Saunders, S. Tuladhar, J. Nelson and M. L. Turner, Sol. Energy Mater. Sol. Cells, 2011, 95, 2186–2193 CrossRef CAS.
- H. Waters, J. Kettle, S.-W. Chang, C.-J. Su, W.-R. Wu, U. S. Jeng, Y.-C. Tsai and M. Horie, J. Mater. Chem. A, 2013, 1, 7370–7378 CAS.
- H. Waters, N. Bristow, O. Moudam, S.-W. Chang, C.-J. Su, W.-R. Wu, U. S. Jeng, M. Horie and J. Kettle, Org. Electron., 2014, 15, 2433–2438 CrossRef CAS.
- J. Kettle, N. Bristow, T. K. N. Sweet, N. Jenkins, G. A. D. R. Benatto, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2015, 8, 3266–3273 Search PubMed.
- F. Guo, S. Chen, Z. Chen, H. Luo, Y. Gao, T. Przybilla, E. Spiecker, A. Osvet, K. Forberich and C. J. Brabec, Adv. Opt. Mater., 2015, 3, 1524–1529 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and mass spectra and polymerization conditions and results. See DOI: 10.1039/c5ra22946d |
| This journal is © The Royal Society of Chemistry 2015 |