
Electrochemical probing of carbon quantum dots: not suitable for a single electrode material
Xinnan Jia and
Xiaobo Ji*
Department of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, China. E-mail: xji.csu.edu@gmail.com; Fax: +86-731-88879616; Tel: +86-731-88879616
First published on 7th December 2015
Abstract
In this article graphene quantum dots (GQDs) were prepared from activated graphene oxide (AGO) for the first time utilizing a reflux route. We focus on the relationship between surface structure and the electrochemical activity of graphene oxide (GO), AGO, GQDs and reduced graphene quantum dots (RGQDs). Surprisingly it was found that GQDs exhibited a much slower electron transfer rate of 1.105 × 10−4 cm s−1 compared with GO of 1.918 × 10−3 cm s−1 and AGO of 6.316 × 10−3 cm s−1. Obviously, a GQDs modified electrode surface can be completely blocked which might result from its low edge-to-basal plane rate though easy aggregation, rapid stacking and high oxygen-functional groups. Contrary to a general view that size-reducing is favorable to electrochemical kinetics, such findings, never reported before, imply that GQDs may not be suitable for applying to a beneficial single electrode material. GQDs, a new type of advanced materials of nanoscale, must be applied in a proper way in order to exert potential superiority to an utmost extent.
Introduction
Graphene-based materials have attracted ongoing enthusiasm in recent years as electrochemical materials in energy storage devices such as their reported use in supercapacitors and batteries. With the advent of nanotechnology, extensive efforts have been focusing on converting graphene to 0D graphene quantum dots (GQDs) which are graphene sheets in the range of a few nm to ∼100 nm.1 Owing to the quantum confinement and edge effect,2,3 GQDs can be simply modified through controlling the size and present many unique properties including high surface area, fine dispersion in water solvents, strong luminescence, low toxicity and excellent biocompatibility.4,5 Consequently, these dots are proposed to be applicable as the composite electrode materials to enhance electrochemical performances in Li or Na ion batteries and supercapacitors.6–9 Chao et al. has elegantly shown that GQDs are successfully coated onto the VO2 surfaces, which demonstrated high Na storage capacity of 306 mA h g−1 at 100 mA g−1, good rate properties and superior cycling life.9 GQDs have also been reported to exhibit enhanced properties towards the devices of bioimaging,10 sensors,11 catalysis12 and photovoltaic.13While many optimistic reports of utilizing GQDs have emerged, it is shown that GQDs may not provide an excellent advantage as single electrode materials. Zhu et al. has clearly indicated that the specific capacitance of GQDs is found to reach at only 53 F g−1, where GQDs are employed as the single electrode materials,6 exhibiting a lower capacitance compare to other carbon materials, such as graphene14 of 104.4 F g−1, and mesoporous carbon15 of 200 F g−1.
Note that in previous studies, two key methods have been explored to obtain GQDs, one of which is directly cutting graphite crystallites or graphene oxide sheets into 0D GQDs via electron beam lithography,16 hydrothermal or solvothermal technique,17 electrochemical route18 and ultrasonic method.19 Another strategy for synthesizing GQDs is in chemical way though pyrolyzing suitable organic precursors or catalyzing fullerene on the surface of ruthenium into small fragments.20–22 However, the extremely tedious process, violent reaction conditions, low yield and long time of these methods limit their application in electrochemical field.
Herein, we report an optimized methodology to obtain GQDs by chemically cutting an ordinarily utilizing flake graphite. After exfoliating the graphite with Hummers method,23 we utilize activated graphene oxide (AGO) rather than graphene oxide (GO) to prepare the final product, which should shorten the reaction time or lower the temperature compared with the previous reported methods. Then we compare the effect of coverage when utilizing GO, AGO, GQDs and reduced graphene quantum dots (RGQDs) respectively as electrode materials for 1 mM potassium ferrocyanide in 0.1 M KCl. Most interestingly, we demonstrate for the first time that GQDs exhibit slow heterogeneous electron transfer (1.105 × 10−4 cm s−1), mainly attributed to easy aggregation, rapid stacking and high oxygen-functional groups at the edges of GQDs. Considering that GQDs, the mini graphene sheets, are similar to graphene that the edge plane contributes to accelerate the electron transfer rather than basal plane.24,25 The disadvantages mentioned above lead to low edge-to-basal plane ratio and consequently GQDs appear to be an inferior single electrode material. Nevertheless, GQDs are promising building blocks for future electrochemical nanomaterials applied for the surfactant, as well as composites with other materials, where the former aim to control structure and the latter to inhibit irreversible process and improve cycling stability. In this perspective, more professional work should be performed to further realize mass production so as to develop the applications of GQDs.
Experimental section
Materials synthesis
RGQDs were prepared through hydrothermal reduction treatment using hydrazine hydrate, because it hardly changed the morphology of substance. Briefly, 30 mg of GQDs were dispersed in deionized water by ultrasonication. Then 2 mL of hydrazine hydrate was added into solution when the solution was heated at 98 °C. The mixed solution was refluxed at 100 °C for 24 h and cooled down to room temperature to form RGQDs.
Material characterization
Transmission electron microscopy (TEM) was obtained by using a JEM-2100F transmission electron microscope. The elemental composition was analyzed with an X-ray photoelectron spectroscopy (XPS, ESCALab250) in order to determine oxygen content of materials. UV-vis absorption spectra (Abs) were collected on a UV-vis spectrophotometer (UV-1801). Photoluminescence (PL) spectra were conducted using a fluorescence spectrophotometer (F-4600).Electrochemical measurements
All the measurements were performed with a three electrode system. A glassy carbon (GC) electrode as a working electrode was prepared for modification by renewing the electrode surface. It was polished with 0.05 mm alumina particles on a cloth and ultrasonicated in ethanol and water for 5 min. This was repeated several times. Then the aliquots of carbon materials dispersed in ethanol (0.4 mg per 1 mL) were carefully pipetted onto the GC electrode surface using a micro-pipette. The solvent was allowed to volatilize at room temperature, when a randomly distributed film is left on the GC electrode surface. A platinum electrode served as a counter electrode, while a Hg/HgCl electrode served as a reference electrode. Cyclic voltammetry (CV) experiments were collected on a Modulab (Solartron Analytical) electrochemical workstation by using KCl (0.1 M) as supporting electrolyte for potassium ferricyanide (1 mM).Results and discussion
The chemical process of GQDs was illustrated in Scheme 1. GO was obtained according to Hummers method. After KOH activation of GO, AGO was prepared with luxuriant pore structure and further cut into mini size GQDs through refluxed by HNO3. Finally, GQDs were reduced by hydrazine hydrate while retaining its morphology. The digital images of GO, AGO, GQDs and RGQDs were displayed respectively in Fig. 1a–d. It was clear that GO turned from red-brown flake into black powder after activation. GQDs, dark brown powder, became black powder after reduction. As could be seen from the digital image of Fig. 1e, AGO and RGQDs were insoluble in aqueous solution while GO was well dispersed in water solution treated by sonication and GQDs were water-soluble without ultrasonic dispersion resulting from different oxygen contents on the surface according to XPS test below. Particularly, GQDs aqueous solution could be conserved more than 6 months without aggregation at room temperature. Fig. 2a showed TEM images of the GO films. Dense 3D pore structure and predominantly atom-thick wall were further confirmed by the TEM of AGO (Fig. 2b and c). In addition, as could be shown in the TEM of GQDs (Fig. 2d and e), the diameters of the GQDs appeared to be in the range of 8–10 nm. Note that the chemical activation with KOH had been widely used to obtain AGO with high specific surface areas (SSA) and dense three dimensional pore structure.27 Therefore, the smaller GO fragments and more pore defects generated in reaction contribute to produce GQDs in gentler reaction conditions than large-thick GO flake and graphite. The activation process using KOH was explained in general as follows:28| 6KOH + 2C → 2K + 3H2 + 2K2CO3 |
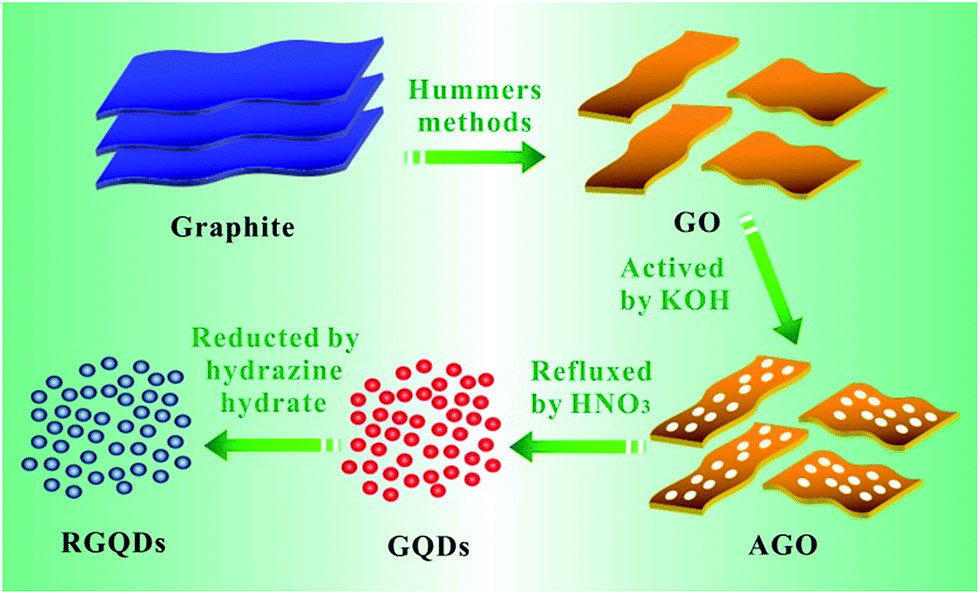 | ||
| Scheme 1 Schematic illustration of the synthesis process of the GQDs. | ||
 | ||
| Fig. 1 Digital images of GO flake (a), AGO powder (b), GQDs powder (c), RGQDs powder (d) and their aqueous solutions (e). | ||
 | ||
| Fig. 2 TEM images of GO (a), AGO (b and c) and GQDs with different magnifications (d and e). | ||
The formation of the GQDs was further confirmed by the optical properties in Fig. 3a. It showed a broad UV-vis absorption from 800 nm to 200 nm with one shoulder around 490 nm and one peak at 214 nm owning to the π → π* transition of aromatic sp2 domains,29 which was also observed in GQDs synthesized by chemical oxidation and hydrothermal methods.17,30 As we could see in the picture, the solution of GQDs emitted strong yellow photoluminescence (PL) under excitation at 365 nm. In addition, the PL spectrum was generally broad and depended on excitation wavelength called excitation-dependent PL behavior. When the excitation wavelength increased from 420 nm to 520 nm, the PL peaks exhibited a red-shift from 520 nm to 580 nm with the remarkable decrease of its intensity and the strongest PL intensity appears under excitation at 460 nm. This phenomenon may ascribe to optical selection by different sizes,31 different emissive sites and surface defects of GQDs, similarly to the excitation-dependent PL of other luminescent carbon-based nanomaterials.32
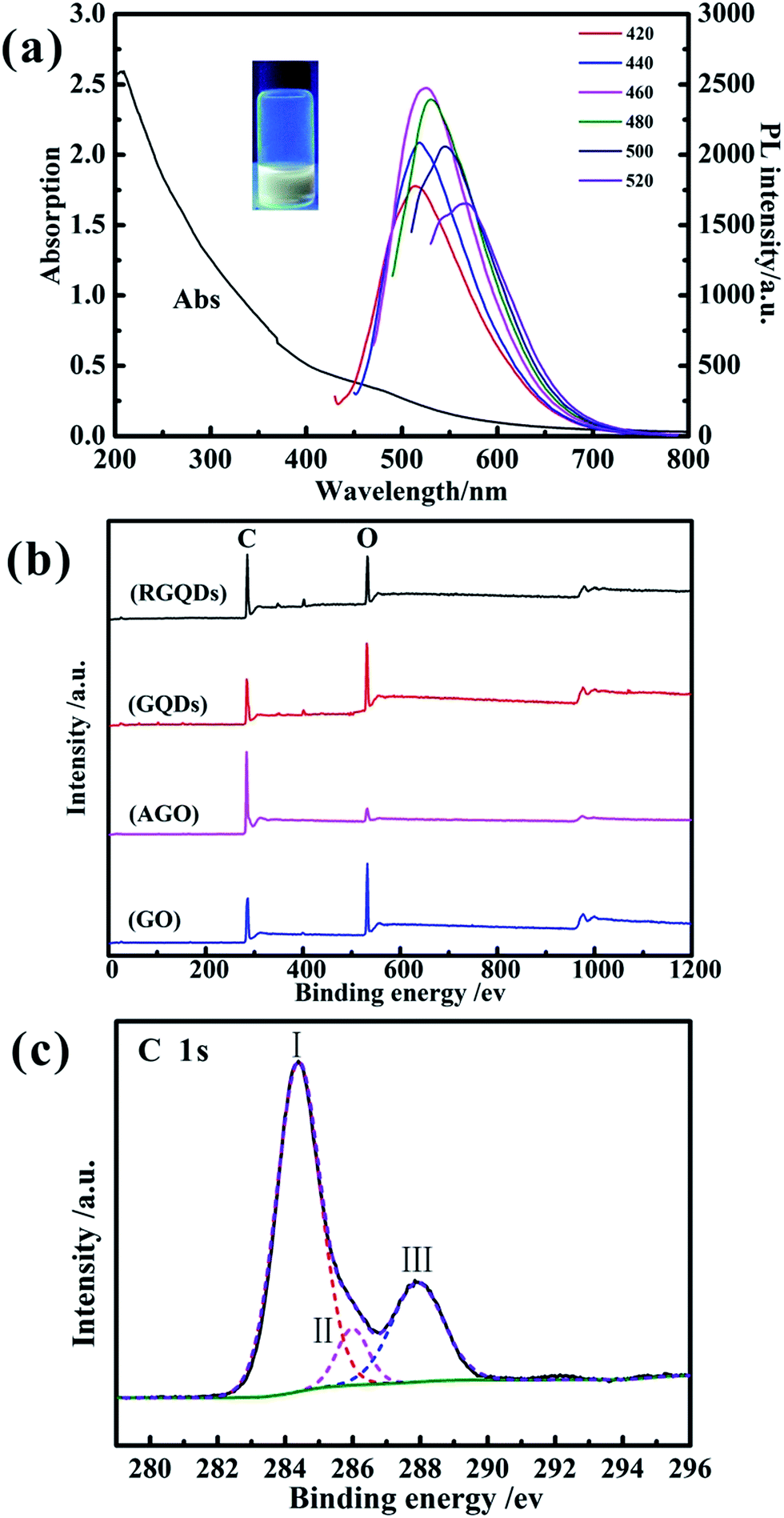 | ||
| Fig. 3 UV-vis absorption and PL emission spectra of GQDs in aqueous solution (a). (Inset: Optical photograph of GQDs solution under UV beam of 365 nm). XPS spectra of GO, AGO, GQDs and RGQDs (b). C 1s core level XPS spectra of GQDs (c). | ||
The X-ray photoelectron spectra (XPS) was conducted to analysis oxygen content of GO, AGO, GQDs and RGQDs and the C/O ratios were calculated based on C 1s and O 1s intensities. The X-ray photoelectron spectroscopy spectra records were shown in Fig. 3b, and the corresponding C and O atom ratios were listed in Table 1. The oxygen content of GQDs was the highest than the other carbon materials, on account of its edge effects3 that can increase more oxygen functional groups during the chemical oxygen process, while the AGO was the lowest one due to carbon–oxygen bond easily broken at activated temperature of 800 °C.28 When the GQDs was reduced to RGQDs, the oxygen content decreased from 34.65% to 23.33%. The C-1s peak of GQDs was consisted of three Gaussian peaks (Fig. 3c) centered at 284.39 eV (peak I), 285.99 eV (peak II) and 287.8 eV (peak III), indicating a considerable degree of oxidation corresponding to carbon atoms in different functional groups, which were sp2 carbon at ≈284.39 eV, C of C–O and/or C–OH bonds at ≈285.99 eV, C of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds at ≈287.8 eV.33,34
O bonds at ≈287.8 eV.33,34
| Sample | GC | GO | AGO | GQDs | RGQDs |
|---|---|---|---|---|---|
| Carbon atom ratio (%) | — | 67.41 | 91.65 | 61.76 | 72.02 |
| Oxygen atom ratio (%) | — | 30.81 | 8.35 | 34.65 | 23.33 |
| Epa (mV) | 220 | 258 | 223 | 301 | 300 |
| Epc (mV) | 142 | 107 | 126 | 22 | 25 |
| ΔEp (mV) | 78 | 151 | 97 | 279 | 275 |
| Ψ | 1.42638 | 0.198915 | 0.65501 | 0.01146 | 0.01396 |
| k0 | 1.375 × 10−2 | 1.918 × 10−3 | 6.316 × 10−3 | 1.105 × 10−4 | 1.346 × 10−4 |
We next turned to investigate the electrochemical properties of a chemical modified GC electrode following modification with 4.8 μg of GO, AGO, GQDs and RGQDs using the ferro/ferricyanide redox probe for comparative purpose. The heterogeneous electron transfer (HET) property was illustrated through determination of the electron transfer rate kinetics (k0) from cyclic voltammetric, which was evaluated using the Nicholson equation for an electrochemically quasi-reversible process as demonstrated by the eqn (1):35,36
| Ψ = k0[πDnνF/(RT)]−1/2 | (1) |
Every symbol has their ordinary meaning, in which Ψ is a kinetic parameter without dimension, D is the diffusion coefficient for [Fe(CN)6]3−/4− in 0.1 M KCl (7.6 × 10−6 cm2 s−1),37 n is the number of electrons working in the electron-transfer process, ν is the scan rate, R is the molar gas constant and T is the temperature. Ψ, the kinetic parameter, is fitted the function of Ψ − ΔEp for a one electron redox reaction (α = 0.5, T = 298 K) as described by the eqn (2):35,36
| Ψ = (−0.6288 + 0.0021X)/(1 − 0.017X) | (2) |
Note that it was ordinary to classify the layered structure of graphite into two graphite plane: the basal plane, which existed all the side of graphite layer as well as the surface paralleling to it, and the edge plane, which was from the peripheral edge perpendicularly to the basal plane.14,38,39 The two planes demonstrated significantly different actions in accelerating electron transfer due to the property of the chemical bonding. When the edge plane was overwhelmingly dominant over the basal plane, the electron transfer was much faster compared to that of being converse. Meanwhile, the oxygen functional groups draw notably attention in influencing electrochemical properties both through the electron-exchange rate and the adsorption of molecules from electrolyte in the redox reaction.40,41
Fig. 4 depicted the cyclic voltammograms curves and Table 1 displayed the calculation of corresponding peak-to-peak separation (ΔEp) and k0. It could be seen that GQDs modified electrode showed high peak separation (ΔEp) of 279 mV at 100 mV s−1 resulting in the slow HET at 1.105 × 10−4 cm s−1, which was similar to that observed at RGQDs (275 mV; 1.346 × 10−4 cm s−1) and significantly slower than that observed at both GO modified electrode and AGO modified electrode (151 mV; 1.918 × 10−3 cm s−1 and 97 mV; 6.316 × 10−3 cm s−1 respectively). It had been reported that GO exhibited slow electron transfer due to the structural effects, where the oxygen-containing groups, low edge plane content and low specific surface area were taken into consideration.14 The AGO reflected a fast electron transfer with great amount of pore defects and relatively low oxygen content, which were beneficial to contain a large ratio of the edge plane to the basal plane. Notably, GQDs exhibited much slower electrode kinetics than GO as if the electrode surface was completely blocked, mainly due to the increasing oxygen-containing groups leading to restrain the electron transfer between GQDs and ferro/ferricyanide. Moreover, GQDs/RGQDs were stacked irregularly in a short time as soon as the solvent evaporated, leading to decrease the specific surface area in fact. Also as Hou et al. had pointed out, the Brunauer–Emmett–Teller (BET) SSA of carbon quantum dots was only 18.6 m2 g−1 calculated from nitrogen adsorption–desorption isotherms.42 In the real experimental case the carbon-based materials were immobilized onto the GC electrode surface so that the materials were accumulated layer upon layer and the SSA of GQDs/RGQDs would likely to be smaller. As a consequence, both GQDs and RGQDs modified electrodes demonstrated effectively blocked because the basal plane substituting the edge plane became the main way to transfer electrons and it was adverse to increase the electron transfer kinetics.
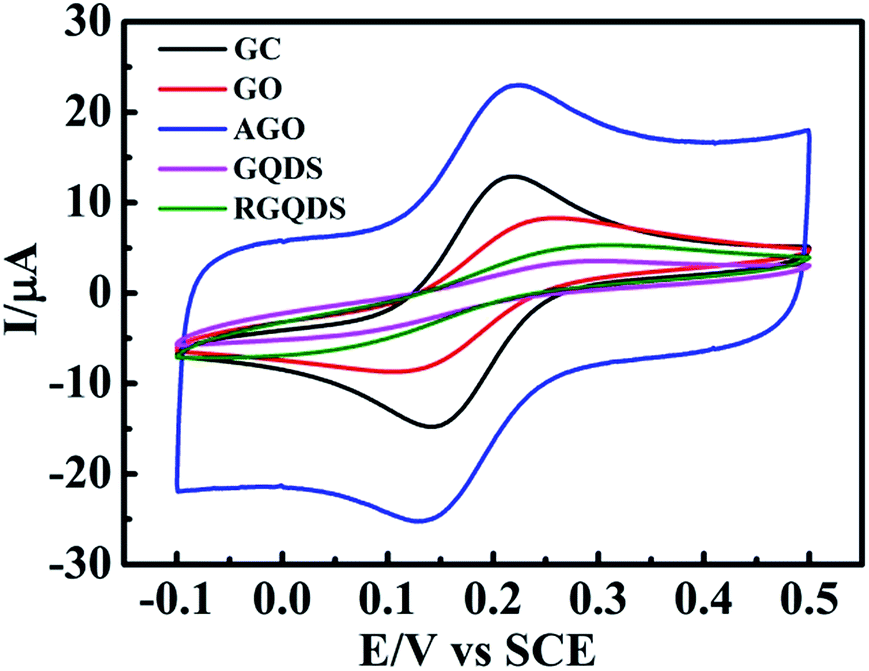 | ||
| Fig. 4 Cyclic voltammetric profiles recorded of GO, AGO, GQDs and RGQDs modified GC electrode utilizing 1 mM potassium ferricyanide in 0.1 M KCl. Scan rate: 100 mV s−1 (vs. SCE). | ||
Fig. 5 depicted the digital images of different amounts of GQDs and RGQDs modified GC electrode. The picture clearly conveyed the significant information that GQDs/RGQDs stacked heavily as increasing their mass. The effect of increasing materials' amount deposition on the GC electrode surface was also depicted in Fig. 6. It showed that with an increasing GO' mass coverage on the GC electrode (Fig. 6a) the peak-to-peak separation tended to be stable, while the peak current descended apparently. This was due to the low conductivity of GO and flakes accumulation.25 Considering the effect of increasing amounts of AGO on the voltammetric curve (shown in Fig. 6b), it was a gradually high current response and a clearly narrow shift between oxidation and reduction peak, owning to the fact that the more porous structures with high specific surface areas14 contributed to a higher number of edge-like structures. We next turned to exploring the effect of increasing GQDs' mass. As was evident in Fig. 6c, the ΔEp had a conspicuously large shift which was different from GO and AGO. It proved that the modified GC electrode surface with a little quantities of GQDs existed incomplete coverage site where the exposed surface remained relatively electrochemically active, thus with increased coverage of GQDs, the “uncovered active sites” disappeared gradually and consequently the heterogeneous electron transfer slowed down. At the same time, the basal plane sites of GQDs completely controlled the electrode reactivity inducing “blocking” on the electrode surface. The “blocking” was ameliorated a little to some degree as GQDs was reduced (displayed in Fig. 6d), resulting from the decreasing oxygen-containing groups. The above interesting findings demonstrated that GQDs were no better than graphene for an excellent single electrode material.
 | ||
| Fig. 5 Digital images of GC electrode surface modified using GQDs (a) and RGQDs (b) with different amounts. | ||
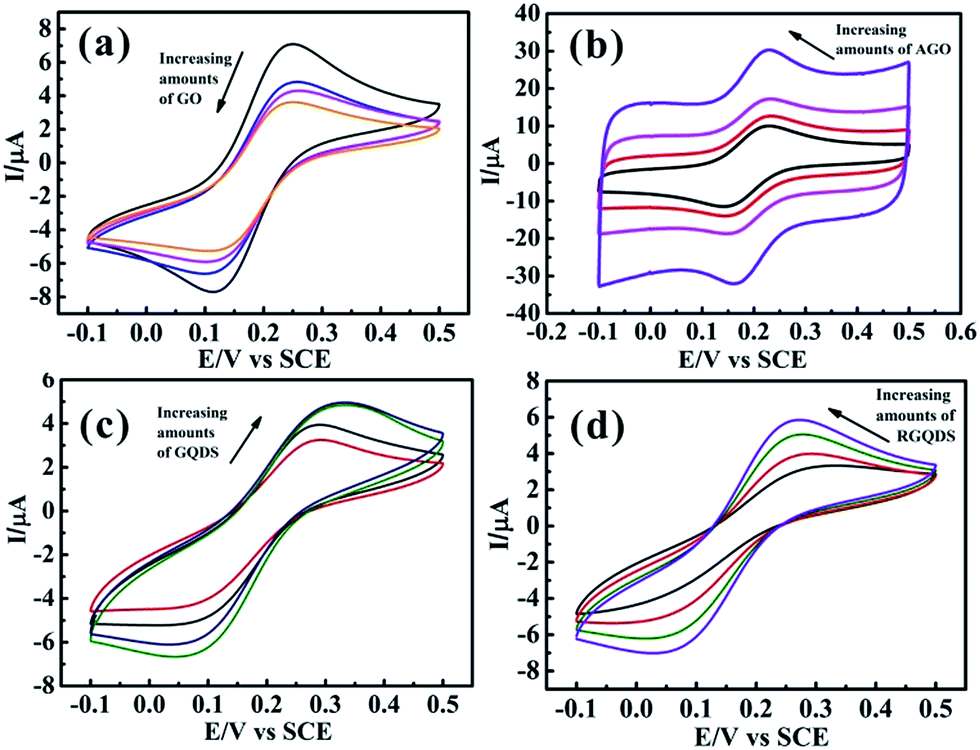 | ||
| Fig. 6 Cyclic voltammetric profiles recorded utilizing 1 mM potassium ferricyanide in 0.1 M KCl, obtained using an GC electrode with the addition of increasing amounts of 2.4, 4.8, 9.6 and 16.8 μg GO (a), AGO (b), GQDs (c) and RGQDs (d). Scan rate: 50 mV s−1 (vs. SCE). | ||
Conclusions
To summarize, we have improved the strategy to prepare GQDs directly by refluxing AGO with the advantages of tender conditions. Moreover, we have compared, for the first time, the electrochemical properties of GO, AGO, GQDs and RGQDs. Interestingly, it is found that GQDs modified electrode surface is blocked, exhibiting slow heterogeneous electron transfer kinetics due to slow edge/basal plane ratio. Easy aggregation, rapid stacking and high oxygen-functional groups of GQDs have a negative impact on the electrochemical properties. Consequently, GQDs, as a single electrode material, exhibit a poor property. Whereas it still has huge potential to be surfactant or composite with other materials, if we keep turning a bright eye on it and use it in a proper way.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (21473258), Distinguished Young Scientists of Hunan Province (13JJ1004), and the Hunan Provincial Innovation Foundation for Postgraduate (CX2015B039).Notes and references
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS PubMed.
- Ç. Ö. Girit, J. C. Meyer, R. Erni, M. D. Rossell, C. Kisielowski, L. Yang, C.-H. Park, M. F. Crommie, M. L. Cohen, S. G. Louie and A. Zettl, Science, 2009, 323, 1705–1708 CrossRef PubMed.
- K. A. Ritter and J. W. Lyding, Nat. Mater., 2009, 8, 235–242 CrossRef CAS.
- J. Shen, Y. Zhu, X. Yang and C. Li, Chem. Commun., 2012, 48, 3686–3699 RSC.
- Z. Zhang, J. Zhang, N. Chen and L. Qu, Energy Environ. Sci., 2012, 5, 8869–8890 CAS.
- Y. Zhu, X. Ji, C. Pan, Q. Sun, W. Song, L. Fang, Q. Chen and C. E. Banks, Energy Environ. Sci., 2013, 6, 3665–3675 CAS.
- J. Pan, Y. Sheng, J. Zhang, J. Wei, P. Huang, X. Zhang and B. Feng, J. Mater. Chem. A, 2014, 2, 18082–18086 CAS.
- H. Ming, Y. Yan, J. Ming, X. Li, Q. Zhou, H. Huang and J. Zheng, RSC Adv., 2014, 4, 12971–12976 RSC.
- D. Chao, C. Zhu, X. Xia, J. Liu, X. Zhang, J. Wang, P. Liang, J. Lin, H. Zhang, Z. X. Shen and H. J. Fan, Nano Lett., 2015, 15, 565–573 CrossRef CAS PubMed.
- S. Zhu, J. Zhang, C. Qiao, S. Tang, Y. Li, W. Yuan, B. Li, L. Tian, F. Liu, R. Hu, H. Gao, H. Wei, H. Zhang, H. Sun and B. Yang, Chem. Commun., 2011, 47, 6858–6860 RSC.
- Y. Dong, G. Li, N. Zhou, R. Wang, Y. Chi and G. Chen, Anal. Chem., 2012, 84, 8378–8382 CrossRef CAS PubMed.
- X. Zhou, Z. Tian, J. Li, H. Ruan, Y. Ma, Z. Yang and Y. Qu, Nanoscale, 2014, 6, 2603–2607 RSC.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014–3017 CrossRef CAS PubMed.
- W. Song, X. Ji, W. Deng, Q. Chen, C. Shen and C. E. Banks, Phys. Chem. Chem. Phys., 2013, 15, 4799–4803 RSC.
- A. B. Fuertes, F. Pico and J. M. Rojo, J. Power Sources, 2004, 133, 329–336 CrossRef CAS.
- L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356–358 CrossRef CAS PubMed.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed.
- J. Zhou, J. Cheiftz, R. Li, F. Wang, X. Zhou, T.-K. Sham, X. Sun and Z. Ding, Carbon, 2009, 47, 829–838 CrossRef CAS.
- S. Zhuo, M. Shao and S.-T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef CAS PubMed.
- J. Lu, P. S. E. Yeo, C. K. Gan, P. Wu and K. P. Loh, Nat. Nanotechnol., 2011, 6, 247–252 CrossRef CAS PubMed.
- X. Yan, X. Cui and L.-s. Li, J. Am. Chem. Soc., 2010, 132, 5944–5945 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2011, 133, 15221–15223 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- C. E. Banks, T. J. Davies, G. G. Wildgoose and R. G. Compton, Chem. Commun., 2005, 829–841, 10.1039/b413177k.
- D. A. C. Brownson, L. J. Munro, D. K. Kampouris and C. E. Banks, RSC Adv., 2011, 1, 978–988 RSC.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS.
- S. Murali, J. R. Potts, S. Stoller, J. Park, M. D. Stoller, L. L. Zhang, Y. Zhu and R. S. Ruoff, Carbon, 2012, 50, 3482–3485 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- Y. Dong, C. Chen, X. Zheng, L. Gao, Z. Cui, H. Yang, C. Guo, Y. Chi and C. M. Li, J. Mater. Chem., 2012, 22, 8764–8766 RSC.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- J. Zhou, C. Booker, R. Li, X. Zhou, T.-K. Sham, X. Sun and Z. Ding, J. Am. Chem. Soc., 2007, 129, 744–745 CrossRef CAS PubMed.
- A. Saha, S. K. Basiruddin, S. C. Ray, S. S. Roy and N. R. Jana, Nanoscale, 2010, 2, 2777–2782 RSC.
- X. Yang, X. Jia and X. Ji, RSC Adv., 2015, 5, 9337–9340 RSC.
- I. Lavagnini, R. Antiochia and F. Magno, Electroanalysis, 2004, 16, 505–506 CrossRef CAS.
- E. P. Randviir, D. A. C. Brownson, J. P. Metters, R. O. Kadara and C. E. Banks, Phys. Chem. Chem. Phys., 2014, 16, 4598–4611 RSC.
- S. J. Konopka and B. McDuffie, Anal. Chem., 1970, 42, 1741–1746 CrossRef CAS.
- B. Šljukić, C. E. Banks and R. G. Compton, Nano Lett., 2006, 6, 1556–1558 CrossRef PubMed.
- R. R. Moore, C. E. Banks and R. G. Compton, Anal. Chem., 2004, 76, 2677–2682 CrossRef CAS PubMed.
- A. Holloway, G. Wildgoose, R. Compton, L. Shao and M. H. Green, J. Solid State Electrochem., 2008, 12, 1337–1348 CrossRef CAS.
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed.
- H. Hou, C. E. Banks, M. Jing, Y. Zhang and X. Ji, Adv. Mater., 2015, 27, 7861–7866 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |