DOI:
10.1039/C5RA11641D
(Paper)
RSC Adv., 2015,
5, 95788-95804
Bacillus amyloliquefaciens-secreted cyclic dipeptide – cyclo(L-leucyl-L-prolyl) inhibits biofilm and virulence production in methicillin-resistant Staphylococcus aureus†
Received
17th June 2015
, Accepted 26th October 2015
First published on 28th October 2015
Abstract
The current study explores the inhibitory efficacy of cyclo(L-leucyl-L-prolyl) (CLP), a cyclic dipeptide from Bacillus amyloliquefaciens on the biofilm and virulence production of methicillin-resistant Staphylococcus aureus (MRSA). The minimal inhibitory concentration (MIC) and maximum bactericidal concentration (MBC) of CLP against three MRSA strains were found to be 256 and 512 μg mL−1 respectively. CLP at its sub-MICs (16, 32, 64 and 128 μg mL−1) exhibited a phenomenal dose-dependent antibiofilm activity against MRSA strains with maximum inhibitions of 85–87%. Confocal and scanning electron microscopic examinations validated the antibiofilm efficacy of CLP. In addition, CLP was proficient enough to greatly modify the surface hydrophobicity and significantly reduced the slime synthesis of MRSA. Appreciable differences noticed in the EPS constituents of CLP treated MRSA signified that the possible antibiofilm mechanism could proceed by impeding the synthesis of EPS and thereby CLP prevents biofilm assemblage and the associated virulence cascade. Interestingly, CLP displayed a prominent disruption (52–54%) on a 48 h preformed biofilm of MRSA. Data from in vivo assays using Caenorhabditis elegans unveiled the non-toxic and anti-infective efficacy of CLP. Down-regulation of all studied virulence genes affirmed the results of the phenotypic and in vivo assays. Thus, the present study exemplifies the use of CLP as a plausible alternative to conventional antibiotics in controlling biofilm associated infections of MRSA.
Introduction
Biofilms are structurally complex and dynamic architectures of sessile bacterial communities entrenched in a self-synthesized matrix of extracellular polymeric substances (EPS), consisting mainly of hydrated polysaccharides, proteins, glycopeptides, extracellular DNA and lipids.1 These agglomerations facilitate the adherence of microbes and firmly attach microbial clusters to the underlying biotic or abiotic surfaces.2 The clinical significance of biofilms and its associated bacterial pathogenesis in several chronic human infections has extensively been acknowledged from the last decade. The bacteria when in a biofilm state are upward with 1000-folds more resistant to the action of conventional chemotherapeutics and host immune defense mechanism than their planktonic counterparts, and thus they ultimately complicate the eradication of infection principally.3,4 This in turn increases the feasibility for gene transfer which causes the emergence of strains with new resistance and/or virulence profiles as well. Hence, the World Health Organization has recently identified antimicrobial resistance as the third greatest threats to human health.
Globally, Staphylococcus aureus has been recognized as one of the most predominant biofilm-forming human pathogens causing both community and nosocomial infections associated with significant morbidity and mortality. This Gram-positive pathogen causes a diverse array of clinical complications that range from minor infections (skin and soft tissue lesions) to life threatening infections (pneumonia, endocarditis, osteomyelitis, septicemia and exotoxins syndromes) through expression of impressive arsenal of various virulence factors, such as protein A, coagulase, hemolysin, TSST-1, enterotoxins etc.5 In recent years, S. aureus has equally become the prime cause of infections related to medical implant devices such as prosthetic joints and vascular catheters as like coagulase-negative Staphylococcus epidermidis.6 Additionally, the ability of S. aureus to form biofilm on the surface of indwelling medical devices like titanium, used during surgery is of particular concern, as the infection is virtually impossible to eradicate once the device is colonized. Importantly, an estimate suggests that the biofilms of pathogenic bacteria accounts for over 80% of bacterial infections in the human body.7
The inappropriate and chronic overuse of potent antimicrobial agents like vancomycin, linezolid and teicoplanin that are opted as the last resort against methicillin-resistant Staphylococcus aureus (MRSA) has fueled the unfortunate rise and sturdy emergence of resistance or reduced susceptible isolates.8 As a consequence, the MRSA is approaching an epidemic level in recent years.9 Thus, these facts emphasize that while there is an urgent need for the discovery of new antibacterial agents, indeed there is parallely a pressing need to develop agents that could prevent adherence or biofilm formation to a therapeutically pertinent extent.
To overcome the issues and pitfalls in conventional antibiotic therapy, an alternative strategy called the ‘antivirulence’ or ‘anti-infective’ therapy has recently been proposed that holds greater promise to treat biofilm associated infections. This novel therapeutic approach specifically inhibits the pathogens' virulence rather than imposing any harm to their free-living planktonic counterpart and thereby it reduces or slows the selection for resistance. In the recent past, several bioactive compounds from microbiota associated to marine, alone or in organization with other marine invertebrates, have reassuringly emerged as potential antibiofilm, anti-quorum sensing and antivirulence agents against both Gram-positive and Gram-negative bacterial pathogens.10–15 For instance, a cyclic dipeptide named cis-cyclo(leucyl-tyrosyl)10 from sponge associated Penicillium sp. F37; exopolysaccharides from both marine Vibrio sp. QY101 (ref. 9) as well as sponge-associated Bacillus licheniformis;11 a novel compound named 4-phenylbutanoic acid from marine Bacillus pumilus S6-15;12 and the exoproducts of marine Pseudoalteromonas13 have been well demonstrated for their effective control and efficient inhibition on the detrimental biofilm and virulence production of a broad range of pathogens causing varied infectious diseases.
Following the same paradigm, we have identified a cyclic dipeptide-cyclo(L-leucyl-L-prolyl) (CLP) from mangrove rhizosphere bacterium-Bacillus amyloliquefaciens that exhibits an inhibitory efficacy toward the cariogenic properties of S. mutans.14 Earlier studies on CLP have reported its secretion by Achromobacter xylosoxidans16 and Streptomyces sp17 that were active against the aflatoxins synthesized by Aspergillus parasiticus as well as the rice blast fungus Pyricularia oryzae, respectively. The current study was intentionally undertaken to demonstrate the in vitro and in vivo (using Caenorhabditis elegans-infection model) antivirulence efficacy of CLP against MRSA and attempts were also made to understand its underlying mechanism through transcriptional analysis.
Results
Determination of MIC and MBC of CLP against MRSA in vitro
The efficacy of CLP on planktonic cells of three MRSA test strains was investigated by determination of MIC and MBC values in MHB medium. CLP inhibited the growth of all the three test strains and their MICs were found to be 256 μg mL−1, irrespective of the strains' antibiotics resistance profile (data not shown). For MBC determination, the inoculum from each well with no visible growth was plated on MHA plates, and 512 μg mL−1 was found to be the minimum concentration that completely inhibited growth on the MHA plates. Thus, the concentrations below MIC were used in all further assays to exclude the inhibition due to bacterial growth. Similarly, the MIC and MBC values for the positive control oxacillin were found to be 64 and 128 μg mL−1 respectively.
Effect of sub-MICs of CLP on MRSA biofilm formation
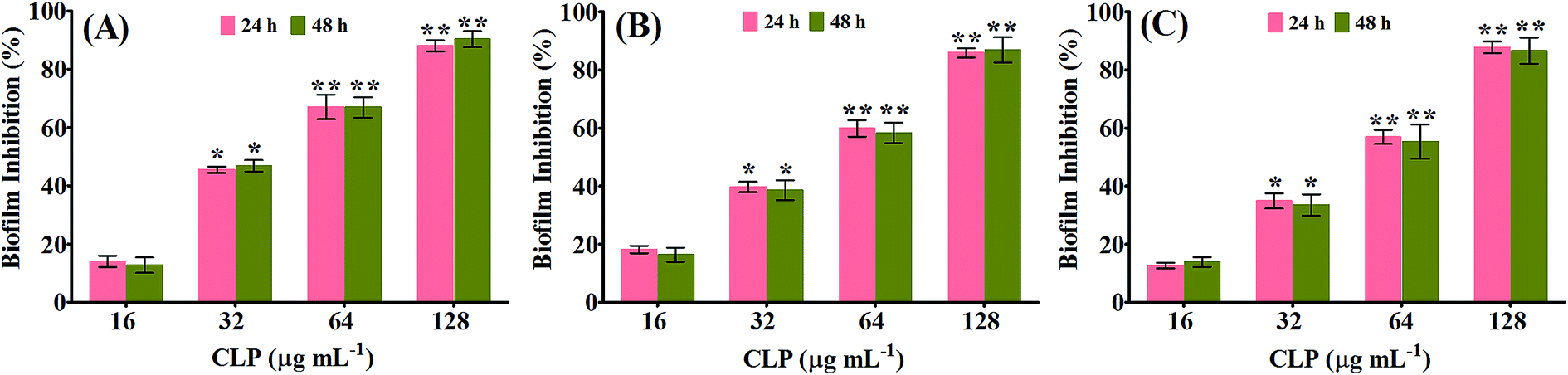
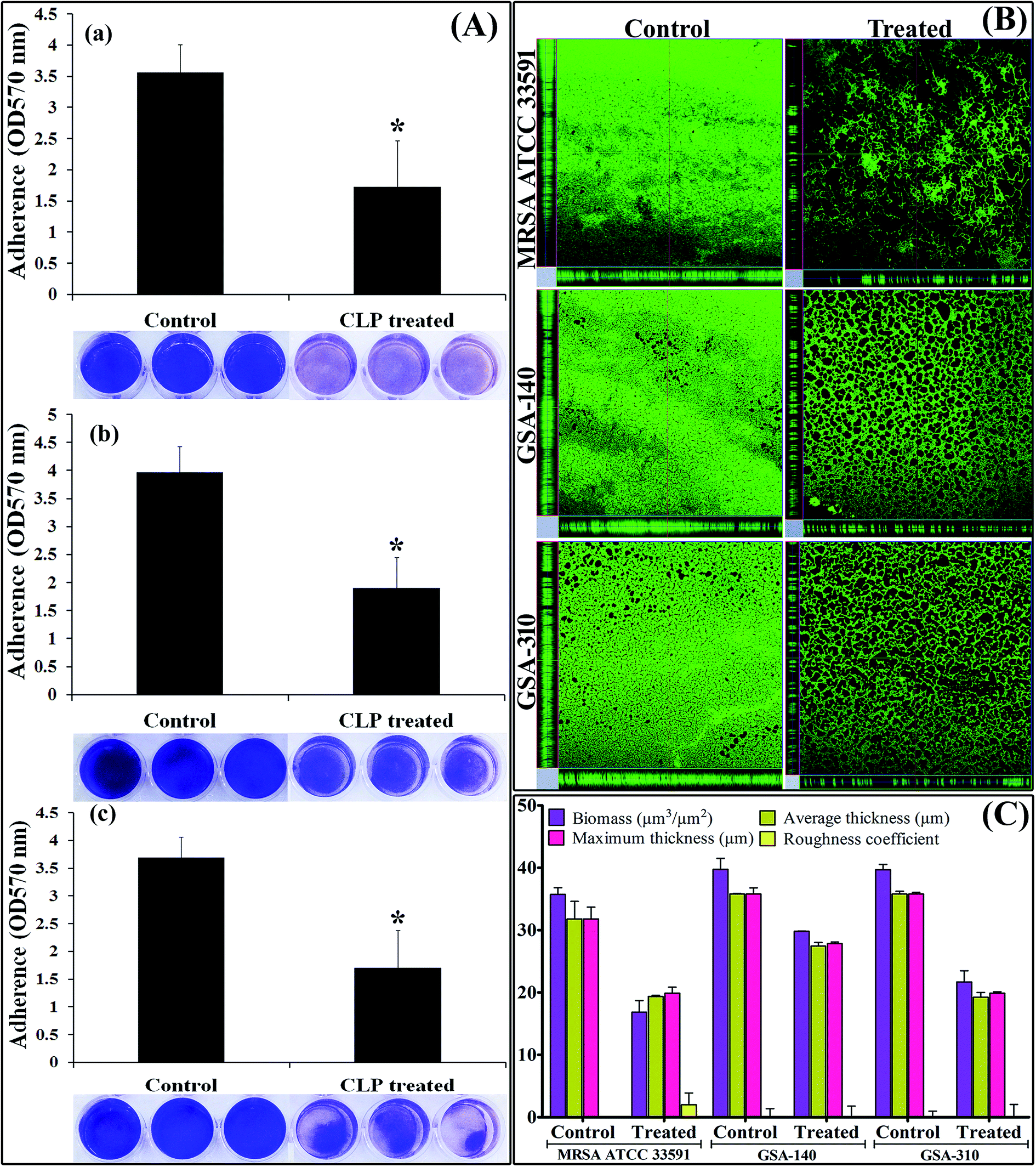
The biofilm inhibitory efficacy of CLP was assessed under in vitro condition by measuring the binding of crystal violet to adherent cells of MRSA on 24-well microtitre plates (MtPs), as it is the most widely used gold-standard method to detect both the biofilm formation as well as antibiofilm efficacy.14,15 Since, the MIC of CLP was determined to be 256 μg mL−1 against all the three tested MRSA strains, the antibiofilm efficacy of CLP was assessed at its sub-MICs, in order to exclude the reduction in biofilm due to antibacterial activity. The antibiofilm activity tested at 1/2 MIC (128 μg mL−1), 1/4 MIC (64 μg mL−1), 1/8 MIC (32 μg mL−1) and 1/16 MIC (16 μg mL−1) against the biofilms of MRSA strains are shown in Fig. 1. As concentration of CLP increases (from 16 μg mL−1 to 128 μg mL−1), statistically significant (p < 0.05) enhancement (from 18% to 87%) in the reduction of biofilm was observed for the test strains MRSA ATCC, GSA-140 and GSA-310 when compared to their respective untreated control groups (Fig. 1). Further, CLP at its sub-MICs was potent enough to retain the antibiofilm efficacy even after 48 h of incubation (Fig. 1), similar to that of 24 h incubation. With these results, it was obvious that CLP exhibits proficient biofilm inhibitory efficacy against MRSA in a concentration-dependent fashion. Quercetin, a known biofilm inhibitor21 was used as a positive control and found to considerably reduce (57–61%) the biofilms formed by the tested MRSA strains at 5 μg mL−1 (data not shown).
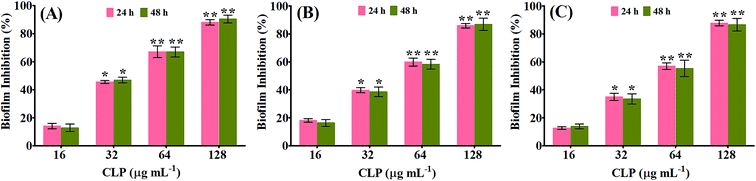 |
| | Fig. 1 Histogram representing the inhibitory effect of CLP at varying concentrations (16, 32, 64 and 128 μg mL−1) on the biofilm formation of (A) MRSA ATCC 33591 (B) GSA-140 and (C) GSA-310 for 24 h and 48 h, quantified by crystal violet adsorption on MtPs and measuring absorbance at 570 nm. Data represent the average of triplicates from independent triplicate assays, and error bars indicate SD. * and ** indicate the statistical significance p < 0.05 and p < 0.01 respectively. | |
The antibiofilm efficacy of CLP-not due to antibacterial activity
To investigate whether the route cause for antibiofilm efficacy of CLP against the MRSA biofilms was directly due to the inhibition of bacterial viability, growth curve analysis was performed using sub-MIC (128 μg mL−1) of CLP. The results of analysis revealed that none of the three tested strains' growth was inhibited (Fig. 2A–C), other than a bit increased growth at certain time hours (which can be neglected as there was no significant increase in growth OD), signifying that CLP at 128 μg mL−1 was not bacteriostatic or bactericidal.
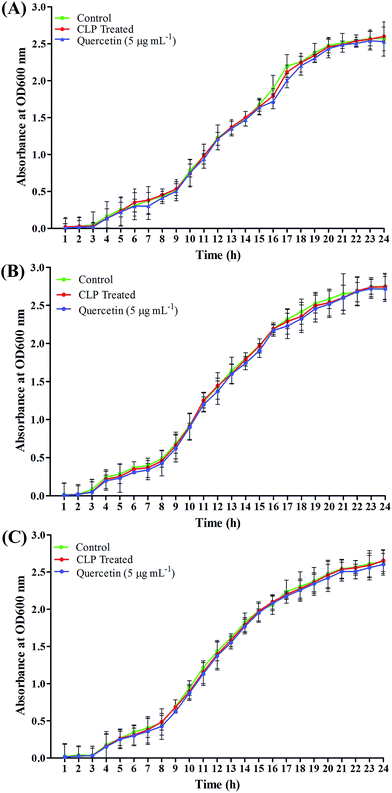 |
| | Fig. 2 Growth curve of (A) MRSA ATCC 33591 (B) GSA-140 and (C) GSA-310 planktonic cells in liquid media with the presence and absence of CLP at sub-MIC (128 μg mL−1). Quercetin was used as a positive control at 5 μg mL−1. The given data represent the mean value of three independent experiments. | |
In situ visualization of biofilm prevention analysis through microscopic image acquisition and analysis
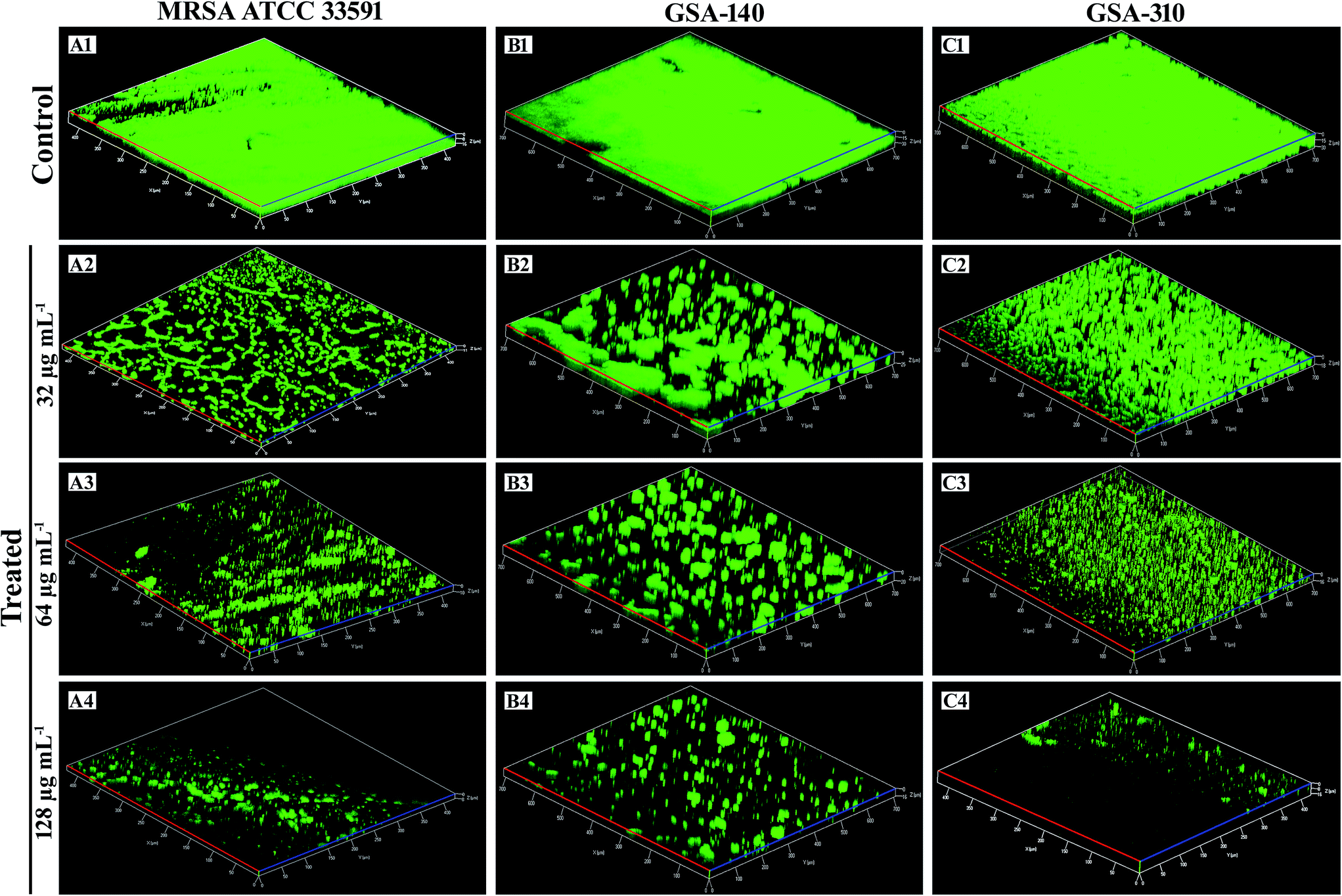
Analysis of biofilms by confocal laser scanning microscopy (CLSM). The surface topology of MRSA biofilm architecture and the CLP's antibiofilm efficacy were visualized and analyzed through CLSM by staining with a nucleic acid fluorescence dye, acridine orange which stains the bacterial cells in biofilm (Fig. 3). Confocal micrographs of the untreated MRSA controls on titanium surface displayed the highly complex multilayered cells and strong adhering ability. Nevertheless, the CLP treated titanium plates depicted the dispersed and disintegrated clumps along with collapsed microcolonies. Furthermore, it was obvious from Fig. 3(A2–A4, B2–B4 and C2–C4) that the biofilm inhibitory efficacy of CLP at its increasing concentrations was extremely dose dependent, as it entirely distorted the microcolony formation and thereby the biomass of MRSA was proficiently decreased. By employing comstat2, a programme used for quantification of three-dimensional biofilm structure which gives insights into biofilms, maximum and average thickness, surface to volume ration etc. (www.comstat.uk), the confocal micrographs were further analyzed and the results revealed that there was a remarkable structural difference in the recalcitrant biofilm architecture of MRSA upon treatment with CLP (Table 1). Substantial reduction in the parameters like biomass, average and maximum thickness of biofilm-treated micrographs inferred the phenomenal antibiofilm efficacy of CLP towards the complex and dynamic architecture of MRSA.
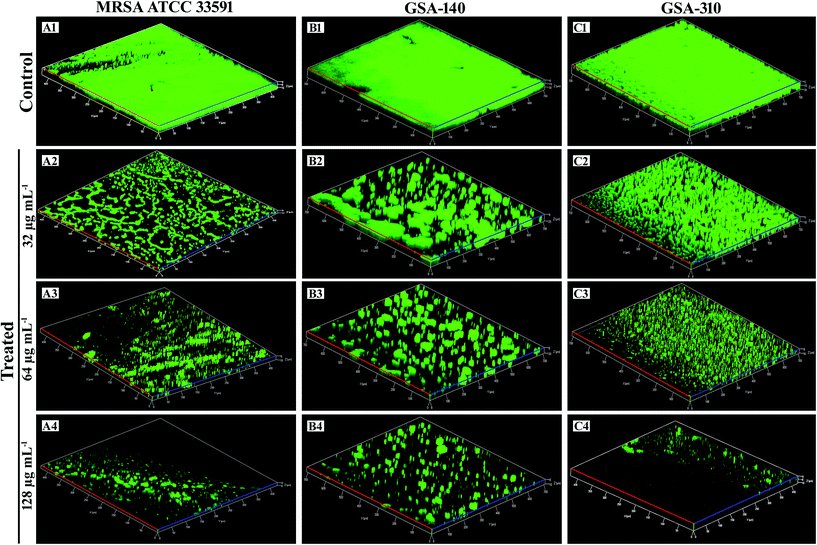 |
| | Fig. 3 Confocal laser scanning micrographs demonstrating the antibiofilm potential of CLP at sub-MICs (32, 64 and 128 μg mL−1) against the biofilms of reference strain [MRSA ATCC 33591 (A1–A4)] and two clinical strains [GSA-140 (B1–B4) and GSA-310 (C1–C4)] grown on titanium plates. | |
Table 1 comstat2 analysis of biofilms formed by three test MRSA biofilms in the presence and absence of CLP. Mean values of triplicate individual experiments and SDs are shown. * indicates the statistical significance (p < 0.05)
| Strain |
Biovolume (μm3 μm−2) |
Average thickness (μm) |
Surface to volume ratio (μm2 μm−3) |
| Control |
T1 |
T2 |
T3 |
Control |
T1 |
T2 |
T3 |
Control |
T1 |
T2 |
T3 |
| MRSA ATCC |
18.03 ± 0.44 |
2.24 ± 0.44* |
1.80 ± 0.11* |
0.12 ± 0.06* |
19.21 ± 1.40 |
3.07 ± 0.80* |
2.55 ± 0.24* |
0.16 ± 0.41* |
0.03 ± 0.17 |
0.04 ± 0.01 |
0.44 ± 0.08 |
0.53 ± 0.05 |
| GSA-140 |
36.26 ± 0.41 |
28.10 ± 0.79 |
21.85 ± 1.16* |
18.46 ± 1.34* |
34 ± 1.53 |
26.35 ± 1.44 |
20.48 ± 1.23* |
17.31 ± 1.17* |
0.03 ± 0.01 |
0.04 ± 0.15 |
0.05 ± 0.01 |
0.06 ± 0.01 |
| GSA-310 |
36.49 ± 0.56 |
22.89 ± 0.91* |
21.14 ± 1.21* |
18.46 ± 1.84* |
34.21 ± 0.61 |
21.44 ± 1.10* |
19.82 ± 1.16* |
17.31 ± 1.19* |
0.03 ± 0.01 |
0.05 ± 0.16 |
0.05 ± 0.01 |
0.06 ± 0.01 |
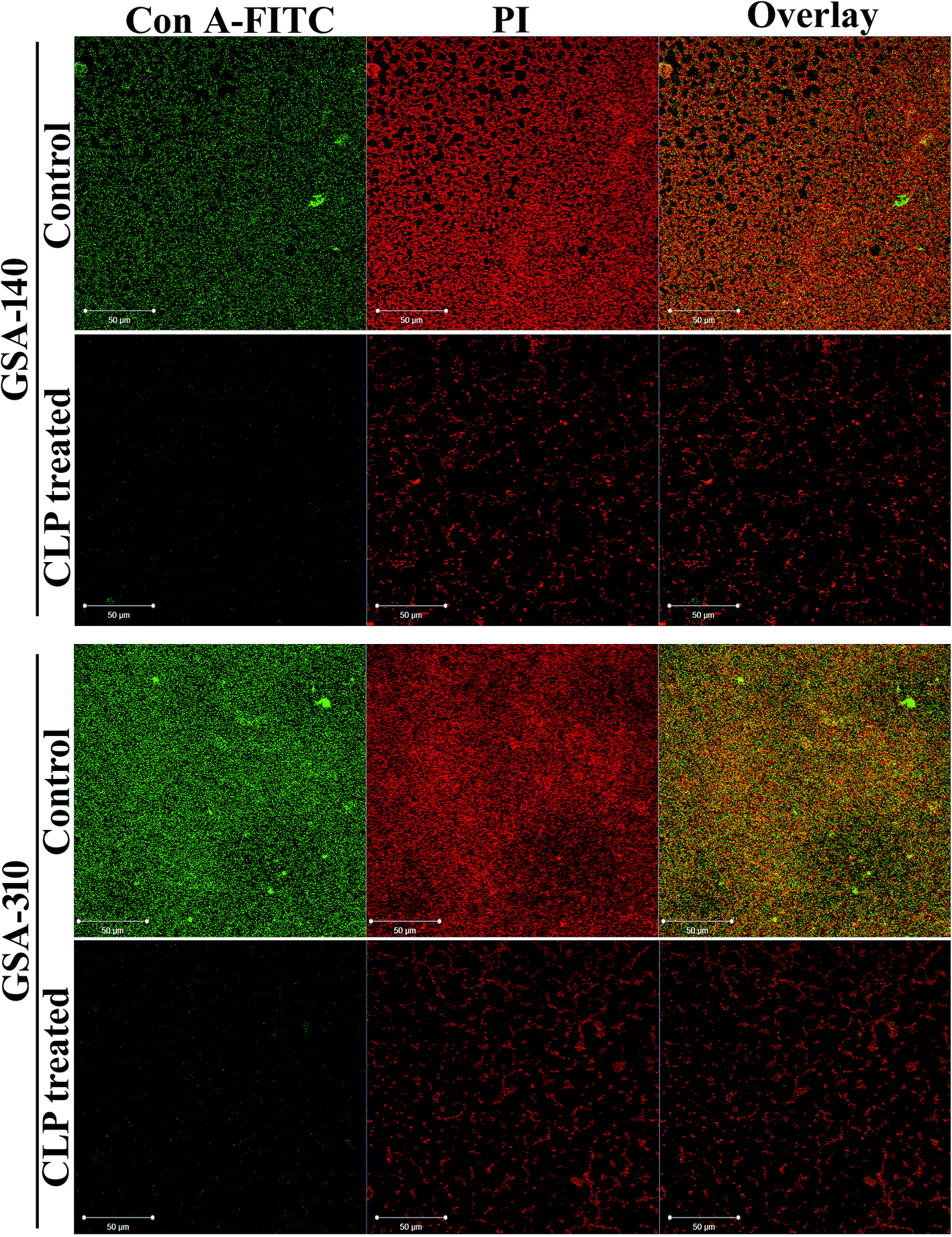
Furthermore, to investigate the effect of CLP on the extracellular polysaccharides, which is a typical constituent of EPS matrix in S. aureus biofilm, we applied polysaccharide specific stain viz. Concanavalin A (Con A) conjugated to fluorescein isothiocyanate (FITC) to intact biofilms formed with and without CLP, and localized the stain within the biofilms using CLSM. S. aureus cells stained with propidium iodide (PI) were easily distinguished from the extracellular matrix by their size and morphology. The superimposed (overlay) confocal micrographs with PI (red) and Con A-FITC (green) fluorescent intensities bring in yellow color, which emulates the polysaccharides being synthesized as a capsular component in biofilm. As can be seen in Fig. 4 (panels representing control), cells were found enmeshed in the matrix of EPS, suggesting a 3-dimensional biofilm architecture of S. aureus. However, such EPS matrix were largely absent in both GSA-140 and GSA-310 strains grown in the presence of CLP (128 μg mL−1) (Fig. 4; panels representing CLP treated).
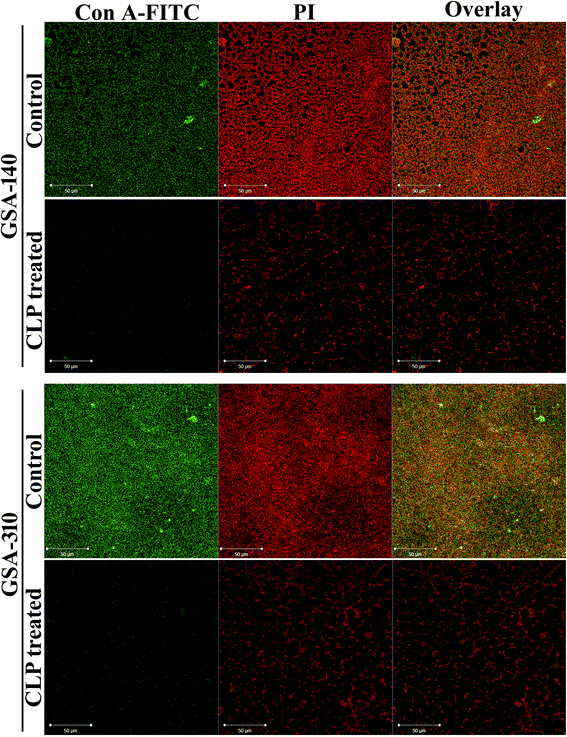 |
| | Fig. 4 Confocal laser scanning micrographs of 48 h grown biofilms of clinical strains (GSA-140 and GSA-310) in the presence and absence of CLP (128 μg mL−1) showing Con A-FITC-stained polysaccharides in green (left sector), propidium iodide-stained bacterial cells in red (middle sector) and capsular components in yellow colour (right sector). Con A-FITC, Concanavalin A-fluorescein isothiocynate; PI, propidium iodide. | |
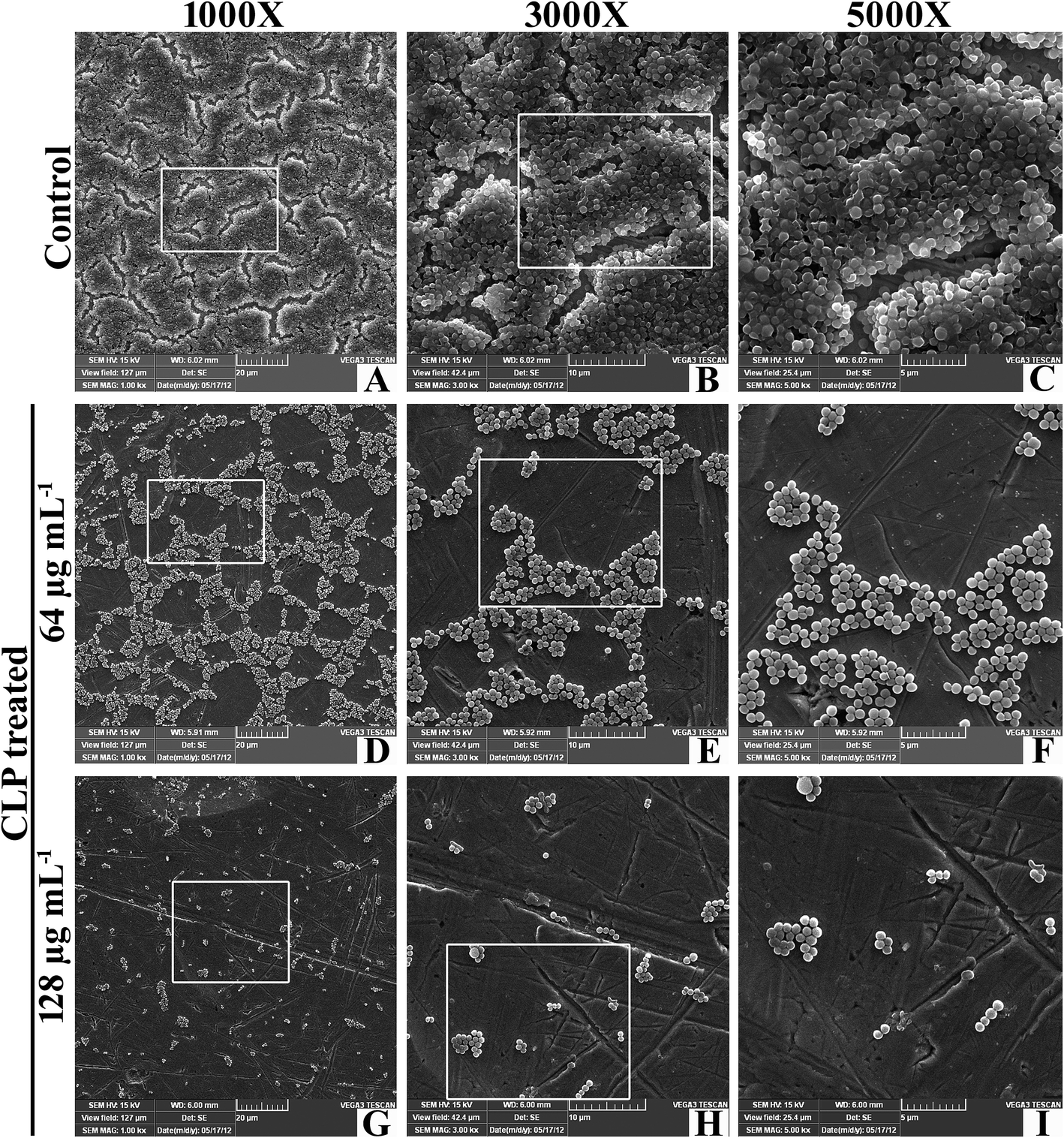
Scanning electron microscopy. SEM analysis was also performed to further elucidate the antibiofilm potential of CLP against biofilms of GSA-140 strain on titanium plates following 24 h of incubation (Fig. 5). The SEM image of untreated GSA-140 on titanium surface depicted a thick heterogeneous layer with conglomerated clusters (Fig. 5(A–C)), which are the characteristics of Staphylococci. While on the contrary, the SEM micrographs of CLP (at sub-MICs) treated titanium plates unveiled the factual disruption and disintegration of recalcitrant biofilm architecture formed by GSA-140 (Fig. 5(D–I)). The poor biofilm development, huge microcolonies and the cell growth as isolated individual colonies (at relatively high concentration of CLP) stand testimony to the remarkable antibiofilm efficacy of CLP against MRSA.
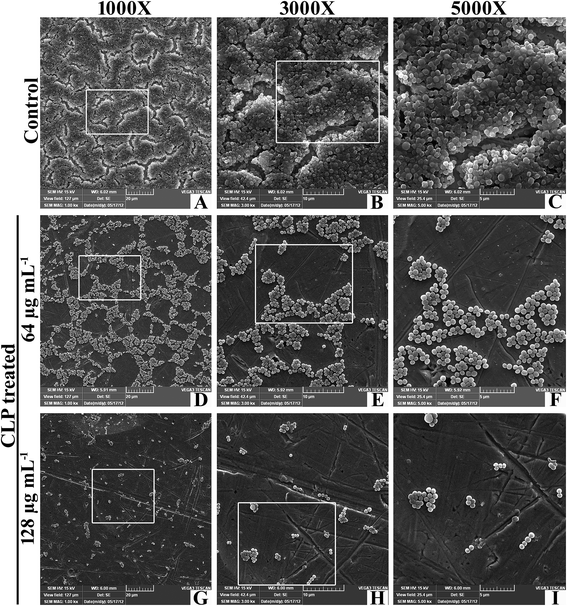 |
| | Fig. 5 Scanning electron micrographs of GSA-140 on titanium plate in the presence and absence CLP at 64 and 128 μg mL−1 concentrations. | |
Inhibitory effect of CLP on biofilm formation of other Gram-positive pathogens
With the intention to know whether CLP endorse broad spectrum of biofilm inhibitory efficacy, the effect of CLP was examined against the biofilms of various Gram-positive pathogens such as Streptococcus mitis (ATCC 6249), Streptococcus salivarius (ATCC 13419), Streptococcus sanguinis (ATCC 10556) by 24-well MtP assay. Biofilms formed by three Streptococcus spp. were significantly (p < 0.05) reduced by the CLP treatment. The percentage biofilm inhibition of CLP at sub-MIC (128 μg mL−1) against S. mitis (Fig. 6A), S. salivarius (Fig. 6B), and S. sanguinis (Fig. 6C) were found to be 85, 87 and 86%, respectively (Fig. 5). Further, the confocal microscopic images (Fig. 6) of control and CLP (128 μg mL−1) treated biofilms of these three pathogens substantiated the result of in vitro MtP assay.
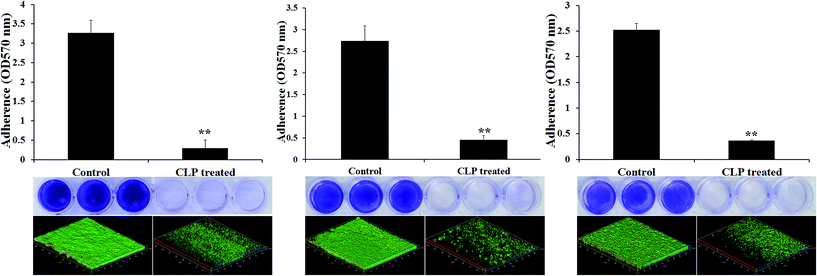 |
| | Fig. 6 Inhibitory effect of CLP at its sub-MIC (128 μg mL−1) on biofilm formation of other Gram-positive pathogens; (A) Streptococcus mitis ATCC 6249, (B) Streptococcus salivarius ATCC 13419 and (C) Streptococcus sanguinis ATCC 10556, quantified by crystal violet adsorption on MtP and measuring absorbance at 570 nm. Confocal micrographs at the lower panel showcase the decreased biofilm in the presence of CLP. ** indicates the statistical significance (p < 0.01). | |
Effect of CLP in inactivating 48 h preformed MRSA biofilm on polystyrene and titanium surfaces
Despite the fact that CLP displayed antibiofilm efficacy against several Gram-positive bacterial pathogens, it was of interest to explore whether the 48 h preformed biofilms of MRSA were also susceptible to CLP. The biofilm biomass assay using 24 well MtPs unveiled that CLP (128 μg mL−1) disrupted 52–54% of preformed biofilms of three test strains (Fig. 7A). In addition, confocal micrographs of control and treated biofilms grown on titanium plates (Fig. 7B) confirmed the biofilm disruption potential of CLP, which was further corroborated through comstat2 software analysis (Fig. 7C). Notably, the biomass and thickness of 48 h preformed biofilms were drastically reduced with the action of CLP (Fig. 7C).
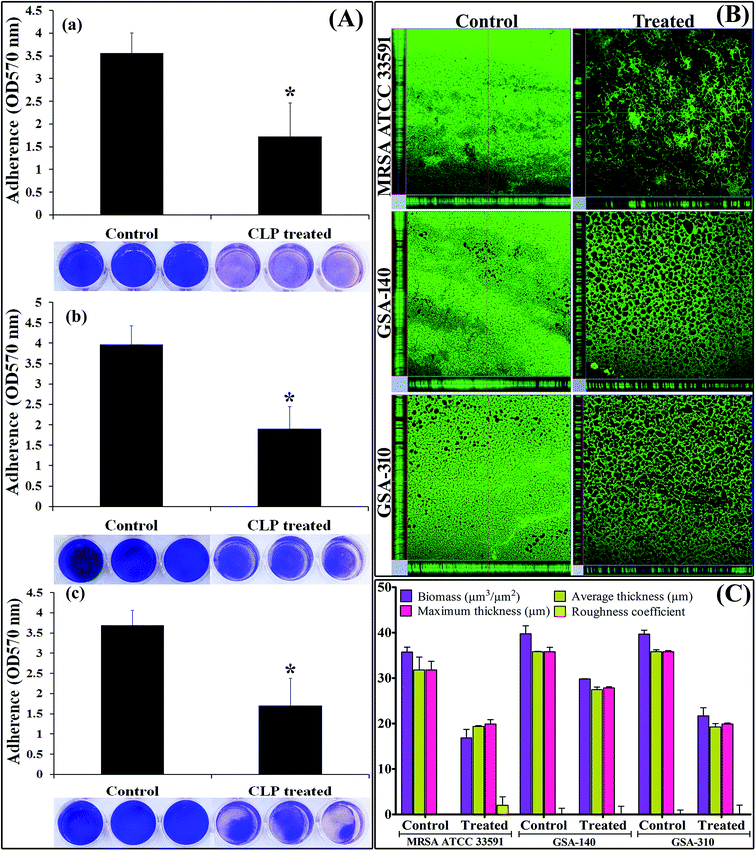 |
| | Fig. 7 Mature biofilm disruption efficacy of CLP (128 μg mL−1) against the recalcitrant biofilms of MRSA ATCC 33591, GSA-140 and GSA-310 divulged by (A) in vitro MtP assay (B) CLSM images and (C) comstat2 software analysis of the obtained CLSM images. * indicates the statistical significance (p < 0.05). | |
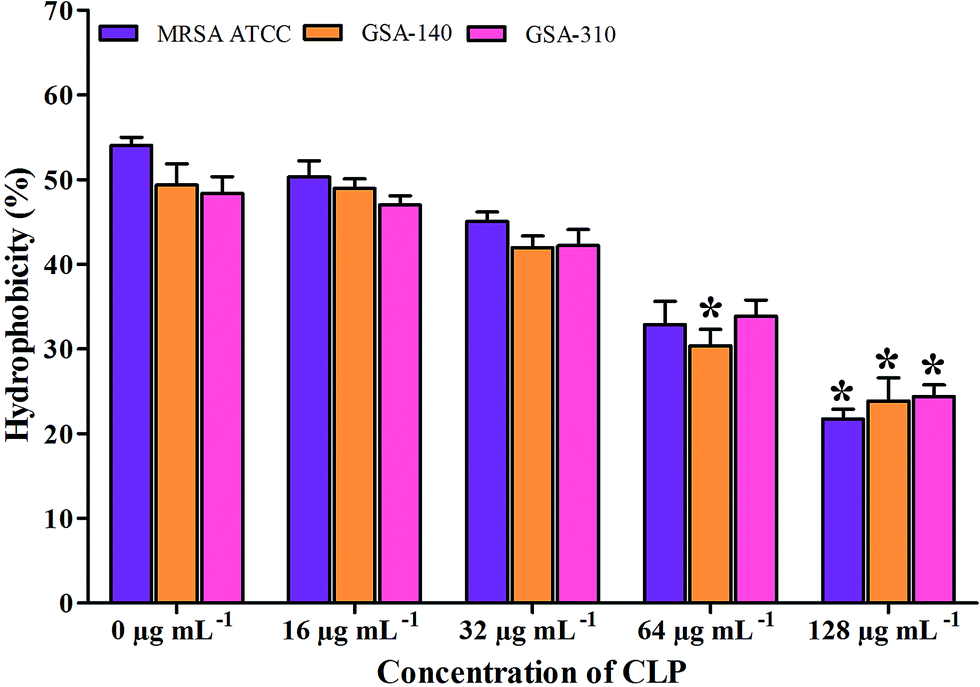
Effect of CLP on cell surface hydrophobicity (CSH) of MRSA
As there are mounting evidences to signify the critical role of CSH in the adhesion property of S. aureus,12,13 we examined the effect of CLP on CSH of test MRSA strains. The CSH of MRSA strains without the action of CLP was found to be 46–50%. Whereas, the strains grown along with CLP (128 μg mL−1) showed significant reduction in CSH (p < 0.05) (Fig. 8). In addition, the hydrophobic nature of MRSA was gradually decreased with increase in CLP concentration (16–128 μg mL−1) (Fig. 8).
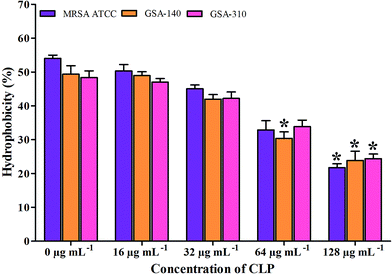 |
| | Fig. 8 Effect of CLP on the cell surface hydrophobicity of MRSA ATCC 33591, GSA-140 and GSA-310 strains. Mean values of triplicate individual experiments and SDs are shown. * indicates the statistical significance (p < 0.05). | |
Effect of CLP on the slime synthesis of MRSA
As a prologue to assess the inhibition of biofilm formation of S. aureus, the ability of CLP to inhibit the S. aureus slime synthesis was qualitatively examined using Congo red agar/broth (CRA/CRB) assays. The stain Congo red is used to show the presence of the exopolysaccharide of aquatic Gram-negative bacilli through light microscopic examination, albeit, the exact mechanism of the Congo red on slime is unclear.24 CLP at sub-MICs was incorporated into CRA plates to know whether the growing colonies show any change in colour from black to red or Bordeaux red. Outcome of assay (observed after 24 h of CRA plates incubation) demonstrated that CLP was potent enough to inhibit the slime synthesized by S. aureus (Fig. 9A). This was confirmed by the appearance of red coloured colonies in the CLP incorporated plates, whereas the colonies in control plate remain black in colour. The Congo red broth (CRB) assay also confirmed the results of CRA assay, stipulating the concentration-dependent inhibitory effect of CLP on the slime synthesis of S. aureus. The gradual colour change from black to red in CRB tubes supplemented with CLP at its sub-MICs is also evident from Fig. 9B. Furthermore, quantification of the culture supernatants from these CRB tubes strongly affirmed the slime inhibiting efficacy of CLP (p < 0.05) (Fig. 9C).
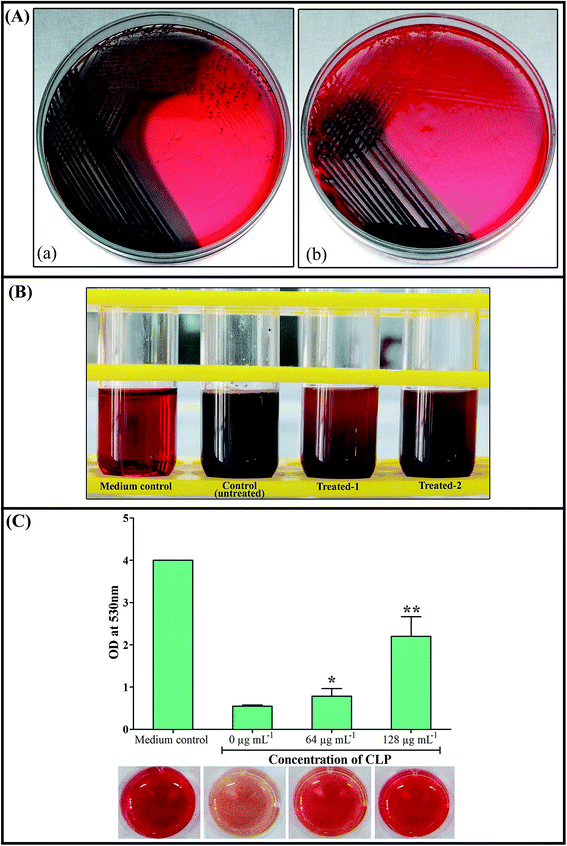 |
| | Fig. 9 Inhibitory efficacy of CLP on the slime synthesis of GSA-140 grown on Congo red agar/broth media: (A) bacterial colonies of GSA-140 grown on CRA plate showing decreased levels of slime production in the presence of CLP at its sub-MIC (b) compared to the control plate depicting the strong black colour colonies (a); (B) GSA-140 grown on Congo red broth, showing gradual reduction in slime synthesis with the presence of CLP at 128 μg mL−1 (treated 1) and 64 μg mL−1 (treated 2), whereas the control tube shows strong black colour; (C) quantification of Congo red broth assay at 530 nm spectroscopically, reflecting change in colour from pale red (untreated control) to bordeaux red (64 μg mL−1) and red (128 μg mL−1) as a result of slime inhibitory efficacy of CLP. * and ** indicate the statistical significance p < 0.05 and p < 0.01 respectively. | |
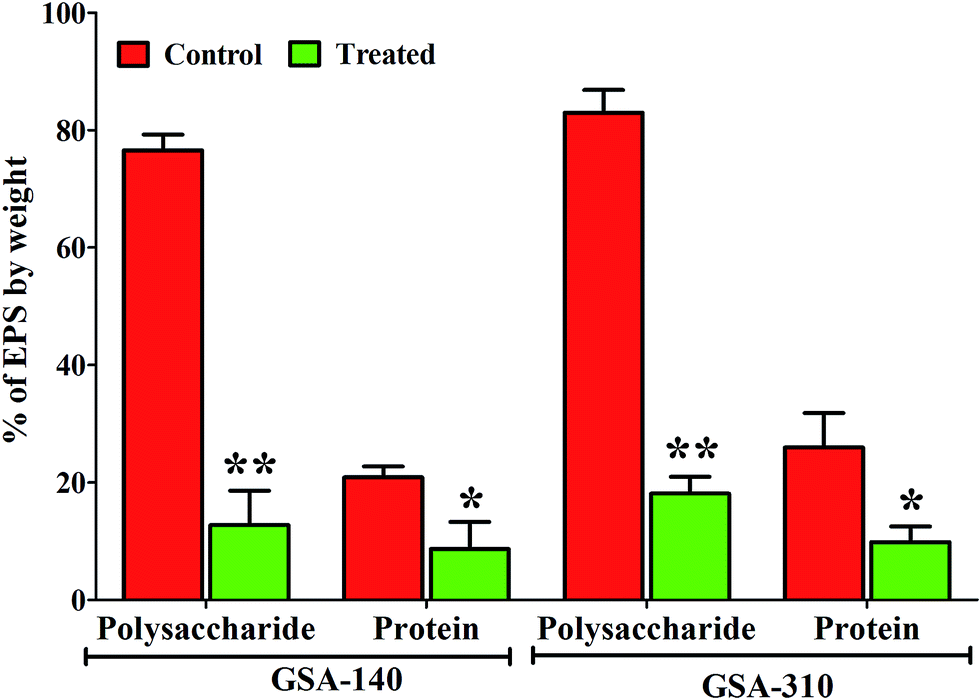
Alteration in extracellular polymeric substances (EPS) of MRSA biofilms by CLP
Quantitative measurement of polysaccharides and proteins from EPS of control and CLP treated MRSA strains indicated a significant (p < 0.05) reduction in the production of both the EPS components. As can be observed from Fig. 10, polysaccharide was predominant in both the strains compared to proteins, and upon treatment with CLP the production of polysaccharides (p < 0.01) as well as proteins (p < 0.05) were decreased to >50% (Fig. 10).
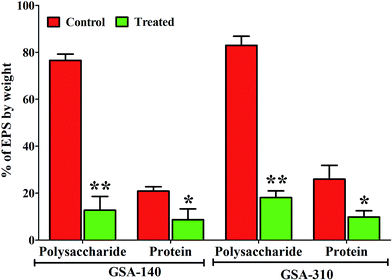 |
| | Fig. 10 Inhibitory effect of CLP at sub-MIC (128 μg mL−1) on the EPS components viz. polysaccharides and proteins in GSA-140 and GSA-310 biofilms. Mean values of triplicate individual experiments and SDs are shown. * and ** indicate the statistical significance p < 0.05 and p < 0.01 respectively. | |
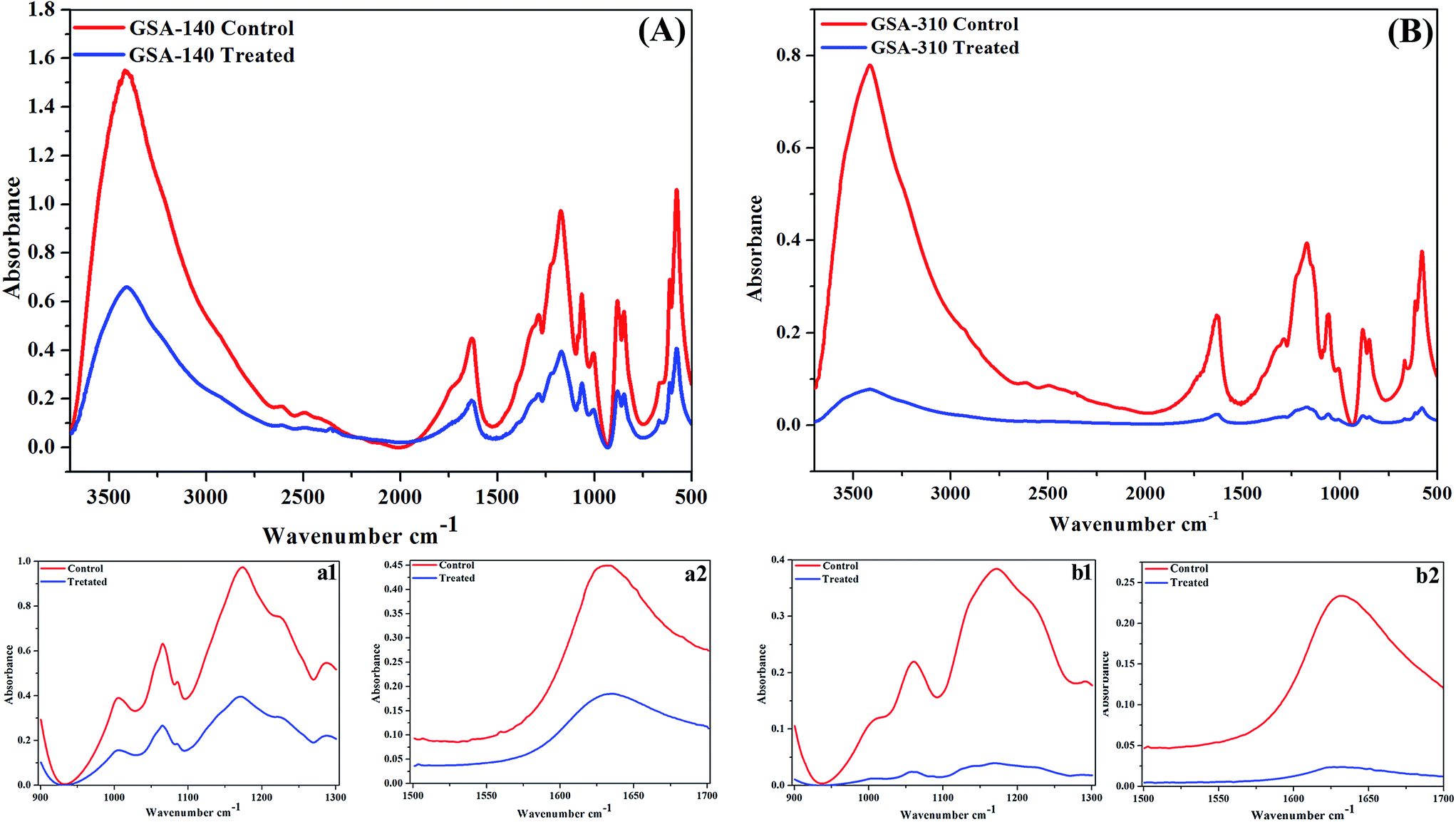
FT-IR spectra of EPS extracted from control and treated strains of two clinical MRSA (GSA-140 (Fig. 11A) and GSA-310 (Fig. 11B)) indicated the presence of polysaccharides, nucleic acids (900 to 1300 cm−1) (Fig. 11(a1 and b1)), as well as proteins (1500 to 1700 cm−1) (Fig. 11(a2 and b2)). The spectra had differences both in shape and in absorbance intensity, indicating that there was variation in the composition and quantity of each individual component. The peaks for both protein and carbohydrate were substantially higher for control EPS of both the clinical strains.
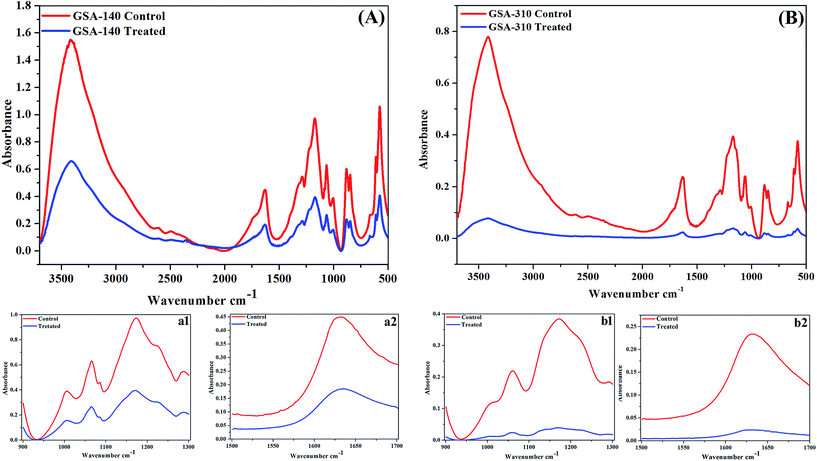 |
| | Fig. 11 FTIR spectra of EPS extracted from (A) GSA-140 and (B) GSA-310 biofilms treated with 128 μg mL−1 of CLP. a1 and b1 represent the –COC– group vibrations in polysaccharides and nucleic acids (900 to 1300 cm−1) region of GSA-140 and GSA-310 respectively; a2 and b2 represent the protein (1500 to 1700 cm−1) region of GSA-140 and GSA-310 respectively. | |
In IR spectral analysis, the peak at 3408 cm−1 of untreated EPS demonstrated the stretching of OH group of water,31,32 whereas the lack of this peak during CLP treatment indicated the dehydration of S. aureus cells. The vibrational modes of esters present in fatty acid (1722 cm−1 and 1284 cm−1) and polysaccharide C–O–C ring (1057 cm−1)32,33 attributed to an increased production of acid sugars containing polysaccharide by biofilm cells. Additionally, the observed O-acetyl group (1722 cm−1 and 1227 cm−1) and glycosidic linkage type of anomeric regions (879 cm−1 and 846 cm−1) are believed to be essential for maintaining the biofilm architecture in the sessile bacterial cells.34 In contrast, the absence of these peaks in CLP treated samples signified the loss of both the production of acid sugars as well as the biofilm integrity that ascribed the reduced virulence of the bacteria.
Rescuing potential of CLP against MRSA infection
Primarily, the toxicity of CLP at 128 μg mL−1 (experimental dosage) was examined in uninfected adult C. elegans by assessing their survival. Even at the tested concentration, the nematodes were found to be healthy, and no significant difference was observed between the CLP treated group and the drug-free group, which evident the non-toxic nature of CLP.
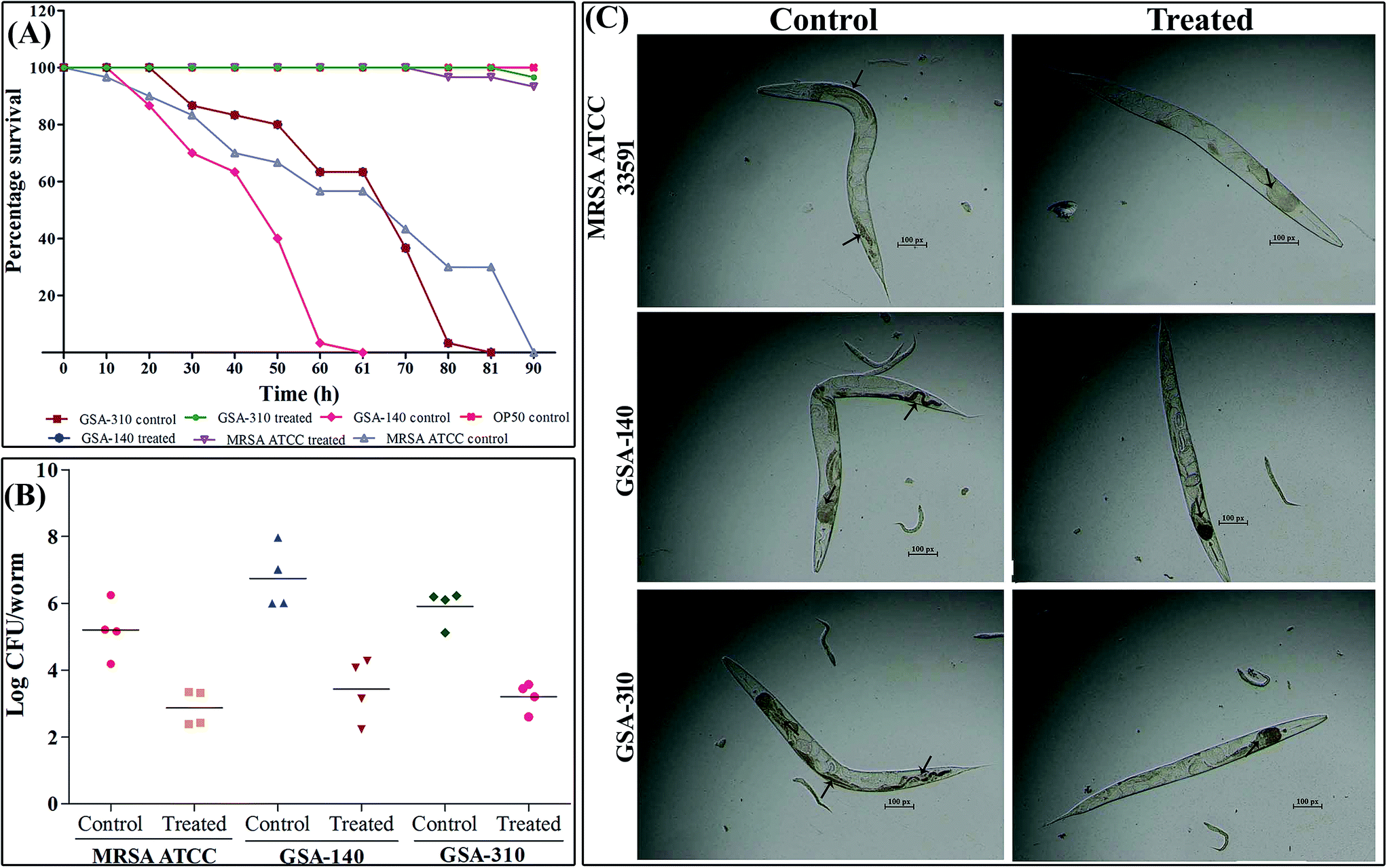
Since CLP was demonstrated to inhibit biofilm in vitro, the most extensively acknowledged pathogenic/virulent trait of MRSA, we further investigated the in vivo antibiofilm efficacy of CLP using C. elegans-MRSA infection model. At tested concentration 128 μg mL−1, CLP significantly (p < 0.005) protected nematodes from MRSA infection (Fig. 12A). More specifically, for the complete killing of nematodes the test strains, MRSA ATCC, GSA-140 and GSA-310 required 90 ± 9, 60 ± 6 and 80 ± 8 h, respectively (Fig. 12A). Every dead nematode had visible bacterial colonization in the pharyngeal and tail regions (Fig. 12C). On the contrary, with supplementation of 128 μg mL−1 of CLP, >95% of the nematodes were found healthy with significantly reduced colonization in the pharyngeal and tail regions till the 96 h up to which the nematodes were observed (Fig. 12A and C).
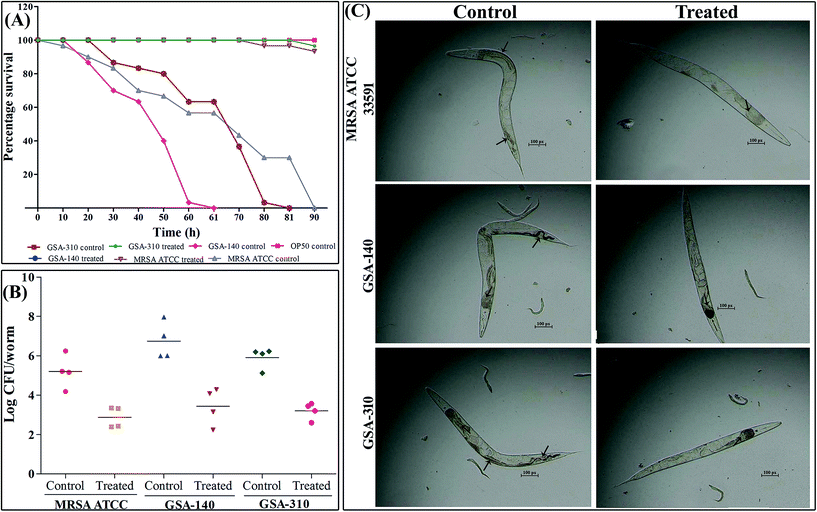 |
| | Fig. 12 In vivo protective efficacy of CLP against S. aureus infection. (A) Survival graph showing the rescued survival of nematodes supplemented with CLP against S. aureus. (B) CLP reduced the intestinal colonization of S. aureus in infected nematodes. (C) Microscopic images showcase the reduced colonization of S. aureus in CLP supplemented nematodes than their respective controls. Arrows in the control panel indicates the dense colonization of S. aureus, whereas arrows in CLP treated panel point to reduced bacterial load inside the nematodes. | |
CLP reduced the bacterial burden in C. elegans
To further confirm the microscopic results on in vivo antibiofilm activity of CLP and to determine the MRSA internalization in C. elegans, a CFU assay was performed. As shown in Fig. 12B, exposing C. elegans to MRSA ATCC, GSA-140 and GSA-310 increased the intestinal bacterial load in nematodes to a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) CFU of 5.21 ± 0.84, 6.00 ± 0.94 and 6.1 ± 0.54, respectively. In contrast, supplementation of CLP reduced the bacterial colonization to a logCFU of 2.4 ± 0.53, 2.2 ± 0.94 and 2.6 ± 0.42 in the nematodes infected with MRSA ATCC, GSA-140 and GSA-310, respectively. The reduced bacterial load inside nematodes' intestine during the gavage of CLP clearly suggested the antibiofilm efficacy of the compound against MRSA infection.
CFU of 5.21 ± 0.84, 6.00 ± 0.94 and 6.1 ± 0.54, respectively. In contrast, supplementation of CLP reduced the bacterial colonization to a logCFU of 2.4 ± 0.53, 2.2 ± 0.94 and 2.6 ± 0.42 in the nematodes infected with MRSA ATCC, GSA-140 and GSA-310, respectively. The reduced bacterial load inside nematodes' intestine during the gavage of CLP clearly suggested the antibiofilm efficacy of the compound against MRSA infection.
Gene expression profile
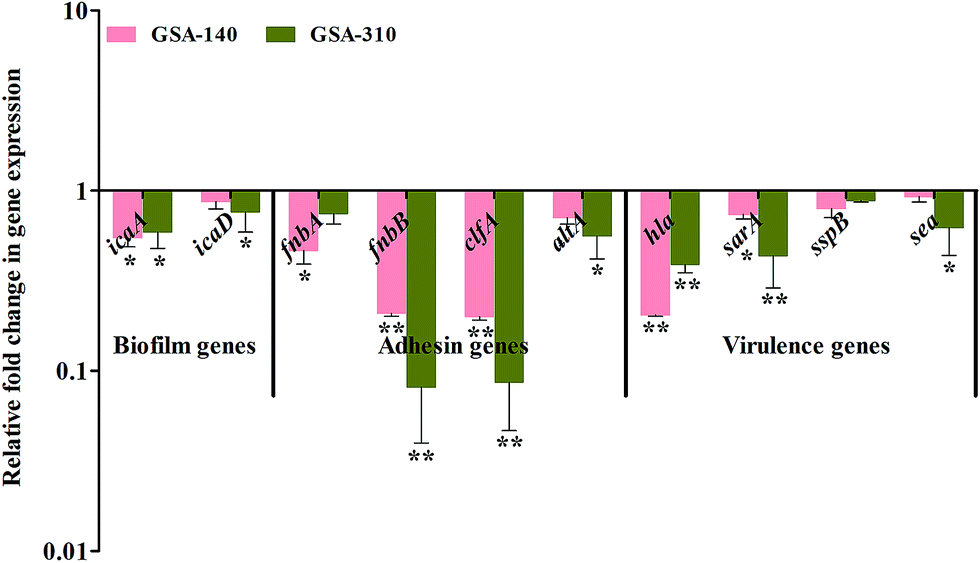
As CLP targets the biofilms and virulence of S. aureus, some of the genes involved in initial attachment, biofilm formation and virulence production were used in the present study. To understand the antibiofilm action mechanism of CLP, we further investigated the differential gene expression of biofilm (icaA and icaD), adhesins (fnbA, fnbB, clfA and altA) and virulence-associated (hla, sarA, sspB and sea) genes with 16S rRNA as an internal control in CLP (sub-MIC) exposed and unexposed MRSA strains (GSA-140 and GSA-310) by real-time PCR (qPCR). The melt curve analysis revealed the absence of non-specific products in all the amplification reactions. Prominently, the qPCR data demonstrated that the treatment of MRSA with CLP has significantly reduced the expression level of all the genes (Fig. 13).
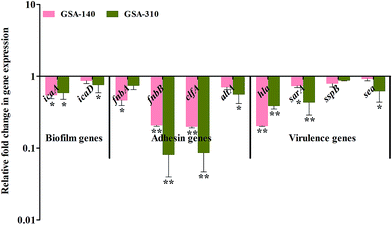 |
| | Fig. 13 Gene expression profile of specific genes involved in biofilm, adhesion and virulence mechanisms. Real time-PCR was carried out in triplicate. Data presented were generated from at least three independent sets of experiments. * and ** indicate the statistical significance p < 0.05 and p < 0.01 respectively. | |
Discussion
Hitherto, the cyclic dipeptides and their derivatives have been well recognized to exhibit versatile bioactive properties for potentially affecting the pertinent biological processes.35 With its privileged structure, this class of molecules obeys the rules of analogue and thereby it binds to diverse array of receptors that endorse them as attractive scaffolds for drug discovery.35 In recent years, the prominence of cyclic dipeptides in interfering with the biofilm and associated virulence of pathogens has offered an alternative approach to antagonize the biofilm-mediated biological activities, which eventually suppress the pathogenicity.10,14,50 Despite the global recognition of CLP as antibacterial, antifungal, antiviral and anticancer agents,35 the antivirulence and/or antibiofilm potential remains unexplored. As this study emphasizes an alternative therapeutic strategy over conventional antibiotics which lead to strong selection for antibiotic resistance, a great deal of attention has been paid on the influence of CLP towards the cellular viability of MRSA. The determined MIC (256 μg mL−1) and MBC (512 μg mL−1) against MRSA are in total agreement with a previous report by Rhee et al., wherein the same molecule was found to exhibit effective antibacterial activity against multi-drug resistant strain of S. aureus and vancomycin-resistant enterococci at a MIC of 256 and 32 μg mL−1, respectively.36 Subsequent growth curve analysis and XTT assay (data not shown) for the sub-MICs of CLP unveiled its non-bactericidal effect against MRSA, and thus, signifies the fact that the phenomenal antibiofilm and/or antivirulence efficacy of CLP at its sub-MICs are not due to its antibacterial effect.
S. aureus resides in various niches of human body as commensal and at times it turns in to an opportunistic pathogen causing many infections (aforementioned).16 This notorious transition of S. aureus from commensal to pathogen is being attributed to its surface colonizing and biofilm forming characteristics.37 Therefore, preventing bacterial adhesion could substantially reduce the risk of developing biofilm and virulence secretion. The sub-MICs of CLP prominently inhibited the biofilms of MRSA at its early stage through reducing the microcolonies. This result falls in line with the findings of a recent study, indicating the remarkable inhibitory efficacy of another cyclic-dipeptide termed cis-cyclo(leucyl-tyrosyl) towards the biofilms of S. epidermidis.10 It is also evident that CLP demonstrated a profound concentration-dependent antibiofilm efficacy against all tested MRSA strains. Further evaluation of CLP for its biofilm inhibitory efficacy toward other pathogens confirmed the broad spectrum antibiofilm ability of CLP. However, the activity of CLP against Gram-negative pathogens was marginal (data not shown), suggesting that the molecule is specifically acting against the biofilms of Gram-positive pathogens. Further investigations at the translational level are needed to shed more light in deciphering the broad spectrum antibiofilm mechanism of this bioactive molecule towards Gram-positive pathogens.
The in situ visualization of characteristic biofilm architecture of S. aureus through CLSM (Fig. 3 and 4) followed by comstat2 analysis and SEM (Fig. 5 and 14) unveiled the dynamic complexicity of S. aureus biofilms by clearly depicting the multilayered matrix structures made up of exopolymeric substance in untreated samples. On the other hand, the titanium plates treated with increasing concentrations clearly explicated the exceptional dose-dependent biofilm inhibitory efficacy of CLP, as it distraught the exopolymeric matrix as well as microcolonies which eventually resulted in poor biofilm development. The mechanism of biofilm formation in S. aureus involves three major stages: initial attachment, maturation of biofilms and dispersion of bacterial cells.38 Therefore, it is envisaged that the CLP inhibits the initial attachment of S. aureus cells to the substratum and also precludes the subsequent stages of biofilm development such as maturation and EPS production.
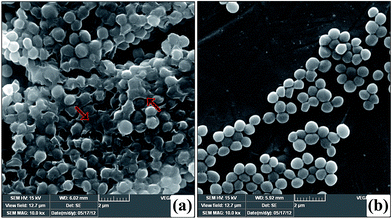 |
| | Fig. 14 Scanning electron micrographs of GSA-140 on titanium plate in the presence (b) and absence (a) of 128 μg mL−1 of CLP. The red arrows in untreated control (a) shows the dense multi-layered EPS forming the matrix that embeds the S. aureus cells; CLP treatment (b) shows the single separate cells without the EPS matrix. | |
Biofilm maturation is the prime phase in comprehensive development of recalcitrant biofilm of any pathogen, which account for the enhanced survival and pathogenicity of that particular pathogen.38 From the confocal micrographs (Fig. 7B) and 24-well MtP assay (Fig. 7A), it is very clear that CLP has a profound inhibitory effect on the recalcitrant mature biofilms of MRSA and thereby the uniform monolayer of cells attached to polystyrene as well as titanium substratum is uncovered. It is envisaged from the current study that CLP could be a promising antibiofilm agent, owing to the fact that mature biofilm dispersion ability of any molecule is a hallmark property of an ideal antibiofilm agent. More specifically, the sub-MIC of CLP inhibited the development of more than 85–87% biofilms (Fig. 1), and dispersed the mature biofilms to about 52–54% (Fig. 7A). Nevertheless, several antibiofilm molecules against the staphylococcal biofilms have been reported in the recent past, many of them failed to have the required efficacy to disrupt the preformed biofilms.9–15,21 Therefore, the present investigation is the first of its kind to offer conclusive evidences for biofilm inhibitory and disrupting efficacies of CLP against MRSA.
Several investigators have signified the vital role of extracellular polymeric substances (EPS) in biofilm assemblage; particularly Hans-Curt et al.39 emphasized the prominence of EPS by stating it as the “house of the biofilm cells”. In addition, EPS acts like a protective sheath in safeguarding the pathogen from the exposure of antibiotics and host immune cells.40 Thus, suppressing the synthesis of EPS could increase the pathogens' vulnerability which in turn ultimately facilitates the eradication of biofilm from the infection site. Here, we demonstrated a similar phenomenon of action by CLP in suppressing the synthesis of EPS, which was evident through the outcomes of CLSM (Fig. 4), SEM (Fig. 14) as well as the quantitation of polysaccharides and proteins of EPS (Fig. 10) extracted from CLP treated and untreated MRSA. To further ascertain the EPS modifications enforced by CLP, FT-IR technique was employed as it effectively probes the interactions and modifications of the biomacromolecules like nucleic acid, proteins, polysaccharides, and lipids.41 Reduction of EPS signals (protein (1800–1500 cm−1) and polysaccharide (1200–900 cm−1)) in IR could be attributed to the decrease of EPS synthesis provoked by CLP. Here, we speculate that CLP might act upon the pathways driving the synthesis of polysaccharides and extracellular proteins to accomplish its inhibitory efficacy towards initial attachment and mature biofilm formation of S. aureus. Furthermore, to corroborate this, qPCR was done for the genes that are responsible for the biosynthesis of polysaccharide intercellular adhesin (PIA) (icaA and icaD), a polymer responsible for cell–cell attachment42 and cysteine protease (sspB), an extracellular enzyme that positively regulates biofilm formation.43 The qPCR data validated the above speculation on CLP by the significant down-regulation of these three genes (Fig. 13). Thus, it is envisaged that CLP may possibly target the multilayered EPS production of MRSA that eventually attributes to its profound efficacies in inhibiting and disrupting the biofilms. Consequently, the remarkable reduction in the slime synthesis of MRSA upon CLP treatment (Fig. 9) substantiated the qPCR results as it was proposed that co-expression of icaA and icaD genes not only regulate the production of N-acetylglucosaminyl transferase (which synthesizes the PIA polymer) but also the synthesis of slime.42
Although, CLP was reported to have plethora of pharmaceutical significance against various bacterial and fungal pathogens,35 initiatives for its application in sub-clinical level is still inadequate. C. elegans, a eukaryotic nematode greatly attracted the attention of many researchers, as it conserves many of the basic physiological processes of human.44 Therefore, this live-animal infection model has been exploited for identifying small molecules with in vivo anti-infective efficacy.45,46 The rescued survival of C. elegans infected with MRSA and decreased bacterial load inside the nematodes' gut clearly delineate the anti-adherence efficacy of CLP in vivo. Further, the observed null mortality in C. elegans exposed with CLP (MIC) affirms its nontoxic nature. To ascertain the anti-adherence efficacy of CLP at the molecular level, expression analysis of adhesin genes (fnbA, fnbB, clfA, cna and altA) were done. For S. aureus to infect host tissue, the initial adhesion and subsequent invasion are crucial, which are being accomplished by the expression of different Microbial Surface Components Recognize Adhesive Matrix Molecules (MSCRAMMs). These MSCRAMMs (collagen binding protein, fibronectin binding proteins A and B, fibrinogen binding protein and clumping factors A and B encoded by genes cna, fnbA & fnbB, fib, and clfA & clfB respectively) have high ability to interact with the host extracellular matrix proteins such as fibrinogen, fibronectin and collagen.47 Autolysin (altA), a major peptidoglycan hydrolase cleaves newly synthesized peptidoglycan components before its integration into cell wall and, notably altA null mutants are unable to perform primary attachment to surfaces.48 In-line with the observed antibiofilm and anti-adherence efficacies of CLP, its treatment also down-regulated the expression of all the adhesin genes, on account of which the S. aureus cells were not be able to adhere and infect C. elegans. As cell surface hydrophobicity of bacteria highly influence the sturdy adhesion and consecutive increase in the biofilm biomass,49 the decreased level of CSH upon CLP treatment augments its prominence to be an anti-adhesive molecule. Of note, some of the virulence genes like hla (alpha toxin gene), sarA (global regulator of many virulence factors including biofilm formation), and sea (staphylococcal enterotoxin A) were significantly down-regulated upon exposure to CLP. This is in agreement with an earlier report wherein, similar dipeptides cyclo(L-Phe-L-Pro) and cyclo(L-Tyr-L-Pro) from Lactobacillus reuteri RC-14 (vaginal isolate) were able to interfere with the quorum-sensing mediated virulence production in S. aureus.50 Based on the gene expression profile changes by CLP, down-regulation of these genes could lead to defects in initial attachment and could also lead to diminished production of virulence.
Conclusion
In summary, our findings for the first time demonstrate the inhibitory effects of CLP, a cyclic dipeptide secreted by B. amyloliquefaciens of marine origin, on the biofilm and virulence production of MRSA. The substantial reduction in expression of multitude of genes involved in initial attachment, biofilm formation and virulence production concurs very well with the antibiofilm activity of CLP in vitro. Furthermore, the mature biofilm disruption and in vivo anti-adherence efficacies suggest the suitability of CLP to be a promising anti-infective agent (alone or in combination with antibiotics) in controlling biofilm-associated MRSA infections.
Experimental section
Extraction and purification of CLP
The cyclic dipeptide – CLP was extracted from the mangrove rhizosphere bacterium-B. amyloliquefaciens (MMS-50) isolated from the mangrove rhizosphere soil of Karankadu mangroves of Palk Strait, Bay of Bengal, India.14 The degree of purity of MMS-50 active fraction (AF) was >98%, as determined by high performance liquid chromatography, gas chromatography/mass spectrometry and Fourier transform-infrared spectroscopy analyses from our previous study.14 The obtained CLP was dissolved in sterile MilliQ water to a final concentration of 1 mg mL−1 for antibiofilm and several other bioassays.
Bacterial strains, strain characterization, culture media and growth conditions
Three MRSA strains used in this study include: two clinical isolates (GSA-140 and GSA-310) and one MRSA ATCC reference strain (MRSA 33591). The two clinical MRSA isolates were selected based on their high degree of biofilm forming ability amongst 63 MRSA isolates, which we had isolated from throat swabs of pharyngitis patients attending the Thoracic Science Department of Government Rajaji Hospital, Madurai.18 We performed multilocus sequence typing (MLST) as described previously by Enright et al. (2000) to ascertain the facts that these two clinical isolates are clonally unrelated.19 The PCR amplicons of seven housekeeping genes (arcC, aroE, glpF, gmk, pta, tpi, and yqiL) were sequenced and the sequences were submitted to the MLST database website (http://saureus.mlst.net) for assignment of allelic profiles and sequence types (STs). The sequence type of MRSA clinical isolates GSA-140 (MLST ID: 5449) and GSA-310 (MLST ID: 5450) was identified as ST772 and ST30, respectively. Besides, three Streptococcus spp. viz. Streptococcus salivarius (ATCC 13419), Streptococcus sanguinis (ATCC 10556) and Streptococcus mitis (ATCC 6249) were also used to evaluate the broad spectrum antibiofilm activity of CLP. Staphylococcus aureus and Streptococcus spp. were grown and maintained on Tryptic soy agar/broth (TSA/TSB) (aerobic condition) and Todd Hewitt agar/broth (THA/THB) (anaerobic condition) (Himedia, Mumbai, India) at 37 °C, respectively. For biofilm assays, TSB supplemented with 0.25% glucose (TSBG) and THB supplemented with 0.25% sucrose (THBS) were used to grow Staphylococcus aureus and Streptococcus spp. respectively.
In vitro antibacterial assays: minimum inhibitory concentration (MIC), minimum bactericidal concentration (MBC)
The MIC and MBC of CLP against the three MRSA were determined by the microdilution susceptibility test according to the Clinical Laboratory Standards Institute guidelines, with required modifications,20 wherein the antibiotic oxacillin was used as the positive control. Briefly, the cell suspension of each MRSA (5.0 × 105 CFU mL−1) was used to inoculate TSB supplemented with 0.25% glucose in 96 well microtitre plate added with serial twofold dilutions of CLP. The lowest concentration of CLP that completely inhibited the visible growth following 24 h incubation at 37 °C was considered as MIC. For MBC determination, 100 μL of broth from clear wells of MIC microtiters was taken and spread onto Mueller-Hinton Agar (MHA) plates. Following 24 h of incubation at 37 °C, MBC was deduced as the lowest concentration of CLP that produced subcultures growing not more than five colonies on each plate (i.e. 99.99% of the initial inoculum killed in a given time using a plate count of viable cells).
Biofilm formation assay in 24-well microtitre plate (MtP)
The effect of CLP on biofilm formation of MRSA was done using 24-well MtP,14 wherein, quercetin, a known biofilm inhibitor of S. aureus was used as the positive control.21 In brief, each of the three MRSA cell suspensions at 100 μL (107 cells per mL) volume were used to inoculate wells containing 1 mL of fresh TSBG supplemented with CLP at its sub-MICs. The MtPs were statically incubated for 24 h at 37 °C. As 24 h incubation was relative short time period to evaluate the biofilm inhibitory potentials of CLP, the biofilm biomass assay using MtPs was performed till 48 h as well. After incubation, the spent medium together with planktonic cells were gently discarded and weakly adherent cells were removed by thoroughly washing twice with sterile 0.1 M phosphate-buffered saline (PBS), and the plates were dried at 55 °C for 1 h before staining. The S. aureus biofilms adhered to the bottom of the polystyrene surface were stained with 1 mL of 0.4% crystal violet (CV) solution (w/v) for 4–5 min. Subsequently, the unstained dye was discarded and wells were rinsed twice with 0.1 M PBS to remove the excess stain. After the MtPs were dried at 40 °C for 1 h, formed biofilms were quantified by solubilization of the CV stain in 1 mL of absolute ethanol for 10 min, wherein the wells devoid of CLP acted as control and wells with both medium and AF served as blank.22 The assay was performed in triplicate and repeated at least thrice. The optical density was determined at a wavelength of 570 nm using the Multilabel Reader (Spectramax M3, USA) and the percentage of biofilm inhibition was calculated using the following formula:
| Percentage of inhibition = ([control OD570 nm − test OD570 nm]/ control OD570 nm) × 100 |
Growth curve analysis
The effect of CLP on cell proliferation of S. aureus was determined as described earlier with required modifications,14 in which quercetin was used as a positive control.21 Single colonies from S. aureus test cultures viz. MRSA ATCC 33591, GSA-140 and GSA-310 were used to inoculate TSBG (0.25%) medium in separate test tubes and cultured for 8 h at 37 °C. Overnight cultures were sub-cultured (1%) in test tubes with fresh TSBG supplemented with or without (control) of CLP (sub-MIC) and incubated at 37 °C. The growth rate was measured using spectrophotometer (UV-VIS Spectrophotometer; Shimadzu) at OD600 nm up to 24 h at 1 h interval. The assay was performed in triplicate with appropriate controls.
In situ visualization of biofilm formation through microscopic techniques
Confocal laser scanning microscopic (CLSM) analysis. Titanium plates (2 mm thick and 6 mm in diameter) were sterilized and placed aseptically onto the wells of MtPs (24-well) containing 10 μL of the MRSA cell suspension (107 cells per mL) in 1 mL fresh TSBG supplemented with sub-MICs of CLP. The MtPs containing titanium plates were incubated at 37 °C for 24 h and then gently washed three times with 0.1 M PBS to remove non-adherent S. aureus cells and stained with 0.1% acridine orange. The excess stain was washed out and the plates were air dried before examination. Titanium plates with cells grown in CLP-free medium were utilized as control. Images of the stained titanium plates were visualized under CLSM (LSM 710, Carl Zeiss, Germany) and processed with Zeiss LSM Image Examiner (Version 4.2.0.121), equipped with an excitation filter 515–560 and magnification at 20×. CLSM images (N = 20) were obtained from triplicates of untreated control and treated biofilms (the experiment was repeated at least thrice), and the Zstack analysis (surface topography and three-dimensional architecture) was done with the Zen 2009 software (Carl Zeiss, Germany).15 Furthermore, the images were analyzed using comstat2 software (kindly gifted by Dr Claus Sternberg, DTU Systems Biology, Technical University of Denmark). Three different parameters i.e. an average and maximum thickness (μm) of the biofilms and the biovolume (μm3), which is the volume of bacteria per μm2 of glass surface used were selected for further analysis.
Extracellular polysaccharides (EPS) staining. Concanavalin A (Con A) conjugated to fluorescein isothiocyanate (FITC; Catalogue no. C7642, Sigma-Aldrich, USA) was used to label extracellular polysaccharide in S. aureus biofilms following the protocol described previously with required modifications.23 Stock solution was prepared using 10 mmol L−1 hydroxyethylpiperazine ethanesulfonic acid (HEPES). The 48 h biofilms of MRSA clinical strains (GSA-140 and GSA-310) grown on the surface of titanium in the presence and absence of 128 μg mL−1 CLP were stained with 15 mM propidium iodide (PI; Product code 81845, Sigma-Aldrich, USA) in dark for 15 min at room temperature. After washing the titanium plates with PBS for thrice, the plates were stained with 50 μg mL−1 of Con A-FITC and incubated for 15 min in dark with at room temperature. The excitation wavelength for PI fluorescence was 568 nm and the emission was monitored at 605 nm. Similarly, the Con A-FITC (green) was excited at 488 nm and fluorescence was detected at an emission wavelength of 522 nm. Images of the stained titanium plates were visualized under CLSM (LSM 710, Carl Zeiss, Germany) and processed with Zeiss LSM Image Examiner (Version 4.2.0.121).
Scanning electron microscopy (SEM). For SEM analysis, the MRSA strain GSA-140 was allowed to form biofilm on the titanium plates together with the presence and absence of CLP at its sub-MICs (64 and 128 μg mL−1) as described in CLSM analysis. After 24 h of incubation, titanium plates were gently washed thrice with 0.1 M PBS to remove non-adherent S. aureus cells, wherein titanium plate in wells containing CLP-free medium served as control. The biofilms on titanium plates were fixed with a solution containing 2.5% glutaraldehyde for 2 h followed by a wash with 0.1 M sodium acetate buffer (pH 7.3). The biofilms on titanium plates were subsequently washed in distilled water and dehydrated at increasing concentrations of ethanol (20%, 50%, 70%, 90% and 100%) for 10 min each. Finally, after critical-point drying and gold sputtering, samples were examined using a scanning electron microscope (Hitachi S-3000H, Japan).
Mature biofilm disruption assay
The three MRSA strains were allowed to form biofilms on 24-well MtPs as described above in biofilm formation assay section for 48 h. After incubation, the spent medium was discarded carefully and thoroughly. To the wells 1 mL of fresh TSBG supplemented with 128 μg mL−1 of CLP was added and incubated for 6 h. Quantification of biofilm biomass using crystal violet dye was done by following the protocol mentioned above (biofilm formation assay section). Similarly, the impact of mature biofilm disruption ability of CLP was also assessed on titanium plates. The test strains were allowed to form biofilms on the titanium plates placed inside the wells of 24-well MtP containing 10 μL of MRSA cell suspension (107 cells per mL) in 1 mL fresh TSBG. The titanium plates with 48 h preformed biofilms of MRSA were further incubated in a fresh MtP containing 1 mL fresh TSBG along with sub-MIC of CLP for 6 h. The staining and CLSM image acquisition were done as described in CLSM analysis section above. Furthermore, comstat2 software was employed to substantiate the results of CLSM analysis.
Phenotypic detection of slime production by Congo red agar (CRA)/broth (CRB) assay
Preliminarily, the clinical MRSA strain GSA-140 was screened for the qualitative slime production by CRA plate assay and modified Congo red broth assay.24 The CRA medium composed of TSB (30 g L−1) (Himedia, Mumbai, India), sucrose (36 g L−1), agar powder (18 g L−1) and Congo red dye (0.8 g L−1), while the same ingredients devoid of agar powder was the composition for CRB medium. Congo red stain was prepared as a concentrated aqueous solution, autoclaved separately and added to the media when the agar/broth had cooled to 55 °C. The plates (CRA) and tubes (CRB) were inoculated and incubated aerobically for 24 h at 37 °C. Biofilm positive strains produced black-coloured colonies on CRA; similarly, they turn the red colour of CRB to black. The CRA/CRB assay was also used to evaluate directly the effect of CLP at sub-MIC (128 μg mL−1) on slime production. CLP at its sub-MICs were mixed together (aseptically) with Congo red and added to the media when the agar/broth had cooled to 55 °C. Plates and tubes without CLP served as control for CRA and CRB assays, respectively. Then, the plates and tubes were inoculated with test strains and were incubated aerobically for 24 h at 37 °C.
Analysis of cellular components in EPS
EPS extraction. The extracellular polymeric substances from the biofilms of two MRSA clinical strains were extracted using the previously described protocol with minor modifications.25 In brief, the 24 h grown biofilms treated with and without CLP at sub-MIC were centrifuged at 15000 × g for 20 min to obtain biofilm pellets. This was further resuspended in 25 mL of ice cold 0.2 M sulfuric acid solution (pH 1.1) and the biofilm matrix was broken using a glass bead homogenizer. The cell suspension was stirred at 4 °C for 3 h before centrifugation at 15000 × g for 20 min. Finally, the supernatant collected was designated further as EPS solution and was stored at −20 °C until further analysis.
Fourier transform infrared (FT-IR) spectroscopy. FT-IR spectroscopy was carried out for control and CLP (128 μg mL−1) treated EPS samples of both GSA-140 and -310 MRSA strains as described by Jiao et al.25 Initially, the collected EPS solution was precipitated by adding 3 volumes of ice-chilled absolute ethanol and incubated at −20 °C for 2 h. The precipitates were then centrifuged at 17500 × g for 20 min at 4 °C. After discarding the supernatant, the pellets were air dried in oven at 50 °C overnight. The infrared spectra were recorded with a FTIR system (Bruker Tensor 27). The spectra were scanned in the 4000–400 cm−1 range using the potassium bromide (KBr) pellet technique. Potassium bromide was dried under a vacuum at 100 °C for 48 h and 100 mg of KBr with 1 mg of sample was taken to prepare the KBr pellet. The absorbance spectrum was plotted as intensity versus wave number.
Polysaccharide estimation. To measure the total carbohydrate content in extracted EPS solution, phenol-sulfuric acid method was employed with glucose as standard with little modification.25 Briefly, 500 μL of EPS solution was mixed with 1.5 mL of concentrated H2SO4 along with 500 μL of phenol (10%), and the mixture was mixed gently before incubation in water bath at 50 °C for 20 min. The mixture was cooled and transferred to a 96-well tissue culture plate. The absorbance at 490 nm was read with a spectrophotometric Multilabel Reader (Spectramax M3, USA).
Protein quantification. To estimate the total protein content of EPS solution, trichloroacetic acid (TCA)/acetone (final concentration, 15%) precipitation method was used with slight modification.25 Briefly, 10 mL of EPS solution was mixed with TCA (dissolved in acetone to a final concentration of 15%) and was incubated on 4 °C for 30 min before centrifugation at 15000 × g for 20 min. The TCA precipitates were washed twice with 15 mL of acetone alone. The protein content was measured using the Bradford assay (Bio-Rad, Hercules, CA) with bovine serum albumin (BSA) as the calibration standard.
Cell surface hydrophobicity assay
Surface hydrophobicity of S. aureus cells were determined by using MATH (microbial adhesion to hydrocarbons) assay as a measure of their adherence to the hydrophobic hydrocarbon (toluene) following the procedure described previously.26 Briefly, 3 mL of overnight S. aureus culture (OD530 nm = 1.0) (initial OD) was taken in glass tubes and 250 μL of toluene along with the CLP (5% v/v) was added. The mixtures were vigorously vortexed for 2 min and left undisturbed at room temperature for better phase separation. Then the lower aqueous phase was pipetted out carefully to measure OD530 nm (final OD). Cells alone incubated with toluene served as control. The percentage of hydrophobicity was calculated according to the formula: % hydrophobicity = [initial OD600 nm − final OD600 nm/initial OD600 nm] × 100.
CLP toxicity assay
As a prelude to assess the in vivo antibiofilm efficacy of CLP, the toxicity assay was performed in a whole biofilm animal model C. elegans to determine whether sub-MIC of CLP used in this study has an effect on the survival of nematodes. The wild-type C. elegans N2 Bristol strain was used in this study. E. coli OP50 was used for C. elegans maintenance and performance of all bioassay. C. elegans was maintained at 20 °C on nematode growth medium (NGM) seeded with E. coli OP50 as a bacterial food source.27 Initially, the nematodes were harvested and age-synchronized to L4 using an alkaline bleach solution (1:1 ratio of house hold bleach and 5 M sodium hydroxide). Thus obtained L4 age-synchronized nematodes were used in all bioassays performed. A batch of 10 nematodes were transferred from the lawn of E. coli OP50 to a sterile 24 well MtP containing sub-MIC of CLP in a M9 liquid medium at 20 °C and the survival of the nematodes was scored in hourly intervals till 100 h, wherein the nematodes fed with E. coli OP50 served as control.28
C. elegans killing assay
For testing the in vivo antibiofilm efficacy, C. elegans killing assay was performed as described previously29 with little modifications. Briefly, a batch of 10 L4 nematodes was transferred to M9 liquid medium containing MRSA ATCC or GSA-140 or GSA-310 (20% inoculum each) in the presence and absence of CLP (128 μg mL−1). The experimental plate was incubated at 20 °C and monitored for the survival of nematodes. The worms which did not show any response to the touch were scored as dead, while C. elegans fed on E. coli OP50 acted as control.
In vivo biofilm formation and microscopic observation
Nematodes were exposed to MRSA ATCC, GSA-140 and GSA-310 in the presence and absence of CLP at sub-MIC for 12 h and thoroughly washed the nematodes to remove the surface attached bacteria and placed on a 1% agar pad containing 1 mM sodium azide to anaesthetize the worms. Anesthetization prevents the expulsion of bacteria from the nematodes' intestine. Further, the internal biofilm formation was documented in the anesthetized worm under inverted light microscope (Nikon, Japan).
Colony forming unit (CFU) assay
To determine the bacterial load inside the worms' gut, a bacterial accumulation assay was performed as described earlier by Kamaladevi et al.30 In brief, a batch of 10 nematodes was exposed to MRSA ATCC, GSA-140 and GSA-310 for 12 h in the presence and absence of CLP at its sub-MIC. After experimental exposure, the surface adhered bacteria was removed by washing at least for 10 times with M9 buffer and approximately 400 mg of 0.1 mm mesh silicon carbide (Himedia, Mumbai, India) was added to the washed nematodes. The mixture was vortexed vigorously for a minute to disturb the worm completely. Finally, the mixture was centrifuged at 94 × g for 1 min and the resulting suspension was serially diluted and plated onto Aureus agar (Himedia, Mumbai, India). The plates were incubated at 37 °C for 12 h and the colonies were counted to determine the CFU.
Total RNA isolation and real time/quantitative-PCR
In order to investigate the effect of CLP on virulence and biofilm genes expression in MRSA, 3 h grown cultures of GSA-140 and GSA-310 were used to inoculate (1%) TSBG medium supplemented with and without CLP (sub-MIC) at 37 °C for 12 h in triplicate. After incubation, the biofilms formed together with cells were harvested by centrifugation at 4600 × g for 3 min and were re-suspended in 1 mL of TRIzol reagent (Sigma-Aldrich, Switzerland) and then transferred to an RNase-free 1.5 mL microcentrifuge tube. Total RNA from both control and treated samples were extracted using the guanidine thiocyanate/phenol extraction method.14 Isolated RNA was dissolved in 25 μL of 0.1% diethylpyrocarbonate (DEPC)-treated water and were stored at −80 °C until required for cDNA conversion. The RNA samples (100 ng) were reverse transcribed using cDNA reverse transcription kit (Applied Biosystems Inc., Foster, CA, USA), following the manufacturer's instructions. The control and treated cDNA samples were quantified by real time PCR using 7500 Sequence Detection System (Applied Biosystems Inc. Foster, CA, USA) and 2(−ΔΔCt) method.14 The PCR primer sequences, their standardized annealing temperature are given in ESI Table S1.† The expression levels of all selected genes were analyzed in triplicate and normalized using 16S rRNA gene, an internal control.
Statistical analysis
Experiments were performed in triplicate and the values were expressed in mean ± S.D. For nematode toxicity and rescue assay, Kaplan–Meier survival analysis (Graphpad prism 5 statistical software) was performed to compare the mean lifespan of control versus CLP exposed nematode group. The significant difference between the survival curves was analyzed by Log-rank (Mantel-cox method) test. For all other experiments, statistical comparisons between treated and untreated control samples were performed with one way analysis of variance (ANOVA) followed by the Dunnett's test using SPSS statistics version 17.0 (SPSS Inc., Chicago, IL, USA).
Acknowledgements
The authors thankfully acknowledge the Department of Biotechnology, Government of India for providing Bioinformatics Infrastructure Facility (Grant No. BT/BI/25/012/2012 (BIF)). The instrumentation facility provided by Department of Science and Technology, Government of India through PURSE [Grant No. SR/S9Z-415 23/2010/42(G)] & FIST (Grant No. SR-FST/LSI-087/2008) and University Grants Commission, New Delhi through SAP-DRS1 [Grant No. F. 3-28/2011(SAP-II)] is gratefully acknowledged. Financial assistance rendered to Shanmugaraj Gowrishankar in the form of Rajiv Gandhi National Fellowship (RGNF) by University Grants Commission, New Delhi [No. F. 14-2 (SC)/2010 (SA-III)] is thankfully acknowledged. The authors also acknowledge Dr Claus Sternberg, DTU Systems Biology, Technical University of Denmark for providing the comstat2 software.
Notes and references
- I. Sutherland, Microbiology, 2001, 147, 3 CrossRef CAS PubMed
 .
. - R. M. Donlan and J. W. Costerton, Clin. Microbiol. Rev., 2002, 15, 167 CrossRef CAS PubMed .
- P. V. Vlastarakos, T. P. Nikolopoulos, P. Maragoudakis, A. Tzagaroulakis and E. Ferekidis, Laryngoscope, 2007, 117, 668 CrossRef PubMed .
- R. E. Kania, G. E. Lamers, M. J. Vonk, E. Dorpmans, J. Struik, P. T. B. Huy, P. Hiemstra, G. V. Bloemberg and J. J. Grote, Laryngoscope, 2008, 118, 128 CrossRef PubMed .
- B. E. Ragle and W. J. Bubeck, Infect. Immun., 2009, 77, 7128 CrossRef PubMed .
- J. Saising, L. Dube, A. K. Ziebandt, S. P. Voravuthikunchai, M. Nega and F. Götz, Antimicrob. Agents Chemother., 2012, 56, 5804 CrossRef CAS PubMed .
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2, 114 CrossRef CAS PubMed .
- V. Khodaverdian, M. Pesho, B. Truitt, L. Bollinger, P. Patel, S. Nithianantham, G. Yu, E. Delaney, E. Jankowsky and M. Shoham, Antimicrob. Agents Chemother., 2013, 57, 3645 CrossRef CAS PubMed .
- P. Jiang, J. Li, F. Han, G. Duan, X. Lu, Y. Gu and W. Yu, PLoS One, 2011, 7, e18514 Search PubMed .
- M. Scopel, W. R. Abraham, A. T. Henriques and A. J. Macedo, Bioorg. Med. Chem. Lett., 2013, 23, 624 CrossRef CAS PubMed .
- S. M. Sayem, E. Manzo, L. Ciavatta, A. Tramice, A. Cordone, A. Zanfardino, M. de Felice and M. Varcamonti, Microb. Cell Fact., 2011, 10, 74 CrossRef PubMed .
- C. Nithya, M. G. Devi and S. K. Pandian, Biofouling, 2011, 27, 519 CrossRef CAS PubMed .
- A. Dheilly, E. Soum-Soutéra, G. L. Klein, A. Bazire, C. Compère, D. Haras and A. Dufour, Appl. Environ. Microbiol., 2010, 76, 3452 CrossRef CAS PubMed .
- S. Gowrishankar, B. Poornima and S. K. Pandian, Res. Microbiol., 2014, 165, 278 CrossRef CAS PubMed .
- S. Gowrishankar, N. D. Mosioma and S. K. Pandian, J. Evidence-Based Complementary Altern. Med., 2012, 862374 Search PubMed .
- P. S. Yan, Y. Song, E. Sakuno, H. Nakajima, H. Nakagawa and K. Yabe, Appl. Environ. Microbiol., 2004, 70, 7466e73 CrossRef PubMed .
- K. H. Rhee, J. Microbiol. Biotechnol., 2003, 13, 984 CAS .
- S. Gowrishankar, R. Thenmozhi, K. Balaji and S. K. Pandian, Infect., Genet. Evol., 2013, 14, 383 CrossRef PubMed .
- M. C. Enright, N. P. Day, C. E. Davies, S. J. Peacock and B. G. Spratt, J. Clin. Microbiol., 2000, 38, 1008 CAS .
- Clinical and Laboratory Standards Institute, Performance Standards for Antimicrobial Susceptibility Testing, Twentieth Informational Supplement, M100–S20, Clinical and Laboratory Standards Institute, PA, USA, 2010 Search PubMed .
- J. H. Lee, J. H. Park, H. S. Cho, S. W. Joo, M. H. Cho and J. Lee, Biofouling, 2013, 29, 491 CrossRef CAS PubMed .
- Z. X. Peng, B. Tu, Y. Shen, L. Du, L. Wang, S. R. Guo and T. T. Tang, Antimicrob. Agents Chemother., 2011, 55, 860 CrossRef CAS PubMed .
- J. A. Banas, K. R. O. Hazlett and J. E. Mazurkiewicz, Methods Enzymol., 2001, 337, 433 Search PubMed .
- D. J. Freeman, F. R. Falkiner and C. T. Keane, J. Clin. Pathol., 1989, 42, 872 CrossRef CAS PubMed .
- Y. Jiao, G. D. Cody, A. K. Harding, P. Wilmes, M. Schrenk, K. E. Wheeler, J. F. Banfield and M. P. Thelen, Appl. Environ. Microbiol., 2010, 76, 2916 CrossRef CAS PubMed .
- H. S. Courtney, I. Ofek, T. Penfound, V. Nizet, M. A. Pence, B. Kreikemeyer, A. Podbielbski, D. L. Hasty and J. B. Dale, PLoS One, 2009, 4, e4166 Search PubMed .
- S. Brenner, Genetics, 1974, 77, 71 CAS .
- S. D. Stowe, A. T. Tucker, R. Thompson, A. Piper, J. J. Richards, S. A. Rogers, L. D. Mathies, C. Melander and J. Cavanagh, Drug Chem. Toxicol., 2012, 35, 310 CrossRef CAS PubMed .
- J. Breger, B. B. Fuchs, G. Aperis, T. I. Moy, F. M. Ausubel and E. Mylonakis, PLoS Pathog., 2007, 3, e18 Search PubMed .
- A. Kamaladevi and K. Balamurugan, Pathog. Dis., 2015, 73 DOI:10. 1093.femspd/ftv021 .
- J. Moreno, M. A. Vargas, J. M. Madiedo, J. Muños, J. Rivas and M. G. Guerrero, Biotechnol. Bioeng., 2000, 67, 283 CrossRef CAS PubMed .
- A. Bosch, D. Serra, C. Prieto, J. Schmitt, D. Naumann and O. Yantorno, Appl. Microbiol. Biotechnol., 2006, 71, 736 CrossRef CAS PubMed .
- D. E. Nivens, D. E. Ohman, J. Williams and M. Franklin, J. Bacteriol., 2001, 183, 1047 CrossRef CAS PubMed .
- A. Synytsya, J. Copíková, P. Matejka and V. Machovic, Carbohydr. Polym., 2003, 54, 97 CrossRef CAS .
- A. D. Borthwick, Chem. Rev., 2012, 112, 3641 CrossRef CAS PubMed .
- K. H. Rhee, Int. J. Antimicrob. Agents, 2004, 24, 423e7 CrossRef PubMed .
- P. G. Christopher, W. Paul and C. Weng, J. Med. Chem., 2013, 56, 1389 CrossRef PubMed .
- M. Otto, Curr. Top. Microbiol. Immunol., 2008, 322, 207 CAS .
- F. Hans-Curt, R. N. Thomas and J. W. Daniel, J. Bacteriol., 2007, 189, 7945 CrossRef PubMed .
- C. A. Fux, J. W. Costerton, P. S. Stewart and P. Stoodley, Trends Microbiol., 2005, 13, 34 CrossRef CAS PubMed .
- T. Gao, Y. Ci, H. Jian and C. An, Vib. Spectrosc., 2000, 24, 225 CrossRef CAS .
- C. R. Arciola, D. Campoccia, S. Gamberini, L. Baldassarri and L. Montanaro, FEMS Microbiol. Lett., 2005, 246, 81 CrossRef CAS PubMed .
- L. N. Shaw, E. Golonka, G. Szmyd, S. J. Foster, J. Travis and J. Potempa, J. Bacteriol., 2005, 187, 1751 CrossRef CAS PubMed .
- M. C. K. Leung, P. L. Williams, A. Benedetto, C. Au, K. J. Helmcke, M. Aschner and J. N. Meyer, Toxicol. Sci., 2008, 106, 5 CrossRef CAS PubMed .
- T. I. Moy, A. L. Conery, J. Larkins-Ford, G. Wu, R. Mazitschek, G. Casadei, K. Lewis, A. E. Carpenter and F. M. Ausubel, ACS Chem. Biol., 2009, 4, 527 CrossRef CAS PubMed .
- M. Dengg and J. C. Van Meel, J. Pharmacol. Toxicol. Methods, 2004, 50, 209 CrossRef CAS PubMed .
- S. S. Atshan, M. N. Shamsudin, A. Karunanidhi, A. Belkum, L. T. Lung, Z. Sekawi, J. J. Nathan, K. H. Ling, J. S. Seng, A. M. Ali, S. A. Abduljaleel and R. A. Hamat, Infect., Genet. Evol., 2013, 18, 106 CrossRef CAS PubMed .
- P. Houston, S. E. Rowe, C. Pozzi, E. M. Waters and J. P. O'Gara, Infect. Immun., 2011, 79, 1153 CrossRef CAS PubMed .
- A. G. Gristina, M. Oga, L. X. Webb and C. D. Hobgood, Science, 1985, 228, 990 CAS .
- J. Li, W. Wang, S. X. Xu, N. A. Magarvey and J. K. McCormick, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3360 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11641d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.