
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-standing polymer-functionalized reduced graphene oxide papers obtained via a UV-process†
I. Roppolo*a,
A. Chiapponea,
L. Boggioneb,
M. Castellinoa,
K. Bejtkaa,
C. F. Pirriab,
M. Sangermanob and
A. Chiolerioa
aIstituto Italiano di Tecnologia (IIT), Center for Space Human Robotics@Polito, Corso Trento 21, 10129 Torino, Italy. E-mail: Ignazio.roppolo@iit.it
bDepartment of Applied Science and Technology – DISAT, Politecnico di Torino, C.so Duca degli Abruzzi 24, 10129 Torino, Italy
First published on 22nd October 2015
Abstract
Graphene based materials are attracting great attention every day due to their outstanding properties. Widening their potentialities through synergic effects in conjunction with other materials represents an intriguing challenge in order to obtain lighter and multi-functional composites. In this paper, novel self-standing graphene-based paper-like sheets are investigated, obtained via a facile dual step UV-induced process. This method, employing graphene oxide as a starting material, allows the obtaining of polymeric functionalized reduced graphene oxide papers that could be easily handled, featuring improved mechanical and peculiar electrical properties. The mechanical and thermal properties were investigated as well as their electrical response under different stimuli, such as temperature and humidity, showing remarkable changes.
Introduction
Graphene-based polymer composites are one of the most promising emerging classes of materials:1,2 they are envisaged to be of fundamental importance in energy storage and conversion applications,3–7 electronics8–10 and sensing.11–14 The homogeneous dispersion of graphene in polymers and good interfacial interaction with the matrix still represent the main challenge.2,15 Furthermore, interesting aspects arise from the interaction between matrix and filler, and, in certain functional applications, the interface itself is considered to be a crucial point.1,15 For this reason, the functionalization of graphene with polymeric chains is important to improve compatibility and to feature new synergic properties.15 Direct graphene functionalization was achieved via covalent bonding16–22 (e.g. ATRP,16 dipolar cycloaddition,21 etc.) or by exploiting van der Waals interactions.23–29Another very promising approach for the large-scale production of polymer-functionalized graphene sheets consists in the chemical modification of graphene oxide (GO).30–32 Generally GO is produced through a well-established oxidative mechanism from graphite,33 which is commercially available and relatively cheap.32 This process brings a large number of reactive groups (e.g. carboxylic acids, epoxy groups and hydroxyl groups) on the surface and at the edges of the GO sheets, which could be used as starting points for chemical reactions or as anchoring points for polymeric chains.33–35 Over the past few decades, a large number of different methods have been proposed with both “grafting to”36,37 or “grafting from”38–40 approaches for GO functionalization with polymeric chains. However, GO does not feature the outstanding properties of graphene such as high electrical and thermal conductivities.32,41 For this reason, the simultaneous reduction of graphene oxide to graphene and its functionalization with polymer chains represent an important goal.
Recently, a facile two-step UV-based process for GO reduction toward reduced graphene oxide (RGO) and the simultaneous covalent grafting of initiating moieties at its surface was proposed. This procedure enables the subsequent photo-grafting of a great variety of monomers for graphene surface functionalization.39,42
In the present work, we applied the cited method to functionalize reduced graphene oxide with dimethylamino ethyl methacrylate (DMAEMA). This monomer has been proposed for graphene functionalization for various application domains such as optoelectronics,43 sensing44–46 and biomedical devices.47–49
By exploiting the attractive properties of poly(dimethyl amino ethyl methacrylate) (PDMAEMA) together with the previously mentioned grafting method, we were able to produce easy-to-handle and self-standing polymer-grafted RGO paper-like sheets. The mechanical and electrical properties of the self-standing materials were then assessed, evidencing the effects induced by the presence of the grafted polymer chains.
Experimental
Materials
Commercial GO (thickness 0.7–1.2 nm; lateral dimensions 300–800 nm) was purchased from Cheap Tubes Inc. (USA) and used without further purification. Benzophenone (BP, Sigma-Aldrich) was used as a reducing and anchoring agent. 2-Dimethyl amino ethyl methacrylate (DMAEMA, Sigma-Aldrich) was used as a functionalizing monomer. Dimethylformamide DMF and ethanol were used as solvents.Functionalization process
The method used in this work is described in detail elsewhere.39 A full scheme of the procedure is reported in Fig. S1 in the ESI.† The amount of each component and the irradiation time were chosen based on previous work.39,42 10 mg of GO in DMF (0.5 mg ml−1 solution) was placed in a 100 ml three necked flask. The mixture was sonicated in an ultrasound bath until a homogeneous dispersion was obtained. 30 mg of BP powder was then added to the solution, which was magnetically stirred and degassed by bubbling nitrogen for 30 minutes. The mixture was UV irradiated under a high-pressure mercury lamp with an intensity of 40 mW cm−2 (Hamamatsu LC8 equipped with an 8 mm light guide) while stirring at room temperature for 5 minutes. Afterwards, the solution was transferred into centrifuge tubes and centrifuged at the speed of 5000 rpm for 10 minutes. The precipitates were then washed with ethanol and centrifuged several times in order to remove the unreacted BP and byproducts. The purified product was dried overnight at 60 °C. For the second functionalization step, 10 mg of the modified powder was dispersed in 20 ml of DMF in the presence of different amounts of the DMAEMA monomer (50, 150 and 500 mg). The solution was magnetically stirred, degassed by bubbling nitrogen for 30 minutes and subsequently UV irradiated for 90 minutes. The mixture was then centrifuged, washed and dried as previously described in order to eliminate the ungrafted polymeric chains. Finally, the solution was filtered using a Whatman Anodisc 47 alumina filter (pore diameter 0.1 μm); a self-standing sample (50 μm thickness) was thus obtained after solvent evaporation (1 h at 60 °C in a vacuum oven).For comparison, in AC electrical measurements, a RGO + 500 DMAEMA paper was subjected to a quaternarization process according to literature results that predicted an improvement of moisture sensitivity.45 The quaternarization process was carried out by laying the membrane for 24 h in an isopropanol/ethylene bromide solution (50/50 wt). The sample was then dried in a vacuum oven and tested.
Characterization methods
FT-IR transmittance spectra were collected using a Tensor 27 FTIR Spectrometer (Bruker). The samples were prepared via casting on Si wafers from ethanol. 64 scans were signal-averaged at a resolution of 2 cm−1 from 4000 to 400 cm−1.A PHI 5000 Versaprobe Scanning X-ray Photoelectron Spectrometer (monochromatic Al Kα X-ray source with a 1486.6 eV energy, 15 kV voltage and 1 mA anode current) was used to investigate the surface chemical composition. A spot size of 100 μm was used in order to collect the photoelectron signal for both the high resolution (HR) and the survey spectra. Different pass energy values were employed: 187.85 eV for the survey spectra and 23.5 eV for the HR peaks. All samples were analyzed with a combined electron and argon ion gun neutralizer system in order to reduce the charging effect during the measurements. All core-level peak energies were referenced to the C 1s peak at 284.5 eV and the background contribution in the HR scans was subtracted by means of a Shirley function.50 The spectra were analysed using Multipak 9.0 dedicated software. The samples were excited with an Ar–Kr laser source (wavelength of 514.5 nm, photon flux ∼300 W cm−2).
Thermogravimetric analysis (TGA) was performed with a Netzsch TG 209 F1 Libra instrument. All samples were previously maintained for 30 minutes at 100 °C in order to eliminate the adsorbed water and then heated between 100 °C and 800 °C at a heating rate of 10 °C min−1 in a nitrogen flow of 60 ml min−1. Afterwards, a purge flow of nitrogen was used (20 ml min−1). For the control of the measurements as well as for data acquisition, NETZSCH PROTEUS 32-bit Software was employed with advanced software packages like c-DTA (calculated DTA-signal), Super-Res (rate-controlled mass change) and Thermokinetics.
Differential scanning calorimetry (DSC) experiments were performed using a Netzsch DSC 204 F1 Phoenix instrument, equipped with a low temperature probe. The experiments were carried out between 0 and 100 °C with a scan rate of 10 °C min−1. For each sample, the same heating module was applied and the final heat flow value was recorded during the second heating cycle. The Tg was defined as the midpoint of the heat capacity change observed in the DSC thermogram.
DMTA measurements were performed with a Triton Technology TTDMA analyser. All of the experiments were conducted with a temperature ramp of 3 °C min−1, applying a force with a frequency of 1 Hz and with 10 μm of displacement.
Morphologic characterization was performed in the top and cross-section views using Field Emission Scanning Electron Microscopy (FESEM, ZEISS Dual Beam Auriga). The cross-section samples were prepared by fracturing them in liquid nitrogen. Prior to FESEM observation, the samples were coated with an 8 nm thick Pt film via sputtering to improve the electrical conductance.
The electrical properties were characterized on free standing membranes at room temperature, using a direct contact setup with stainless steel blocking electrodes, both in DC and in AC regimes, up to 2 MHz; the measurements were performed using a Keithley 2635A multimeter (DC) and an Agilent E4980A precision LCR meter (20 Hz up to 2 MHz).
Results and discussion
In this work, RGO paper-like samples covalently functionalized with different amounts of PDMAEMA using GO as the starting material were synthesized through a UV-mediated two step method previously proposed (Fig. S1 in ESI†).39The process involves in its first step the UV irradiation of a GO solution in the presence of BP. In fact, the latter is known to be able to abstract, under UV irradiation, extractable hydrogen atoms from the surroundings, creating semipinacol radicals.51 It was demonstrated that the BP abstraction of hydrogen atoms from GO surfaces, which are rich in H-containing groups, induces a spontaneous reduction of GO. Simultaneously, some of the generated semipinacol radicals recombine with latent radicals on the reduced GO surface, inducing the grafting of these molecules.42
During the second step, these bonds can be homolitically broken through a second UV irradiation step, generating an initiating point for the grafting of acrylic and methacrylic monomers.
At the end of this process, polymer-functionalized RGO is obtained, featuring polymer brushes grown from the RGO surface. After a washing step (performed in order to get rid of the ungrafted chains), self standing functionalized papers were obtained after filtering the solutions, producing easy-to-handle and mechanically robust samples (see Fig. 1a).
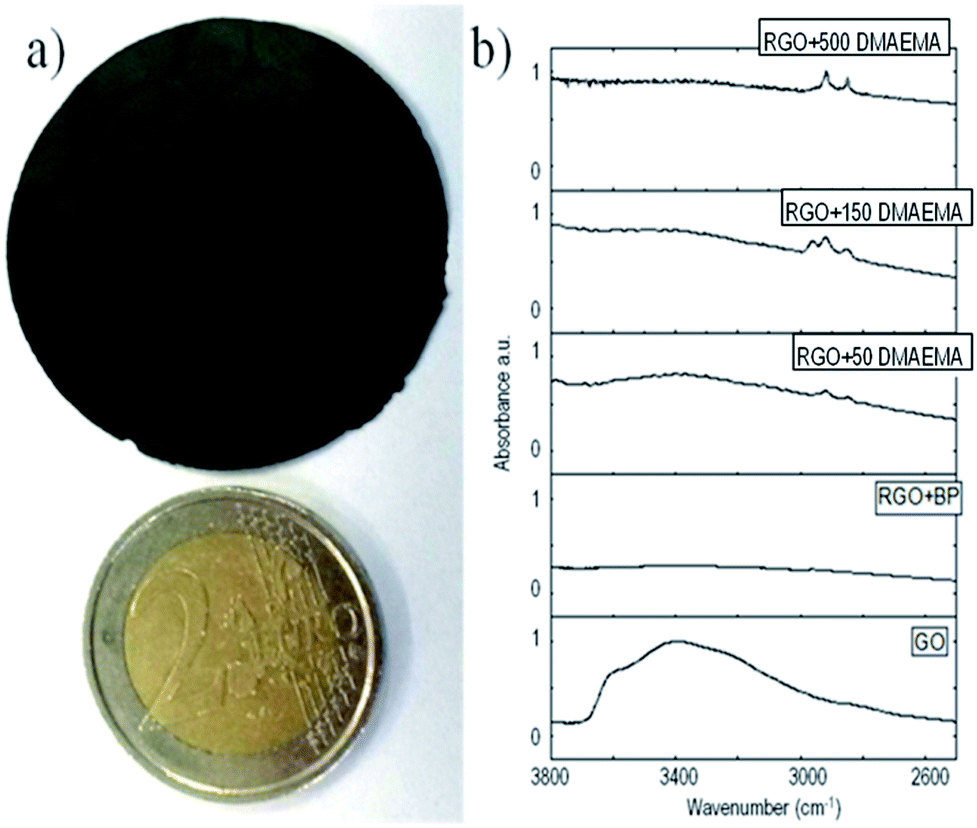 | ||
| Fig. 1 (a) Figure of the self-standing RGO paper compared with a 2€ coin and (b) IR spectra of the synthesized samples. | ||
The presence of grafted polymeric chains on RGO was confirmed using different methods. Fig. 1b shows a partial FT-IR spectra of GO, where RGO was reduced in the presence of benzophenone and functionalized with different amounts of DMAEMA (full spectra are reported in ESI†). In all of the samples irradiated in the presence of methacrylic monomers, the evidence of C–H stretching peaks around 2900 cm−1 is clear. They could be attributed to grafted polymer chains since the same peaks are not present in the pristine GO and in the GO reduced in the presence of BP. This measurement clearly shows the presence of polymeric chains grafted on the RGO sheets; however it was not possible to quantify the extent of polymer functionalization, leading to a further experimental search.
XPS measurements were performed in order to better qualify the grafted polymer on the RGO samples. In Fig. 2, the HR spectra of C 1s are reported (see the relative survey spectra in ESI†). Fig. 2a reports the starting material (RGO + BP) which shows a high degree of reduction since the peaks relative to C–O (peak 2) and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (peak 3) represent only 15.5% of the total area, while the rest is due only to C–(C,H) (peak 1) species. On the other hand, by increasing the amount of DMAEMA in the initial mixture, an increase in peak 2 and peak 3 was observed (Table 1). This can be attributed to a larger presence of C–(O,N) and O–CO bonds belonging to the monomers confirming a higher grafting degree (Table 1). Moreover, it is possible to evidence that N is present only in the sample in which DMAEMA was grafted and an increase in the relative abundance of O and N was measured in the samples in which more polymer was grafted. The N 1s peak (Fig. S11 in ESI†) is a clear fingerprint of the amine in the DMAEMA monomers. It is made up by the superposition of a higher peak at a lower binding energy (peak 1) and a lower peak at a higher chemical shift (peak 2). The first component is due to the tertiary amine signal, while the second one is due to the protonated tertiary amine signal.52 The relative abundance of the two chemical bonds is reported in Fig. S12 in ESI.† While in the RGO + 50 and RGO + 150 samples the tertiary amine signal is higher and increases with the increase in the initial monomer amount, it is clearly noticeable that in the RGO + 500 DMAEMA sample, the protonated amine peak has reached the amine intensity. This can be explained considering that an increase in the amount of polymer grafted on the RGO surface increases also the probability of inter-chain H abstraction induced by the amine moiety and thus the amount of protonated tertiary amines.
O (peak 3) represent only 15.5% of the total area, while the rest is due only to C–(C,H) (peak 1) species. On the other hand, by increasing the amount of DMAEMA in the initial mixture, an increase in peak 2 and peak 3 was observed (Table 1). This can be attributed to a larger presence of C–(O,N) and O–CO bonds belonging to the monomers confirming a higher grafting degree (Table 1). Moreover, it is possible to evidence that N is present only in the sample in which DMAEMA was grafted and an increase in the relative abundance of O and N was measured in the samples in which more polymer was grafted. The N 1s peak (Fig. S11 in ESI†) is a clear fingerprint of the amine in the DMAEMA monomers. It is made up by the superposition of a higher peak at a lower binding energy (peak 1) and a lower peak at a higher chemical shift (peak 2). The first component is due to the tertiary amine signal, while the second one is due to the protonated tertiary amine signal.52 The relative abundance of the two chemical bonds is reported in Fig. S12 in ESI.† While in the RGO + 50 and RGO + 150 samples the tertiary amine signal is higher and increases with the increase in the initial monomer amount, it is clearly noticeable that in the RGO + 500 DMAEMA sample, the protonated amine peak has reached the amine intensity. This can be explained considering that an increase in the amount of polymer grafted on the RGO surface increases also the probability of inter-chain H abstraction induced by the amine moiety and thus the amount of protonated tertiary amines.
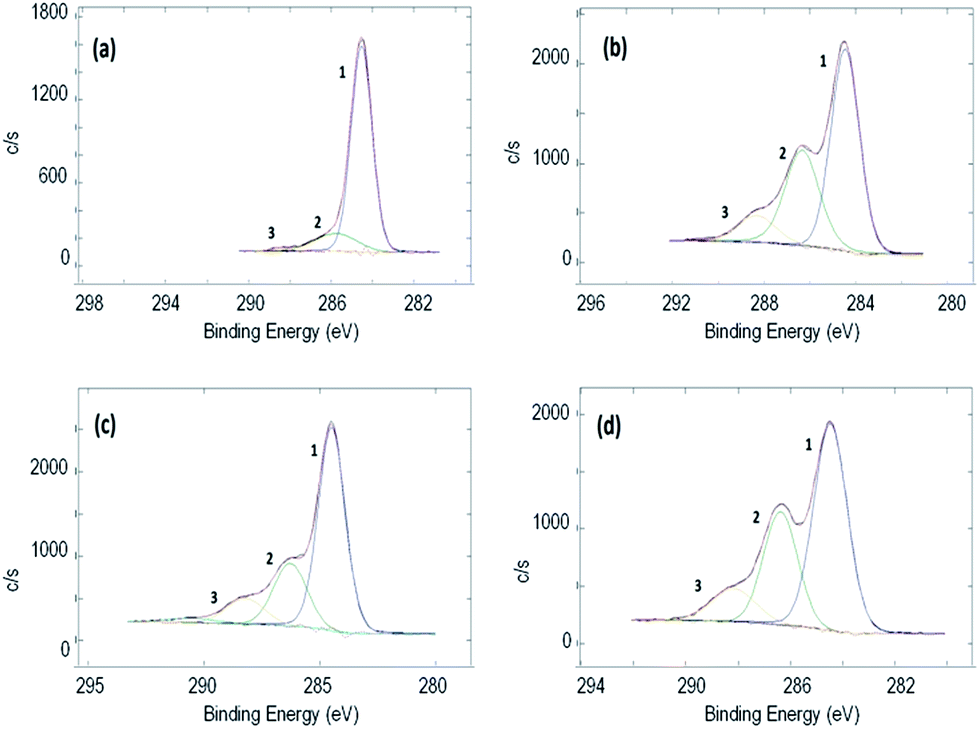 | ||
| Fig. 2 XPS spectra of C 1s of (a) RGO + BP, (b) RGO + 50 DMAEMA, (c) RGO + 150 DMAEMA, and (d) RGO + 500 DMAEMA. | ||
| Sample | Atomic% | C 1s | ||||
|---|---|---|---|---|---|---|
| C | O | N | C–(C,H) | C–(O,N) | O–CO |
|
| RGO + BP | 85.8 | 14.2 | — | 84.5 | 14.5 | 1.0 |
| RGO + 50 DMAEMA | 72.4 | 25.5 | 2.1 | 55.6 | 33.9 | 10.5 |
| RGO + 150 DMAEMA | 70.3 | 27.0 | 2.7 | 64.5 | 24.3 | 11.2 |
| RGO + 500 DMAEMA | 68.1 | 29.0 | 2.9 | 57.2 | 30.8 | 12.0 |
TGA experiments were performed to evaluate the extent of grafting via weight drop, and D-TGA curves are reported in Fig. 3. As already demonstrated,39,42 the peak at 180 °C is related to the presence of unreacted semipinacol groups while the second peak at about 350 °C is related to the degradation of the PDMAEMA polymeric chains (see Fig. S11 in ESI†). It is evident that, by increasing the amount of monomer, the second peak becomes more intense while the first one decreases. This is an indication that, by increasing the amount of monomer in the formulations, at the end of the second irradiation step, a lower number of unreacted semipinacol radicals remain on the RGO sheets and a higher number of polymer chains grow from the sheet surface. Through the evaluation of the weight drop between 200 °C and 450 °C (values between which the polymer undergoes complete thermal degradation) and excluding the measurement drift, it is possible to estimate the amount of polymer grafted. The drop of weight values, reported in Table 2, show that by increasing the amount of monomer in the initial solution, a higher degree of polymer functionalization was achieved. However, those values should be taken as an approximation since it is not possible to compare directly the thermal behaviour of neat polymers and of polymeric chains grafted on a surface due to the synergic effects during thermal degradation.
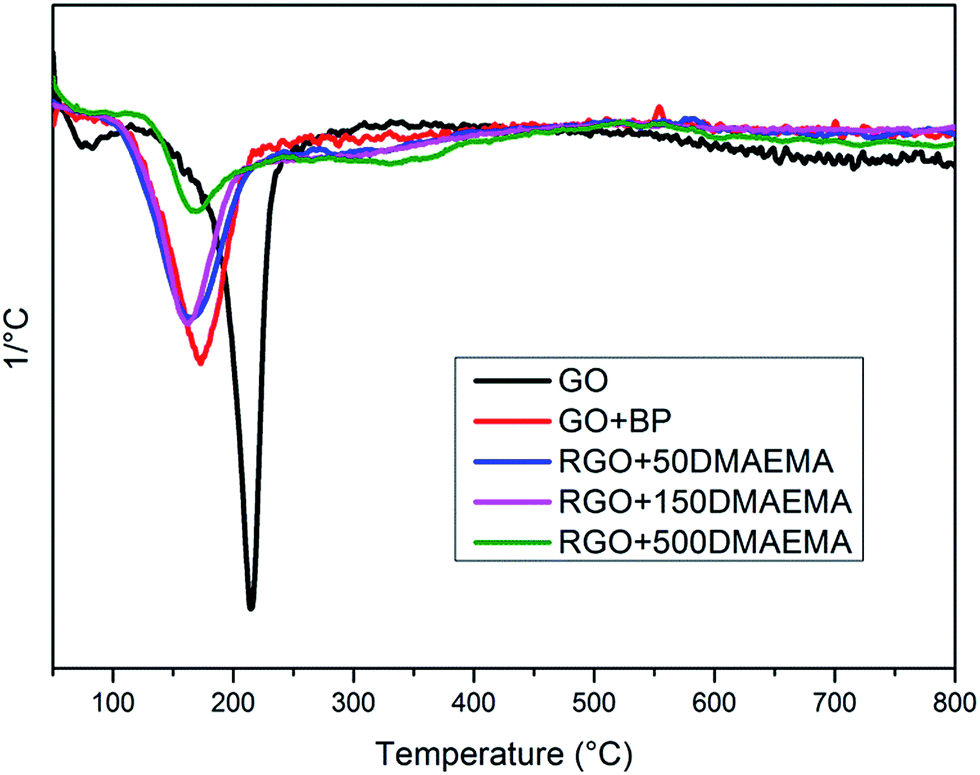 | ||
| Fig. 3 D-TGA for GO, RGO + BP and RGO with different amounts of DMAEMA. | ||
| Sample | Tdegr1 [°C] | Tdegr2 [°C] | Polymer drop of weight [%] | Tg (DSC) [°C] | Tslipping (DMTA) [°C] |
|---|---|---|---|---|---|
| RGO + BP | 172 | — | — | — | — |
| RGO + 50 DMAEMA | 162 | 334 | 3 | 64 | 18 |
| RGO + 150 DMAEMA | 162 | 334 | 5 | 75 | 50 |
| RGO + 500 DMAEMA | 168 | 334 | 7.5 | 65 | 60 |
The increase in the grafted polymer has a clear influence on the thermal behaviour of the free standing RGO papers. The data reported in Table 2 show that a glass transition temperature (Tg) appears to be evident only in the grafted samples and it is more and more evident upon increasing the amount of monomer (see ESI, Fig. S14†). This is a further confirmation that increasing the monomer content in the initial solution proportionally scales the grafting degree. Tgs measured in the RGO + PDMAEMA samples are higher than those of the neat polymer (54 °C, see Fig. S16 in ESI†), indicating that the relaxation of the grafted chains is hindered by the grafting on the RGO sheets.
The higher number of grafted polymer chains have an important influence on the mechanical properties of the RGO papers. First of all, macroscopically, after grafting, the RGO papers are easier to handle and quite robust while the GO and RGO + BP papers are brittle and difficult to manipulate. To better quantify this property, DMTA measurements were performed. In Fig. 4, the DMTA curves for the samples produced after the filtering step are shown. It is clear that both the GO and RGO + BP papers present very poor mechanical properties (E′GO = 2 MPa, E′RGO+BP = 4 MPa), related to simple secondary force interactions between the GO or RGO sheets. Those values are stable to temperature variation, confirming the absence of any transitions, as measured using DSC. For the sample grafted with PDMAEM, an improvement of the mechanical properties at low temperature was found (E′RGO+50 DMAEMA = 15 MPa, E′RGO+150 DMAEMA = 40 MPa, E′RGO+500 DMAEMA = 130 MPa), showing the positive influence of polymer grafting on the mechanical properties.53 These E′ values make our paper compatible with applications such as artificial skin, which is of particular interest in certain robotic frameworks.53 At higher temperatures, an important decrease in the storage moduli is also found for all of these samples. These transition values (Tslipping in Table 2) could be seen as the temperatures at which the grafted polymer chains have enough mobility to disentangle and thus slip, reaching E′ values comparable with unfunctionalized papers. This means that at temperatures lower than Tslipping, the mechanical response of the papers is controlled by interchain interactions while at higher temperatures only secondary forces are operating. This transition occurs at higher temperatures by increasing the number of grafted polymer chains; this could be explained considering that the higher the number of polymer chains (and their length too), the higher the temperature necessary to disentangle them. The Tslipping values are quite different from the Tgs measured using DSC but the fact that the two measurements are based upon the evaluation of different properties must be taken into account. While DSC measures differences in heat absorbance, DMTA evaluates the mechanical properties under a sinusoidal force. So, as previously mentioned, Tslipping could be seen as the temperature at which the chains grafted on the RGO sheets disentangle and not as the Tg of the material. On the other hand, the Tgs measured using DSC could be considered to be more similar to the final fast moduli-decrease observed at temperatures higher than 85 °C, when a typical polymer flow occurs.
 | ||
| Fig. 4 E′ DMTA curves obtained for the free standing samples. | ||
The presence of polymeric chains is particularly evident by observing the top view and cross-section FESEM images shown in Fig. 5 and S17 in ESI,† respectively. The pristine RGO + BP (Fig. 5a) shows mostly wrapped flakes resulting in rough surfaces, while in the presence of the polymer (Fig. 5b) the surface appears to be smoother. This is an indication that the polymeric chains completely cover the sheets and help them to extend smoothly. When the content of the polymer chains is further increased, polymeric aggregation occurs on the surface of the sheets, resulting in white particles on the surface. Moreover, it is possible to foresee that the presence of polymeric chains increases the distance between RGO sheets (see membrane cross-section images in Fig. S17 in ESI†). This is in agreement with the DMTA studies in which the effect of the presence of polymeric chains on the mechanical properties of the papers was demonstrated.
 | ||
| Fig. 5 FESEM images of (a) RGO + BP, (b) RGO + 50 DMAEMA and (c) RGO + 500 DMAEMA. | ||
Electrical characterization
Electrical measurements were performed on graphene membranes using the two point contact method in order to assess any interface effect due to the presence of the polymeric chains (Fig. 6a). It was observed that the conductivity of the RGO + BP paper is enormously lower than the conductivity of the single RGO + BP sheet (13 S cm−1).39 This may be expected, considering that the conductivity of the whole material is affected by the sheet to sheet contact resistance, in other words by the difficult inter-sheet hopping of charges.54 Moreover, it is evident that the increase in the amount of polymer grafted on the sheets strongly affects the electrical conductivity, decreasing σ by 3 orders of magnitude with respect to the RGO + BP. This is in good agreement with our previous evaluation: the presence of a polymeric chain on the graphene sheets further increases the distance between the sheets and thus decreases the conductivity.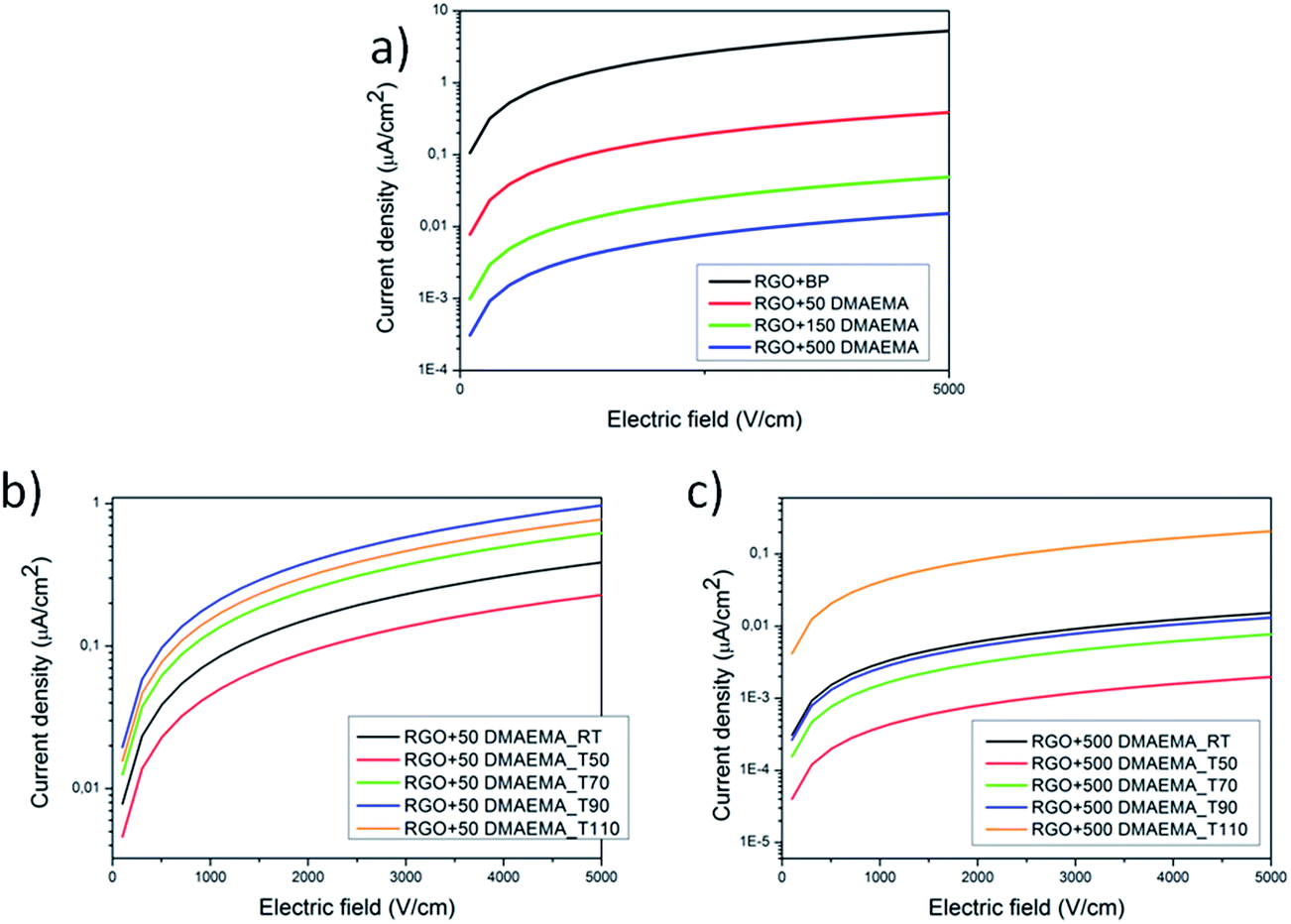 | ||
| Fig. 6 I–V curves of the synthesized materials at room temperature (a), and I–V curves collected after thermal treatments at different temperatures for RGO + 50 DMAEMA (b) and RGO + 500 DMAEMA (c). | ||
In order to assess the effect of the temperature on the polymer grafted RGO papers, the samples were first heated at different temperatures up to 110 °C and then tested at room temperature. With this approach, the effect of a thermal treatment on the papers was evaluated. The RGO + BP paper conductivity is not strongly affected by the thermal treatments (see Fig. S17 in ESI†) while in the presence of the polymer chains the conductivity increases by more than one order of magnitude. This could be explained considering that by increasing the temperature at T > Tg, the polymer chains could undergo relaxation and thus a higher hopping probability could be induced in the samples, according to a mechanism that we propose in Fig. 7. The comparison between the cross-section images of the papers before (see Fig. S18 ESI) and after thermal treatment (Fig. S19 in ESI†) in fact confirms that the latter are more closely packed, indicating a reduction of the distance between the RGO sheets. Subsequently, FTIR experiments were performed on the tested samples in order to investigate whether the polymeric chains were degraded or not after thermal treatment. The graphs (reported in Fig. S20 in ESI†) confirm the presence of the polymeric chains at any temperature, indicating that the structures were preserved.
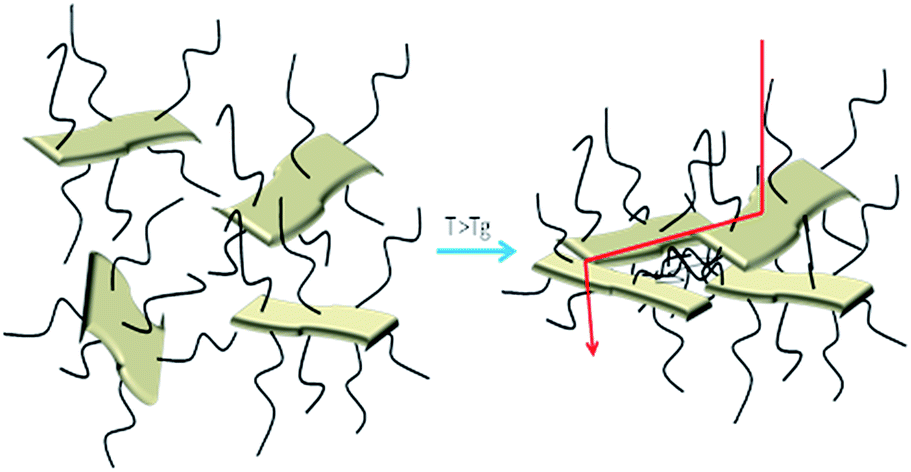 | ||
| Fig. 7 Mechanism proposed as an explanation of the electrical behaviour of the polymer functionalized RGO paper samples. | ||
AC electrical measurements were also performed on two families of samples: standard samples always kept under room conditions and vacuum-dried ones (kept overnight in a vacuum oven to remove adsorbed moisture). Fig. 8 shows the impedance spectroscopy measurements (panel a: resistance; panel b: reactance), for all of the tested samples. Sample RGO + 500 DMAEMA shows no remarkable difference between the measure after exposure to either vacuum (RGO 500 vac sample) or to standard atmosphere (25% RH, RGO 500 atm sample). Sample RGO + 50 DMAEMA also shows overlapping impedance responses, in both conditions. GO + BP features a response very similar to the other samples when subjected to vacuum. However, when subjected to the standard atmosphere, its conductivity is much higher in the low frequency regime (below 1 kHz) and slightly lower in the high frequency regime (above 10 kHz).
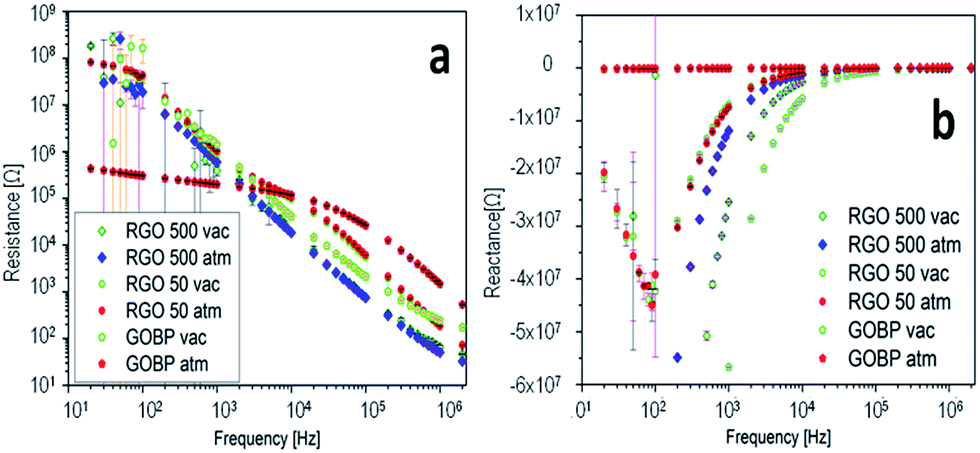 | ||
| Fig. 8 Impedance spectroscopy measurements in the range from 20 Hz to 2 MHz at room temperature, resistance (panel a) and reactance (panel b) versus frequency. | ||
To better understand the frequency response of the most relevant samples (GO + BP, the one with the highest amount of polymer RGO + 500 DMAEMA and a sample submitted to the quaternarization process), we propose a comparison between the Nyquist plots in bilogarithmic scale (Fig. 9a), summarizing the impedance curve in one single plane. Two curves are compared for each material: one recorded after exposure to primary vacuum-drying and one after exposure to standard atmosphere (40% R.H.). We tested the membranes with two signals, one featuring a small amplitude (100 mV) and another one featuring a higher rms (1 V); since there were no relevant differences we show only the 1 V-related measurements, featuring a lower noise. The removal of water results in a different conduction mechanism, depending on the tested material. The impedance locus evolution is such that we do not see the diffusive part anymore (change in slope appearing at frequencies in the kHz regime (red dots), standard atmosphere with 40% R.H.) and we just see the capacitance associated with the membrane (that is the single sloppy line). This means that humidity in the GO + BP membrane provides a mechanism for the diffusion of charge carriers, without which no more diffusion occurs. In the case of RGO + 500 DMAEMA, we have a reversed mechanism, since the change in slope associated with the diffusive part is only seen when moisture is present in the film. Furthermore, an extremely interesting property was found: in the kHz regime, the two responses overlap, providing a clue for practical applications: by spanning the frequency range from this point on, one may find a much reduced impedance modulus as a function of the signal frequency. All of the other responses show a more constant difference, which is not dependent on the frequency. We find the greatest difference between the two electronic states of RGO + 500 DMAEMA at 2 MHz, suggesting that we are not simply looking into the ionic conduction channel, but rather at a structural difference induced by water molecules. Finally a RGO + 500 DMAEMA sample after the quaternarization process is proposed, showing that we recover the same phenomenology of the GO + BP paper, with a much lower intensity with respect to the same sample before quaternarization. In conclusion, fixing the signal frequency to 1 MHz, for example, we will find that: GO + BP shows a difference between the dry conditions and 40% R.H. of one order of magnitude in resistance and 0.3 orders of magnitude in reactance; RGO + 500 DMAEMA shows a difference of one order of magnitude in both resistance and reactance (best performance); and RGO + 500 DMAEMA after the quaternarization process shows no difference in resistance and 0.1 orders of magnitude in reactance.
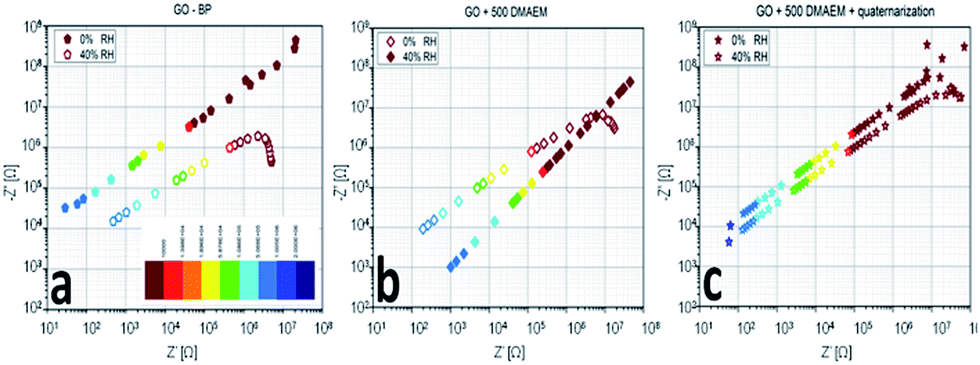 | ||
| Fig. 9 Nyquist plots of RGO + BP (a), RGO + 500 DMEAM (b) and the same as (b) after the quaternarization process (c) membranes in response to humidity variations. | ||
We can thus conclude that the presence (absence) of moisture affects the AC response at frequencies above a threshold value, comprised between 10 Hz and 2 MHz, hindering (enhancing) the conductive and admittive components of impedance; in other words, moisture entraps some ions, excluding them from transport. This only happens in the polymer-grafted papers, as in RGO + BP the effect is reversed and the presence of moisture helps the conduction between adjacent sheets.
The effect of humidity on the functionalized RGO papers presents also a huge consequence at the macroscopic scale: holding a sample in the hand induces an immediate folding, similarly to other work present in the literature.55 This effect is isotropic since it can be reproduced by turning the paper on the other side and is completely reversible within a short time. This effect can be better visualized in the video reported in the ESI.†
Conclusions
In this work, self-standing RGO papers were obtained by means of UV-irradiation, applying a two step process leading to both an extensive reduction of pristine GO and its covalent functionalization with DMAEMA. The grafting was demonstrated to have great influence on the mechanical and electrical properties of the synthesized samples. The functionalized papers showed improved mechanical properties, making these materials easy to handle, contrary to the pure RGO samples. Moreover, using DMTA experiments, the behaviour of the papers was demonstrated to be dependent on the amount of polymer grafting, showing a thermoplastic-like behaviour for the samples with the highest degree of functionalization. The degree of functionalization influences also the electrical response of the papers: the samples featuring a higher amount of polymer show a very insulating behaviour, with a conductivity three orders of magnitude lower than that of the corresponding neat RGO sample. On the other hand, the polymer functionalized samples showed an electrical response dependent on the environmental conditions, exhibiting different conductivities after different thermal treatments and a mechanical response to humidity.References
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- C. X. Guo, M. Wang, T. Chen, X. W. Lou and C. M. Li, Adv. Energy Mater., 2011, 1, 736–741 CrossRef CAS.
- X. Jiang, Y. Cao, P. Li, J. Wei, K. Wang, D. Wu and H. Zhu, Mater. Lett., 2015, 140, 43–47 CrossRef CAS.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- Z. Song, T. Xu, M. L. Gordin, Y.-B. Jiang, I.-T. Bae, Q. Xiao, H. Zhan, J. Liu and D. Wang, Nano Lett., 2012, 12, 2205–2211 CrossRef CAS PubMed.
- Q. Wu, Y. Xu, Z. Yao, A. Liu and G. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- G. Eda and M. Chhowalla, Nano Lett., 2009, 9, 814–818 CrossRef CAS PubMed.
- S. Porro, E. Accornero, C. F. Pirri and C. Ricciardi, Carbon, 2015, 85, 383–396 CrossRef CAS.
- A. Zopfl, M.-M. Lemberger, M. Konig, G. Ruhl, F.-M. Matysik and T. Hirsch, Faraday Discuss., 2014, 173, 403–414 RSC.
- H. J. Salavagione, A. M. Diez-Pascual, E. Lazaro, S. Vera and M. A. Gomez-Fatou, J. Mater. Chem. A, 2014, 2, 14289–14328 RSC.
- L. Al-Mashat, K. Shin, K. Kalantar-zadeh, J. D. Plessis, S. H. Han, R. W. Kojima, R. B. Kaner, D. Li, X. Gou, S. J. Ippolito and W. Wlodarski, J. Phys. Chem. C, 2010, 114, 16168–16173 CrossRef.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027–1036 CrossRef CAS.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061–1105 CrossRef CAS.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC.
- S. Niyogi, E. Bekyarova, M. E. Itkis, H. Zhang, K. Shepperd, J. Hicks, M. Sprinkle, C. Berger, C. N. Lau, W. A. deHeer, E. H. Conrad and R. C. Haddon, Nano Lett., 2010, 10, 4061–4066 CrossRef CAS PubMed.
- T. A. Strom, E. P. Dillon, C. E. Hamilton and A. R. Barron, Chem. Commun., 2010, 46, 4097–4099 RSC.
- S. Vadukumpully, J. Gupta, Y. Zhang, G. Q. Xu and S. Valiyaveettil, Nanoscale, 2011, 3, 303–308 RSC.
- J. Choi, K.-j. Kim, B. Kim, H. Lee and S. Kim, J. Phys. Chem. C, 2009, 113, 9433–9435 CrossRef CAS.
- M. Quintana, K. Spyrou, M. Grzelczak, W. R. Browne, P. Rudolf and M. Prato, ACS Nano, 2010, 4, 3527–3533 CrossRef CAS PubMed.
- L.-H. Liu, M. M. Lerner and M. Yan, Nano Lett., 2010, 10, 3754–3756 CrossRef CAS PubMed.
- Y. Wang, X. Chen, Y. Zhong, F. Zhu and K. P. Loh, Appl. Phys. Lett., 2009, 95, 063302 CrossRef.
- V. K. Kodali, J. Scrimgeour, S. Kim, J. H. Hankinson, K. M. Carroll, W. A. de Heer, C. Berger and J. E. Curtis, Langmuir, 2011, 27, 863–865 CrossRef CAS PubMed.
- X. An, T. W. Butler, M. Washington, S. K. Nayak and S. Kar, ACS Nano, 2011, 5, 1003–1011 CrossRef CAS PubMed.
- M. Lopes, A. Candini, M. Urdampilleta, A. Reserbat-Plantey, V. Bellini, S. Klyatskaya, L. Marty, M. Ruben, M. Affronte, W. Wernsdorfer and N. Bendiab, ACS Nano, 2010, 4, 7531–7537 CrossRef CAS PubMed.
- H.-C. Cheng, R.-J. Shiue, C.-C. Tsai, W.-H. Wang and Y.-T. Chen, ACS Nano, 2011, 5, 2051–2059 CrossRef CAS PubMed.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- W. Tu, J. Lei, S. Zhang and H. Ju, Chem.–Eur. J., 2010, 16, 10771–10777 CrossRef CAS PubMed.
- H. Yang, F. Li, C. Shan, D. Han, Q. Zhang, L. Niu and A. Ivaska, J. Mater. Chem., 2009, 19, 4632–4638 RSC.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- H. Li and C. Bubeck, Macromol. Res., 2013, 21, 290–297 CrossRef CAS.
- A. Badri, M. R. Whittaker and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2981–2992 CrossRef CAS.
- M. Steenackers, A. M. Gigler, N. Zhang, F. Deubel, M. Seifert, L. H. Hess, C. H. Y. X. Lim, K. P. Loh, J. A. Garrido, R. Jordan, M. Stutzmann and I. D. Sharp, J. Am. Chem. Soc., 2011, 133, 10490–10498 CrossRef CAS PubMed.
- L. Kan, Z. Xu and C. Gao, Macromolecules, 2010, 44, 444–452 CrossRef.
- S. H. Lee, D. R. Dreyer, J. An, A. Velamakanni, R. D. Piner, S. Park, Y. Zhu, S. O. Kim, C. W. Bielawski and R. S. Ruoff, Macromol. Rapid Commun., 2010, 31, 281–288 CrossRef CAS PubMed.
- I. Roppolo, A. Chiappone, K. Bejtka, E. Celasco, A. Chiodoni, F. Giorgis, M. Sangermano and S. Porro, Carbon, 2014, 77, 226–235 CrossRef CAS.
- Y. Yang, J. Wang, J. Zhang, J. Liu, X. Yang and H. Zhao, Langmuir, 2009, 25, 11808–11814 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- I. Roppolo, A. Chiappone, S. Porro, M. Castellino and E. Laurenti, New J. Chem., 2015, 39, 2966–2972 RSC.
- A. Kuila, N. Maity, R. K. Layek and A. K. Nandi, J. Mater. Chem. A, 2014, 2, 16039–16050 RSC.
- J. Wang, D. Song, S. Jia and Z. Shao, React. Funct. Polym., 2014, 81, 8–13 CrossRef CAS.
- Y. Li, Y. Chen, C. Zhang, T. Xue and M. Yang, Sens. Actuators, B, 2007, 125, 131–137 CrossRef CAS.
- S. Gupta, M. Agrawal, M. Conrad, N. A. Hutter, P. Olk, F. Simon, L. M. Eng, M. Stamm and R. Jordan, Adv. Funct. Mater., 2010, 20, 1756–1761 CrossRef CAS.
- Y. Q. Yang, B. Zhao, Z. D. Li, W. J. Lin, C. Y. Zhang, X. D. Guo, J. F. Wang and L. J. Zhang, Acta Biomater., 2013, 9, 7679–7690 CrossRef CAS PubMed.
- D. Lin, Q. Jiang, Q. Cheng, Y. Huang, P. Huang, S. Han, S. Guo, Z. Liang and A. Dong, Acta Biomater., 2013, 9, 7746–7757 CrossRef CAS PubMed.
- S. Maji, F. Mitschang, L. Chen, Q. Jin, Y. Wang and S. Agarwal, Macromol. Chem. Phys., 2012, 213, 1643–1654 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- H. Ma, R. H. Davis and C. N. Bowman, Macromolecules, 1999, 33, 331–335 CrossRef.
- N. Graf, E. Yegen, T. Gross, A. Lippitz, W. Weigel, S. Krakert, A. Terfort and W. E. S. Unger, Surf. Sci., 2009, 603, 2849–2860 CrossRef CAS.
- A. Chiolerio, P. Rivolo, S. Porro, S. Stassi, S. Ricciardi, P. Mandracci, G. Canavese, K. Bejtka and C. F. Pirri, RSC Adv., 2014, 4, 51477–51485 RSC.
- D. Yu and L. Dai, J. Phys. Chem. Lett., 2010, 1, 467–470 CrossRef CAS.
- D.-D. Han, Y.-L. Zhang, H.-B. Jiang, H. Xia, J. Feng, Q.-D. Chen, H.-L. Xu and H.-B. Sun, Adv. Mater., 2015, 27, 332–338 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sketch of the functionalization mechanism, FT-IR spectra, XPS survey spectra and N high resolution spectra, TGA and DSC curves, cross-section FESEM images of the samples as prepared and after thermal treatment, IR spectra of the functionalized samples after thermal treatment, and video showing the effects of humidity on the samples. See DOI: 10.1039/c5ra21521h |
| This journal is © The Royal Society of Chemistry 2015 |