
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical deposition of Fe2O3 in the presence of organic additives: a route to enhanced photoactivity†
Dereje H.
Taffa
*a,
Ines
Hamm
b,
Christian
Dunkel
a,
Ilya
Sinev
b,
Detlef
Bahnemann
cd and
Michael
Wark
*a
aInstitute of Chemistry, Technical Chemistry, Carl von Ossietzky University Oldenburg, Carl-von-Ossietzky-Str. 9-11, 26129 Oldenburg, Germany. E-mail: dereje.hailu.taffa@uni-oldenburg.de; Michael.wark@uni-oldenburg.de
bFaculty of Chemistry and Biochemistry, Laboratory of Industrial Chemistry, Ruhr-University Bochum, Universitaetsstr. 150, 44801 Bochum, Germany
cInstitute of Technical Chemistry, Leibniz University Hannover, Callinstr 3, 30167 Hannover, Germany
dLaboratory for Nanocomposite Materials, Department of Photonics, Faculty of Physics, Saint-Petersburg State University, Ulianovskaia Str.3, Peterhof, Saint-Petersburg, 198504, Russia
First published on 16th November 2015
Abstract
The photoelectrochemical activity of hematite films prepared by electrochemical deposition (ED) in the presence of organic additives is discussed. The studies focus on the role of small organic additive molecules in the tuning of the morphology of the films and their influence on the photoelectrochemical oxidation of water. The organic additives, namely, coumarin 343 (C343), γ-glucuronic acid (GA) and sodium dodecyl sulfonate (Sds), possess functional moieties to interact with iron ions in the ED bath electrostatically or through metal–ligand complexation reactions. XPS measurements prove that the organic additives are incorporated, and the oxidation state of Fe3+ rules out the presence of mixed valences in the films. SEM and XRD measurements present morphological and structural evidence, respectively. The photoelectrochemical study shows that organically modified hematite films exhibit enhanced photoactivity; the photocurrent density at 1.4 V vs. RHE on a GA-modified electrode is up to 5–6 times higher than on the unmodified electrode. Electrochemical impedance results reveal the role of the organic additives in reducing the charge transfer resistance from the hematite surface to the solution. In addition, a simple Ti post treatment greatly enhances the photoactivity of all electrodes under investigation.
Introduction
Solar energy has been realized as one of the most promising renewable energy sources which can potentially meet the increasing need of energy globally.1 Since the seminal work of Fujishima and Honda the direct splitting of water using a photoelectrochemical cell (PEC) has become an elegant way of storing energy in chemical bonds which is available on demand.2–4 The solar irradiated semiconductor surface is a crucial part of the PEC device with stringent requirements. These criteria include chemical stability (in alkaline electrolytes), significant visible light harnessing ability, suitable band edge alignment to perform the water oxidation or reduction reactions, low production cost and good catalytic activity.5 Among the promising candidates hematite (α-Fe2O3) fulfils most of the decisive factors and has received considerable research interest.6–10 With a band gap of 2.0–2.2 eV hematite is able to absorb a significant part of the solar spectrum (∼40%), it is quite stable under illumination in aqueous solution and the valence holes are energetic enough to drive the water oxidation reaction.11 However, the performance of hematite photoanodes is considerably low in contrast to the theoretical predictions.8,12 Factors limiting the efficiency include a short hole diffusion length, a long light absorption cross section, and the mismatch of the conduction band edge for the water reduction.13,14 These shortcomings are addressed by nanostructuring to improve light absorption and charge collection15–19 and doping20,21 to enhance charge carrier density. Additionally, oxygen evolution catalysts22,23 and passivation layers24,25 are employed to decrease the overpotential of the multistep 4e− oxygen evolution reaction which is inherently sluggish on hematite photoelectrodes.Hematite photoanodes can be prepared following several strategies such as sol–gel methods,26 spray pyrolysis27,28 chemical vapour deposition (CVD)15,29 sputtering,19 atomic layer deposition (ALD)30,31 and electrodeposition (ED).32–34 ED presents numerous attractive features over the other techniques such as the use of non-toxic iron precursors, simple instrumentation, high flexibility in terms of composition and experimental parameters, and easy scale up for the fabrication of large area electrodes. Both cathodic and anodic ED routes for the preparation of α-Fe2O3 are reported.35,36 The co-deposition of dopants such as Mo, Cr, Zr and Ni using the same α-Fe2O3 ED bath has been demonstrated by several research groups.37–39 Inverse opal α-Fe2O3 photoanodes prepared via ED show outstanding performance.40
In this study we present the anodic ED of hematite thin films in the presence of small organic molecules (capping or complexing agents) (Scheme 1). Previously we demonstrated the use of different dye and surfactant molecules in the ED of ZnO films to tune the porosity, surface area and morphology of the resulting films.41,42 However, to the best of our knowledge such a synthesis route and the effect on the photoactivity of the hematite photoanode was not reported. The molecules used in this study possess functional moieties, i.e. carboxylic acid groups, to interact with iron ions in the ED bath electrostatically or via metal–ligand complexation reactions. As the deposited films are amorphous they can be converted to transparent crystalline α-Fe2O3 by annealing. The organic molecules influence the size and shape of the crystallites thereby affecting the photoelectrochemical performance. Structural and morphological evidence is presented from XRD and SEM measurements.
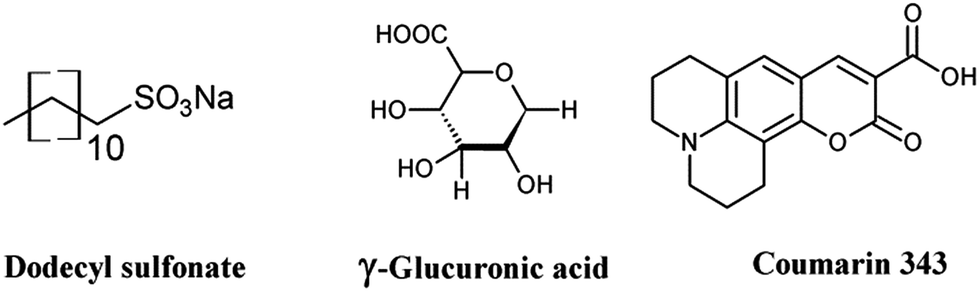 | ||
| Scheme 1 Structure of the organic additives used for the ED of hematite films. | ||
Experimental
Materials and reagents
Ammonium iron(II) sulfate hexahydrate, sodium acetate, cobalt(II) nitrate tetrahydrate, coumarin 343 (C343), and γ-glucuronic acid (GA) were analytical grade, purchased from Sigma-Aldrich and used as received. Sodium dodecyl sulfonate (Sds), potassium hydrogen phosphate, and potassium dihydrogen phosphate were purchased from Roth, and titanium(IV) butoxide (Ti(OBu)4) from Fluka. Conductive fluorine doped tin oxide (FTO) coated glass (15 Ω sq−1) was purchased from Pilkington Germany.Preparation of hematite photoelectrodes
FTO substrates were cleaned by sequential sonication in 0.1 M NaOH, 0.1 M HCl, ethanol, and acetone, each for 10 min in an ultrasonic bath. Prior to each sonication step the FTO substrates were rinsed with distilled water and finally dried by N2 blowing. The ED of hematite thin films was carried out with an AMEL Potentiostat/Galvanostat 7050 interfaced with a computer running under Junior Assist software for Windows, version V3 (AMEL, Italy). For the ED experiments a conventional three electrode electrochemical cell with FTO glass as a working electrode, coiled Pt wire as a counter electrode and an Ag/AgCl (XR300 Radiometer Analytical, sat. KCl) electrode separated by a salt bridge as a reference electrode was used. The deposition bath consisted of 0.1 M CH3COONa, 1 mM of the organic additives and was bubbled with N2 for 10–15 min prior to addition of (NH4)Fe(SO4)2·6H2O (0.02 M). The ED was performed potentiostatically at 0.4 V and the bath temperature was kept at 45 °C. The amount of charge was fixed to 0.3 C to obtain the same amount of photoactive material. Then the films were rinsed with distilled water and dried with N2. The as prepared films were annealed at 600 °C for 1 hour with a temperature ramp of 2 °C min−1.Ti doping was performed by drop casting a solution of 25 μL Ti(OBu)4 in absolute ethanol onto the electrodeposited iron oxide films prior to thermal treatment. The cobalt phosphate (Co-Pi) was deposited potentiostatically at 0.9 V vs. Ag/AgCl from a solution of 0.5 mM Co(NO3)2·6H2O in 0.1 M phosphate buffer.
Structural characterization
Photoelectrochemical measurements
All photoelectrochemical measurements were performed with a Zahner Zennium potentiostat using Thales software. The electrolyte was either 1 M NaOH (pH = 13.6) or 0.1 M NaOH containing 0.5 M H2O2 (pH = 12.5). Platinum wire and Ag/AgCl (sat. KCl) were used as the counter electrode and reference electrode, respectively. Hematite coated FTO served as the working electrode; electrical contact was made using the bare FTO surface with copper tape and the illumination area (2.54 cm2) was defined with an O-ring. Light enters the cell from the electrolyte side through a quartz window (front side illumination). The light source is equipped with a calibrated white LED controlled by a second power potentiostat (Zahner PP211). The output power can be varied between 10–100 mW cm−2. Current–voltage (I–V) curves were recorded at a rate of 10 mV s−1 from −0.4 V to +1.0 V vs. Ag/AgCl (sat. KCl) in the dark and under illumination. Potential values were converted to a potential relative to the reference reversible hydrogen electrode (RHE) using the relationship (eqn (1))ERHE = ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) E + EoAg/AgCl + 0.59pH E + EoAg/AgCl + 0.59pH | (1) |
Incident photon to current conversion efficiency (IPCE) measurements were carried out using a white LED (TLS, Zahner) coupled with a USB controlled monochromator between 430–730 nm. During the measurement a constant background light source is maintained and a small light excitation (ca. 10%) was superimposed at 0.2 Hz. The data were collected every 10 nm and simulated curves were obtained using the CIMPS software.
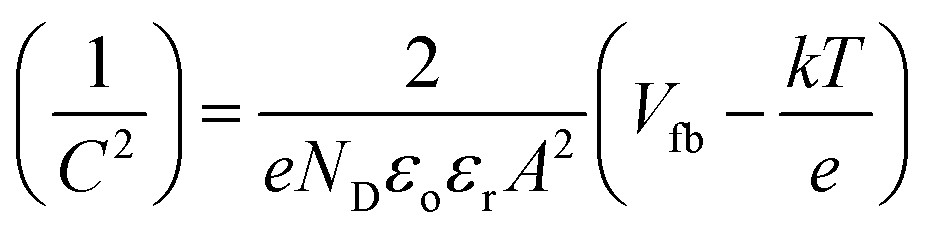 | (2) |
Results and discussion
Fig. 1 shows typical SEM images of the electrodeposited hematite thin films with and without different organic additives. The morphologies of the films can be tuned by adding 1 mM of the organic additives into the deposition bath. The as prepared films without additives feature a ‘corn flake’ structure (Fig. 1a inset) which converts to elongated plate like nanoparticles with average lengths of 110–130 nm and diameters of ca. 45–60 nm upon annealing at 600 °C. Addition of Sds results in similar nanoplate morphologies of reduced dimensions: 30–40 nm in diameter and ca. 100 nm in length. This similarity arises due to the very weak interaction of Fe2+ and sulfonate in the presence of excess carboxylate ions in the electrolyte. Films with GA are composed of ‘worm’ like aggregates of domain size ranging from 220–330 nm after thermal treatment. The aggregates have branches and beneath these aggregates there are round shaped nanoparticles with diameters of 40–60 nm. C343 containing films consist of nanoplates of ca. 50–60 nm which are sintered together. Different capping agents (CH3COO−, F−, Cl−, C2H8N2) are used to control the shapes and morphologies of the electrodeposited metal oxide films such as ZnO, Cu2O, α-FeOOH.43–45 In general these capping agents can preferentially adsorb on certain crystal planes and block or slow down the growth which results in anisotropic growth. As the applied potentials, and the concentrations of acetate and Fe ions are kept constant in the current study the morphology variations likely arise from the organic additives and a similar mechanism can be envisaged. The films are smooth and homogeneous over wide areas and have good adhesion to the FTO support without any cracks (Fig. 1S, ESI†). On the other hand, the film thickness is controlled by the deposition time and potential used. A linear increase of absorbance with the deposition time is observed (Fig. 2Sa†). There is a slight variation of film thicknesses according to the type and concentration of the organic additive. Films without organic additives (Fe2O3) have a thickness of 150–160 nm and those with additives are slightly thinner (140–150 nm) as evidenced using cross sectional SEM (Fig. 1b inset). The amount of photoactive material is cross-checked spectrophotometrically by dissolving the same area of the films in a 2 M HCl solution and following the absorbance of the [FeCl4]−/[FeCl2]+ complex at 340 nm (Fig. 2Sb and c†).46 The amount of ‘FeOOH’ deposited by passing 0.30 C is estimated to be 0.17–0.20 mg which gives a deposition efficiency of 75–82%. The deposition time varies in order to have the same amount of charge and it decreases in the order of Fe2O3/C343 > Fe2O3/GA > Fe2O3/Sds > Fe2O3. This suggests that the organic additives decrease the rate of film formation either by adsorbing or forming soluble complexes which compete with the film growth. | ||
| Fig. 1 SEM images of Fe2O3 films electrodeposited in 3 min at 0.4 V vs. Ag/AgCl and annealed at 600 °C: (a) without additives (Fe2O3) and in the presence of (b) 1 mM Sds (Fe2O3/Sds), (c) 1 mM GA (Fe2O3/GA), and (d) 1 mM C343 (Fe2O3/C343). Insets are (a) the as prepared Fe2O3 films and (b) the cross-sectional view of the Fe2O3 films. | ||
The UV-vis absorption spectra of the annealed films are obtained both in the absorbance and diffuse-reflectance modes. The as deposited films with additives display a light yellow colour and turn orange after annealing. The films without additives appear in general more reddish. All films exhibit similar absorption features (Fig. 2). Typically, a shoulder at 540 nm and a peak between 380–400 nm are observed, which are characteristics of Fe2O3 films and originate from the indirect Fe 3d to 3d and direct O 2p to Fe 3d transitions.47 The optical band gap is calculated by converting the respective diffuse-reflectance spectra into Tauc plots (Fig. 2Sd†). Values of 2.10–2.15 eV were obtained by fitting data points near the absorption onset for the indirect band gap transition. These values are in good agreement with other reported values for Fe2O3.35,37 The Fe2O3/GA and Fe2O3/C343 films exhibit slightly extended absorption in the region from 400 nm to 500 nm which may be due to the larger grain sizes of the films.
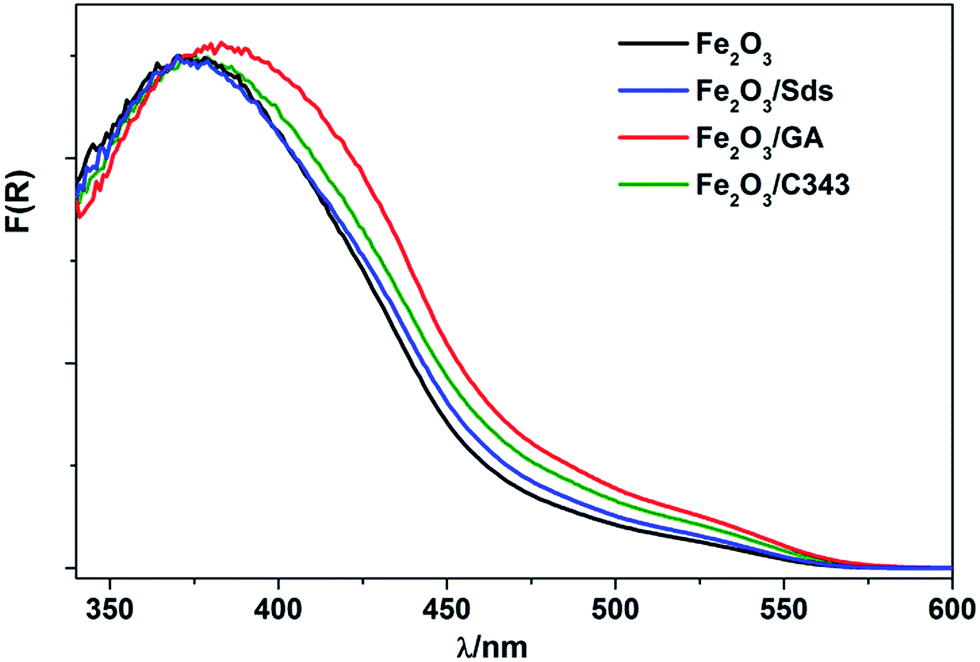 | ||
| Fig. 2 UV-vis spectra of α-Fe2O3 with and without the organic additives. | ||
Fig. 3a compares the X-ray diffraction patterns of the films with and without the organic templating agents after thermal treatment at 600 °C. The formation of the hematite phase is evidenced by the comparison of the standard α-Fe2O3 XRD patterns (PDF 99-000-0131) with the peaks associated with the films. We observe reflexes associated with the (012), (110) and (024) planes of which the intensity of the (110) reflex is the highest. This suggests that the Fe2O3 films have a preferred (110) orientation which favours a higher electron conductivity.48 Additionally, looking at the ratio of the peaks’ FWHM ((110)/(024)) of the films, values of 0.86, 0.72, 0.95 and 0.90 are obtained for Fe2O3, Fe2O3/Sds, Fe2O3/GA and Fe2O3/C343, respectively. The result suggests that GA and C343 containing films have a large fraction of crystals which are oriented in the 110 plane. This will significantly affect the measured photocurrents (see below). Furthermore, the as-prepared films are amorphous and no noticeable reflections are observed except those arising from the FTO substrate.
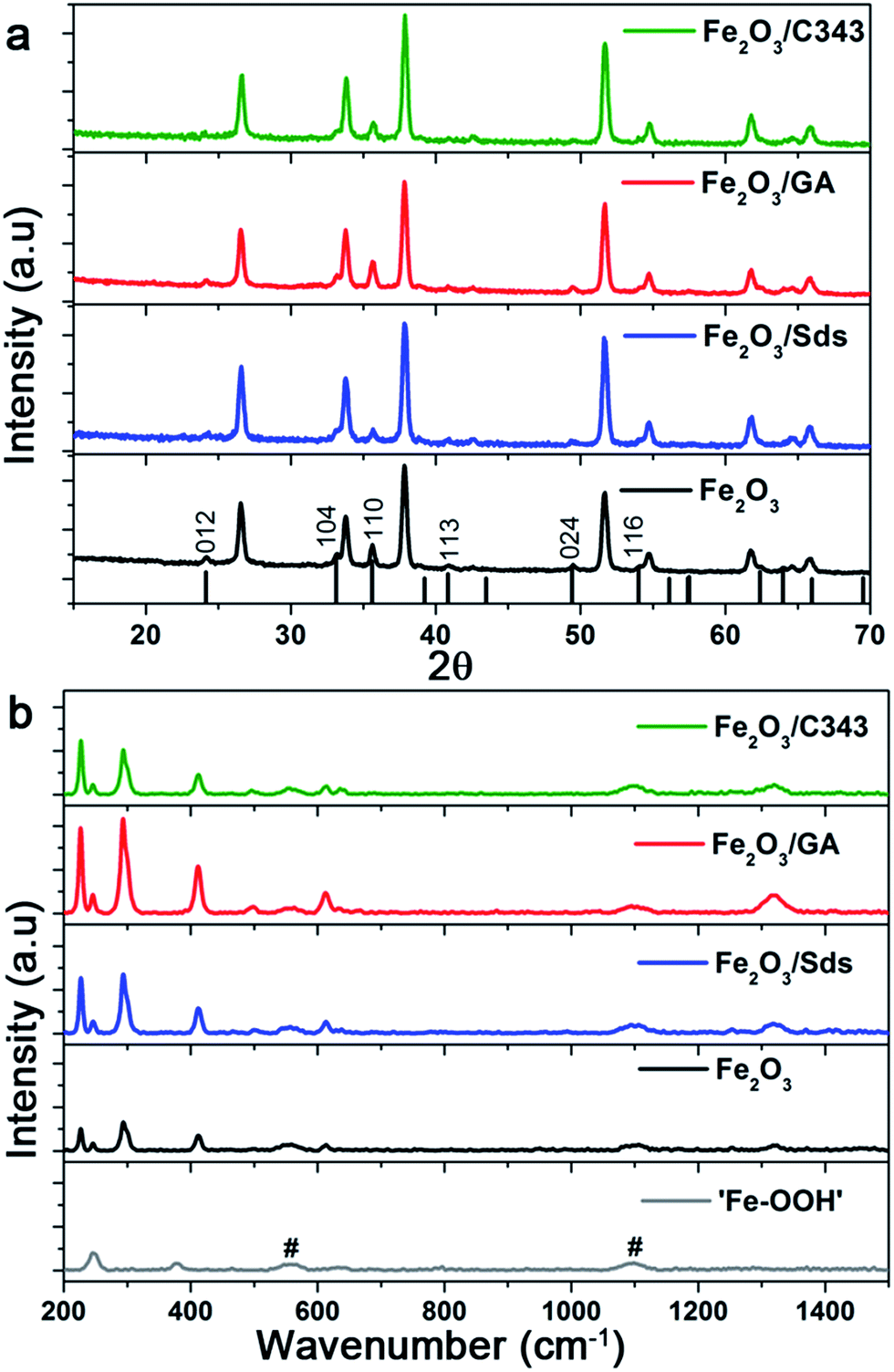 | ||
| Fig. 3 X-ray diffraction patterns (a) and Raman absorption bands (b) of the α-Fe2O3 films electrodeposited at 0.4 V vs. Ag/AgCl and annealed at 600 °C. Signals from the FTO substrate are indicated by #. | ||
As can be seen from the XRD reflexes the hematite peaks are dominated by FTO reflexes and further analysis on the XRD patterns is difficult. To get further structural insights we took Raman measurements as Raman is inherently a surface sensitive method. The as prepared films indicated as ‘Fe–OOH’ (Fig. 3b bottom) show two characteristic peaks at 245 and 377 cm−1 and these peaks match to the two intense peaks of γ-FeOOH.49 The annealed hematite films show seven Raman peaks at 227, 246, 293, 412, 498, 613, and 1320 cm−1 which can all be indexed to reported hematite peaks.50,51 Additionally, weak peaks at 558 and 1100 cm−1 are also observed which belong to the FTO substrate. The absence of the peak at 660 cm−1 which is characteristic for Fe3O4, suggests that our hematite films are pure and agrees well with the XPS measurements (see below). The features of the Raman lines such as peak intensity, and broadness or narrowness of the line give information relating to the grain size and degree of crystallinity. In general the full width at half maximum (FWHM) increases as the feature size of the crystals decreases due to phonon confinement.50 For the most intense peak at 294 the FWHM varies slightly between the films, and is 12.1, 10.6, 11.3 and 11.2 cm−1 for Fe2O3, Fe2O3/Sds, Fe2O3/GA, and Fe2O3/C343, respectively. This partly suggests that the organic additives lead to larger crystal sizes as indicated by the SEM. Additionally, the ratio of peak intensities (498/294) has the highest value for Fe2O3/GA (0.48) compared to Fe2O3/Sds (0.38) indicating a high degree of crystallinity for the GA modified hematite electrodes.
XPS measurements were recorded in order to determine the oxidation state of Fe and to verify the incorporation of the organic additives in the films. The XPS spectra presented in Fig. 4 show the Fe 2p2/3 peaks for the as-prepared films and the Fe2O3 films after annealing. The annealed films exhibit peaks located at 710.5 eV and 709.5 eV which is consistent with Fe3+ ions in hematite. The weak satellite peak 8 eV above the main 2p3/2 line provides additional evidence for the Fe3+ state which is consistent with the previously reported values.52 The Fe 2p3/2 peak for the as-prepared samples is slightly shifted towards higher binding energies and is centred at 710.9 eV indicating the formation of Fe hydroxides. The O 1s peaks also indicate the formation of Fe2O3 (Fig. 3Sa†). The C:Fe atomic ratios were estimated from the C 1s (C–O) and Fe 2p peaks; for the as-prepared films values of 0.48 and 0.34 were obtained for ‘FeOOH’ films with and without the organic additive GA, respectively. As the films were prepared from an acetate containing electrolyte the inclusion of a considerable amount of C is expected.
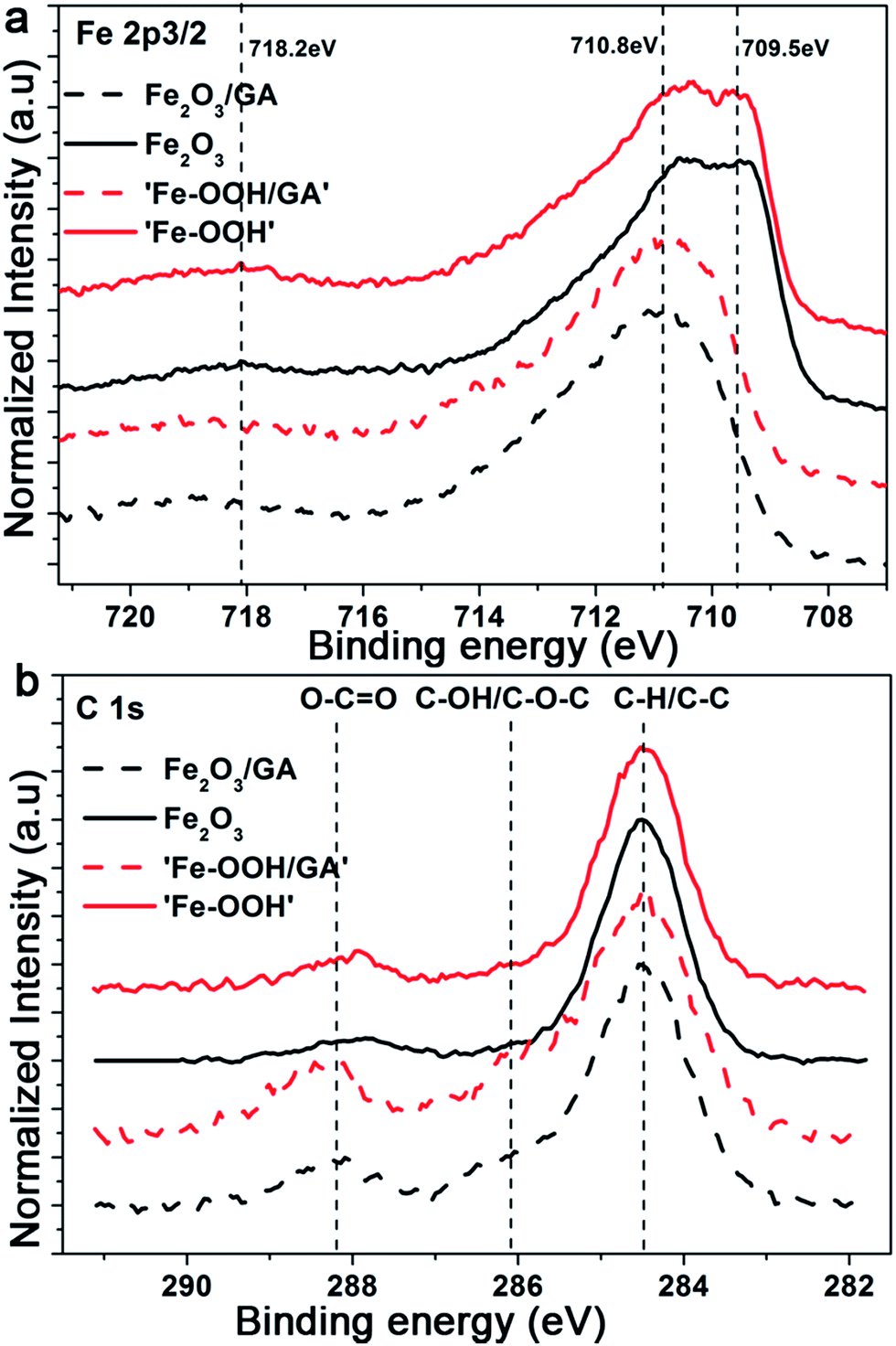 | ||
| Fig. 4 XPS spectra of Fe2O3 films electrodeposited without and with GA showing the (a) Fe 2p and (b) C 1s regions films, respectively. | ||
However, the difference in the C content indicates the successful incorporation of the organic additives to the film. However, after annealing, the majority of the C will decompose and burn out. The weak peak observed at around 288.2 eV is related to C 1s (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). As the adsorbed carbonate species also appear in the same region unambiguous assignment would be difficult. Additionally, the peak intensities are comparable and the absence of C 1s (Fe–C) which appears at a low binding energy compared to the C 1s reference peak also supports the claim that there is no preferential doping effect from the organic additives. We also investigate the possible N doping effect arising from the Fe source or from the addition of C343. The survey spectra collected for Fe2O3/GA and Fe2O3/C343 show no traces of N (Fig. 3Sb†). This suggests C343 does not induce N doping and the observed photocurrent difference.
O). As the adsorbed carbonate species also appear in the same region unambiguous assignment would be difficult. Additionally, the peak intensities are comparable and the absence of C 1s (Fe–C) which appears at a low binding energy compared to the C 1s reference peak also supports the claim that there is no preferential doping effect from the organic additives. We also investigate the possible N doping effect arising from the Fe source or from the addition of C343. The survey spectra collected for Fe2O3/GA and Fe2O3/C343 show no traces of N (Fig. 3Sb†). This suggests C343 does not induce N doping and the observed photocurrent difference.
The photoelectrochemical activities of the films were investigated using current–voltage curves, chopped light voltammetry and IPCE measurements. Since there is no significant photoelectrochemical activity for films annealed below 600 °C, all photoelectrochemical measurements were performed for films annealed at this temperature. Fig. 5 shows the current–voltage curves of the hematite films under light illumination from a white LED at 100 mW cm−2. The potential was scanned from −0.4 V to +1 V vs. Ag/AgCl at a scan rate of 10 mV−1 s. The onset of the dark photocurrent for all electrodes is at around 1.7 V vs. RHE and no significant variation is observed (Fig. 5 solid lines). Under illumination the photocurrent is negligible below 1.0 V vs. RHE and starts to rise only above 1.0 V. As the bias potential increases anodically, the photocurrent shows a significant increase and continues increasing up to the dark current onset at 1.7 V. The magnitude of the photocurrent slightly depends on the direction of illumination (front vs. back side illumination) for all electrodes except Fe2O3/GA. In the case of Fe2O3/GA there is a marked difference in the photocurrent from front and back side illumination (Fig. 5c dashed line vs. dashed dotted line). A significant enhancement was observed under back side illumination even though there is a loss in light intensity due to the FTO. Back side illumination is preferable because the pathway for electrons to the high conducting FTO is shorter. But effective photocurrents can only be obtained if the electrode is porous enough to allow suitable electrolyte penetration. The obtained result suggests that the Fe2O3/GA electrode especially provides the needed high porosity which allows the easy penetration of the electrolyte thereby decreasing the diffusion pathway for the photogenerated electron/hole pairs. Our observation agrees with a recent report by Toussaint et al. which shows the effect of the illumination direction for mesoporous Fe2O3 films formed by soft templating via a sol–gel route.53 However, the other organic additives (Sds as well as C343) appear to lead to relatively compact hematite films for which only electron/hole pairs generated at the electrolyte semiconductor interface are effectively collected irrespective of the direction of illumination.
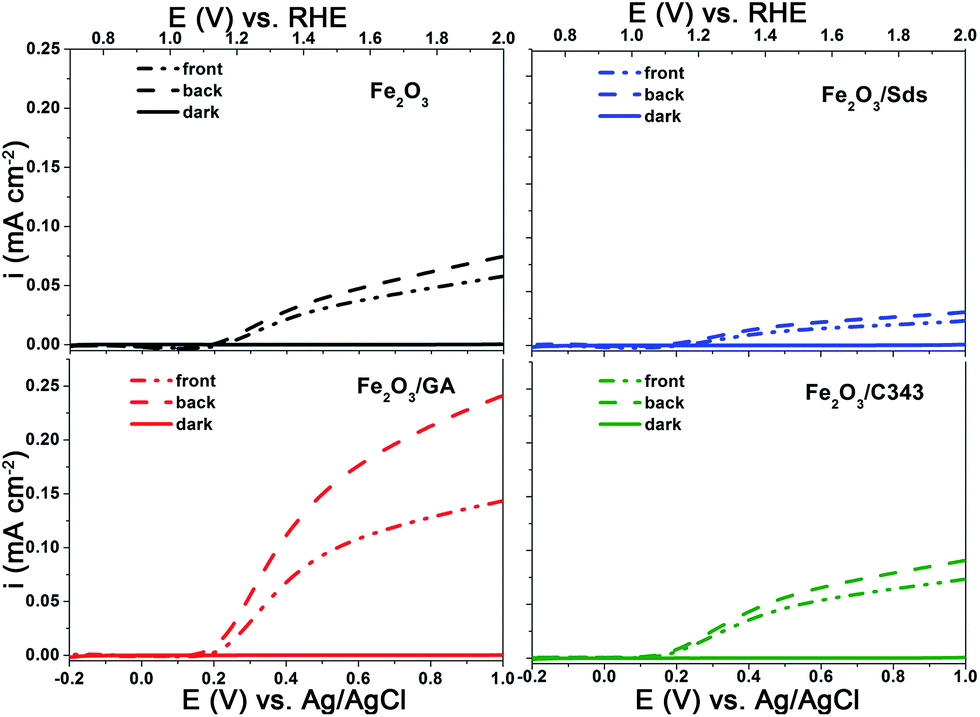 | ||
| Fig. 5 I–V curves of the electrodeposited Fe2O3 films with and without organic additives in 1 M NaOH showing the effect of the light illumination direction. | ||
Comparison of the photoanode performance of the different electrodes shows that Fe2O3/GA exhibits the highest photocurrent and is followed by Fe2O3/C343. The photocurrent of the unmodified Fe2O3 is higher than Fe2O3/Sds which exhibits the lowest photocurrent of all. Evidence is given by Raman measurements showing a lower degree of crystallinity likely combined with an increased number of grain boundaries for Fe2O3/Sds. The low photoresponse of Fe2O3/Sds is probably due to charge recombination at these grain boundaries. At an applied potential of 1.40 V vs. RHE the photocurrent for Fe2O3/GA is 5–6 times higher than that achieved for the unmodified Fe2O3. The obtained photocurrents are generally small compared to the state-of-the-art APCVD hematite films15 but are comparable with the recently reported value by Fu et al. in which ethylene glycol was used as an additive for the ED of hematite films.54
The photoactivity differences between the different hematite films become more evident when an effective hole acceptor molecule is used during the photoelectrochemical experiments. H2O2 is used as a probe molecule to evaluate the efficiency and the kinetic limitations of the different photoanodes as the oxidation of H2O2 is kinetically much easier than water oxidation.55,56 We performed photoelectrochemical experiments in the presence of 0.5 M H2O2 (Fig. 4Sa and b†). The anodic photocurrent is significantly increased and the onset of the photocurrent is shifted to a value close to the flat band potential of the hematite photoanode. The photoactivity of the electrodes can be easily compared by calculating the hole transfer efficiency taking the photocurrent ratios measured with and without H2O2 at a given bias potential (Fig. 6a). The result clearly shows that the organically modified hematite electrodes, particularly Fe2O3/GA, outperform those prepared in the absence of the additives. When the kinetic barrier for the oxidation reaction is removed the variation in the activity of the different electrodes arises from the influence of the organic molecules on the morphology of the films leading to better charge separation or hole transfer to the solution.
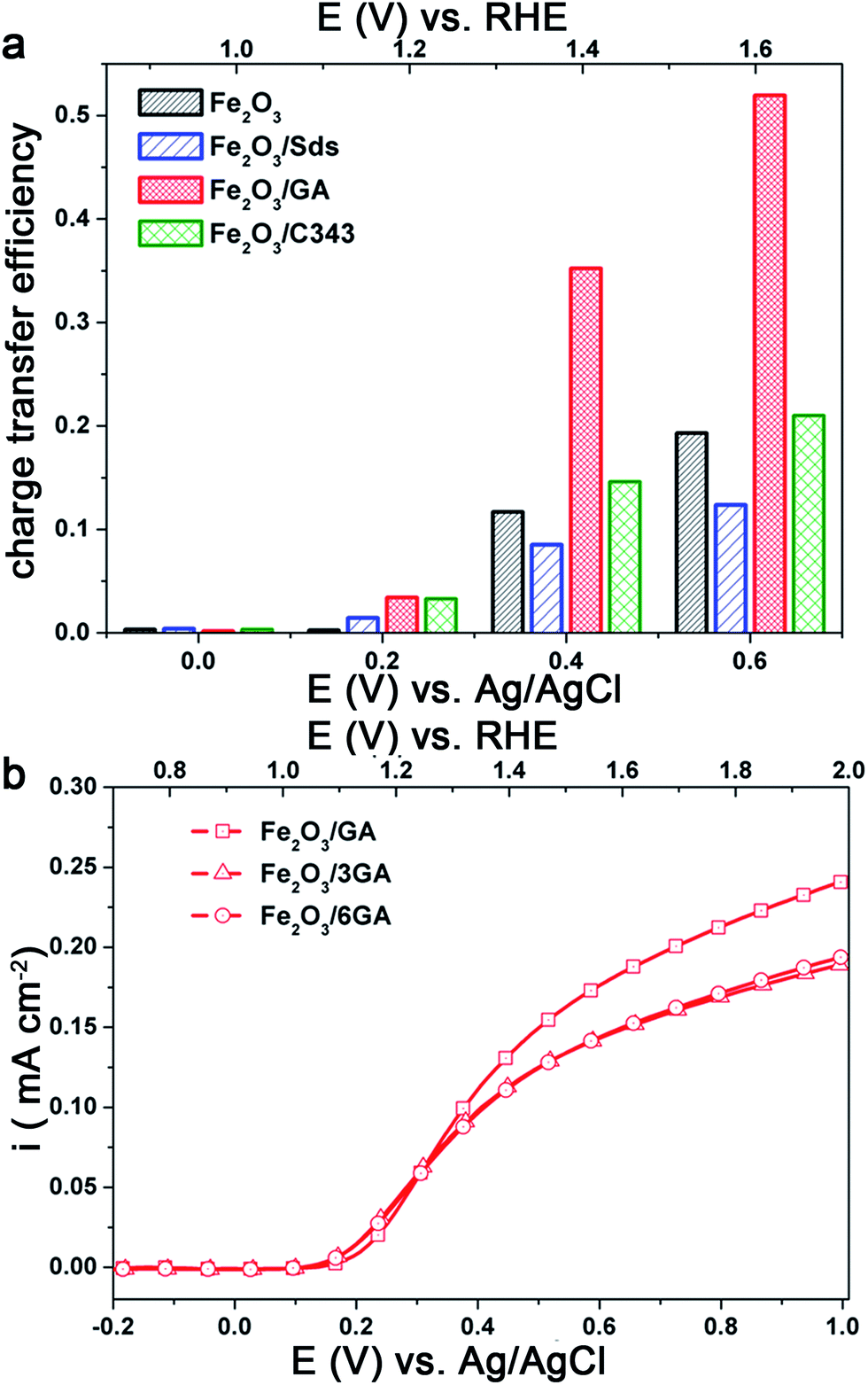 | ||
| Fig. 6 (a) Charge transfer efficiency calculated taking the ratio of the photocurrent densities (iH2O/iH2O2), (b) the effect of GA concentration on the photocurrent under back side illumination. | ||
In light of the above results we attempted to improve the photoresponse of Fe2O3/GA by preparing Fe2O3 electrodes from a bath containing different concentrations of GA. As the concentration of GA increased from 1 mM to 6 mM the photocurrent slightly decreases under back side illumination (Fig. 6b). The SEM images of Fe2O3/GA with different concentrations of GA are presented in Fig. 5S† and show similar morphologies. However, there are differences in the way the grain domains are interconnected. As the GA concentration increases the domains become large and loosely packed. This probably leads to a photocurrent loss due to a reduction in the available reaction sites and the need for the charge carriers to travel a longer distance to reach the interface. Based on these results further experiments were conducted on electrodes prepared from solutions containing 1 mM GA.
Effect of Ti treatment
Ti doping has been shown to improve the performance of the hematite photoanodes significantly and the role of the Ti is to control the crystal size and in some cases lead to a preferential orientation of the crystals.57 Additionally, a thin underlying TiO2 layer improves the photoresponse of the hematite electrodes by blocking the electron–hole recombination.58 Following a simple and facile post treatment procedure reported by Franking et al.18 the hematite photoanodes were treated with 25 μL of 25 mM Ti(OBu)4 and dried in air prior to annealing. Higher concentrations of Ti(OBu)4 were tested but the optimum was 25 mM and data are presented for the optimized concentration. The Ti treatment has no significant influence on the morphology of the films, but the presence of Ti is confirmed by EDXS (Fig. 4Sc and d†).Fig. 7a shows the current–voltage behaviour of the Ti treated hematite photoanodes under light illumination. The photocurrent increases significantly and the onset of the photocurrent shifts ca. 30 mV cathodically for Fe2O3 and Fe2O3/GA after Ti loading (Fig. 7b). The photocurrent density at 1.4 V RHE increased by a factor of 2 for Fe2O3/GA/Ti (0.20 mA cm−2) and Fe2O3/C343/Ti (0.12 mA cm−2). Both Fe2O3/Ti and Fe2O3/Sds/Ti show a pronounced increase in photocurrent density giving values of 0.12 mA cm−2 and 0.06 m Acm−2, respectively, which are 4–5 fold higher than those without Ti treatment. The Ti treatment has a modest influence on the Fe2O3/GA and Fe2O3/C343 electrodes compared to the Fe2O3 and Fe2O3/Sds electrodes. This shows that the organic additives GA and C343 already form hematite morphologies with fewer grain boundaries so the Ti treatment is of lower importance. Interestingly, the Fe2O3/GA/Ti photoanode still maintains a photocurrent density which is 1.6 times higher than that for Fe2O3/Ti. Previous studies on the surface modification of hematite photoanodes with very thin oxide over layers (Ga2O3 and Al2O3) demonstrated improved photo responses.59,60 The oxide over layers passivate the surface states which act as recombination centres or hole traps. Thus in the presence of the oxide over layers a reduced capacitive response (due to the charging of the surface states) is expected. Following the method of Bertoluzzi et al.61 we performed cyclic voltammetry (CV) investigations on the Ti modified Fe2O3 and Fe2O3/GA electrodes to ascertain the function of the Ti oxide over layer. The electrodes are first polarized at a higher anodic potential (1.8 V vs. RHE) under illumination and cyclic voltammograms are recorded at different scan rates. As shown in Fig. 7Sa and c,† only one cathodic capacitive peak is observed without the accompanying anodic peak which shows that the hematite films are suffering from high recombination (as Bertoluzzi et al. described in detail). But after the Ti treatment (Fig. 7Sb and d†) the corresponding anodic peak is partially recovered and the magnitude of the cathodic peaks is reduced. The cathodic peak current measured at 50 mV s−1 is reduced from 16.2 to 6.3 μA cm−2 and from 8.7 to 4.9 μA cm−2 for the Fe2O3 and Fe2O3/GA electrodes after the Ti treatment, respectively. The evolution of the anodic peak is an indication that the Ti oxide over layer facilitates charge detrapping during the reverse scan. The peak capacitive current linearly increases with the scan rate (Fig. 7Sa–d inset†) which suggests that the current is not diffusion controlled. The improved photoactivity brought about by the Ti treatment is probably due to the passivation of the grain boundaries (trap states).
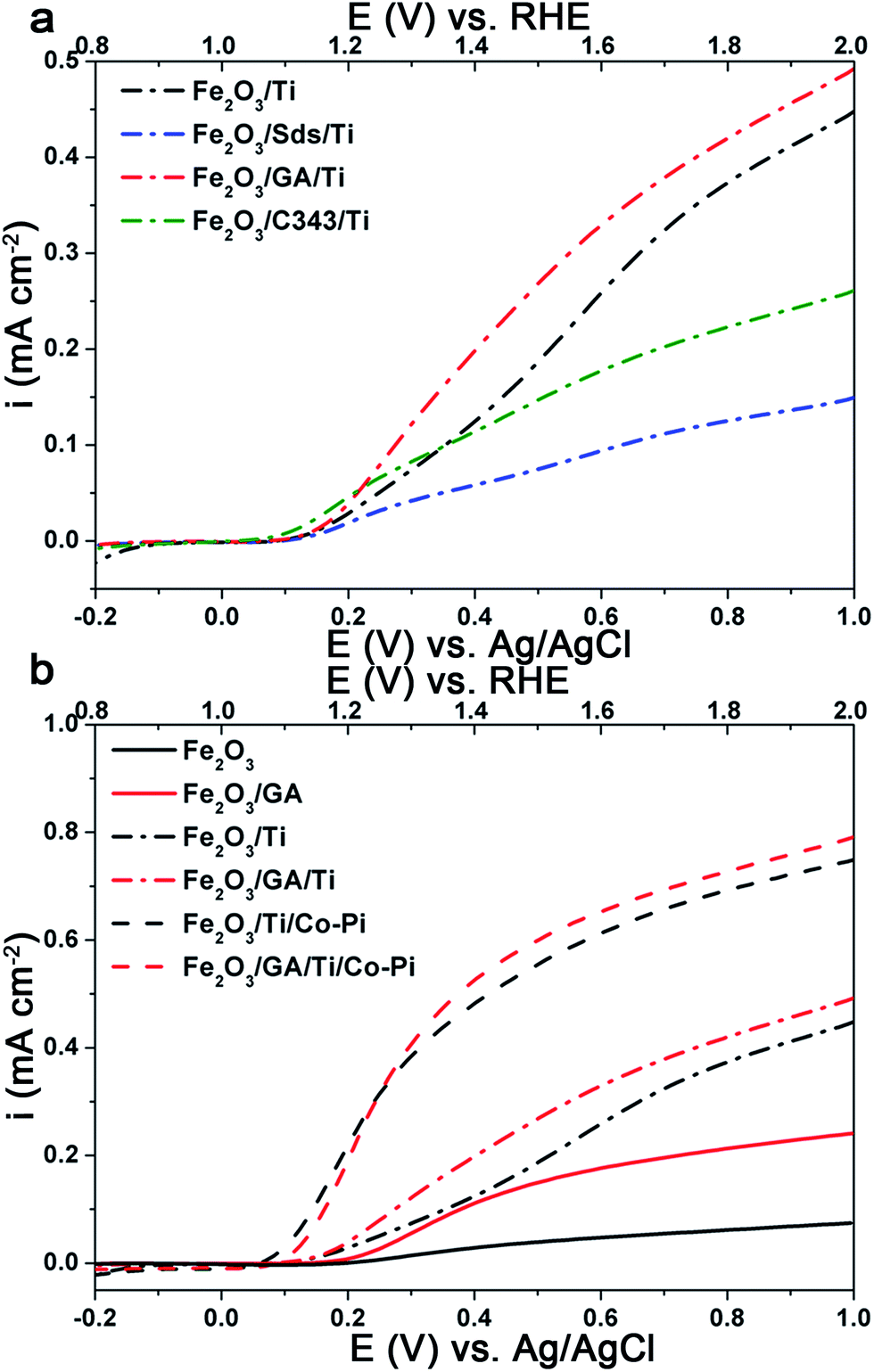 | ||
| Fig. 7 Effect of the Ti and Co-Pi treatments: (a) I–V curves of the Ti doped electrodeposited Fe2O3 films with and without organic additives in 1 M NaOH and (b) I–V curves showing the shift of the onset for Fe2O3 and Fe2O3/GA after the loading of Ti and Co-Pi. | ||
Effect of Co-Pi treatment
Cobalt phosphate (Co-Pi) has been extensively studied as an alternative and cheap oxygen evolution (OER) catalyst compared to catalysts containing more rare elements such as IrO2 and RuO2.62,63 It has been proven as an effective water oxidation co-catalyst in several oxide-based PEC systems. Thus, Co-Pi was deposited onto the Ti-doped hematite electrode using electrodeposition at 0.9 V vs. Ag/AgCl for 210 s from a phosphate buffer solution containing 0.5 mM Co(NO3)3. Fig. 7b shows the selective photocurrent densities obtained on the Fe2O3 and Fe2O3/GA electrodes after the Ti and Co-Pi treatments. After the Co-Pi treatment the photocurrent density at 1.4 V vs. RHE was increased to 0.53 mA cm−2 and 0.48 mA cm−2 for Fe2O3/GA and Fe2O3, respectively. Remarkably, the onset of the photocurrent is cathodically shifted by ca. 50–70 mV for both electrodes. This confirms the role of Co-Pi as a catalyst which facilitates the hole transfer and reduces surface recombination.61,64,65 The treatment of Ti and Co-Pi also improves the photocurrent of the Fe2O3/Sds (0.25 mA cm−2) and Fe2O3/C343 (0.27 mA cm−2) electrodes (Fig. 6Sa†).IPCE measurements
The incident photon to current conversion efficiency (IPCE) of the electrodeposited hematite films was measured as the direction of illumination was changed (Fig. 8). In general the IPCE spectra follow the absorption spectra of the films (Fig. 2) very well but they decrease more rapidly at longer wavelengths. The IPCE values also demonstrate that the hematite films with the organic additives, with the exception of Fe2O3/Sds, are able to convert the absorbed photons into current more effectively than Fe2O3. As observed in the I–V curves in Fig. 5 the back side illumination results in higher IPCE values particularly in the short wavelength regime. The penetration depth of the high energy photons is much shorter and creates electron–hole pairs at the FTO/hematite interface which is remote from the hematite/solution interface and hence the majority of them are lost through recombination and do not contribute to the photocurrent.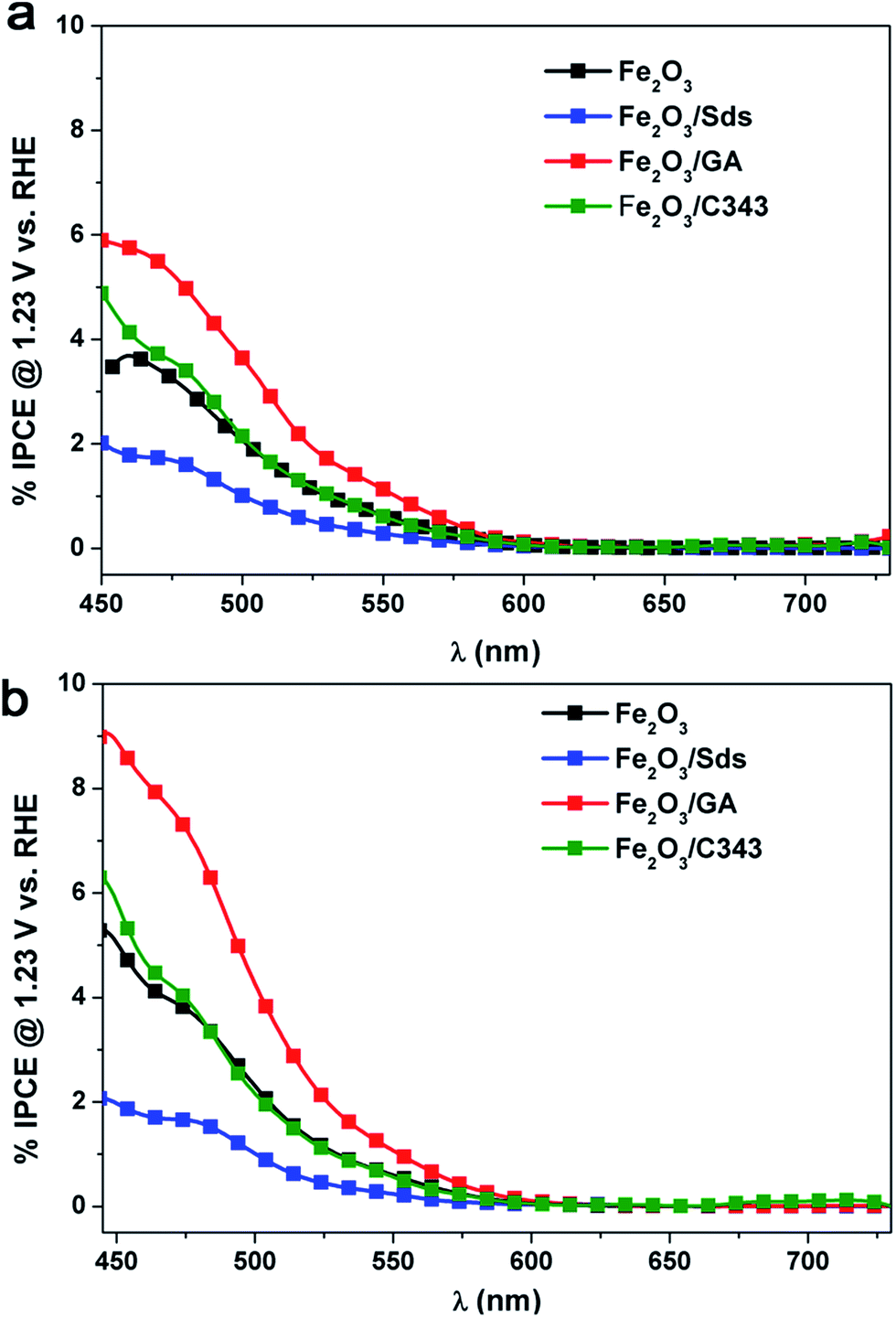 | ||
| Fig. 8 IPCE spectra of Fe2O3 films deposited with and without additives under (a) front and (b) back side illumination. | ||
Thus back side illumination results in a lower photocurrent (a lower IPCE) compared to front side illumination and the situation is prevailing if the hematite layer is compact and/or thick.15 However, if the hematite layer exhibits a certain degree of porosity which extends to the FTO back contact, back side illumination is beneficial as the holes from the high energy photons have the chance to reach the electrolyte and participate in the oxidation process.53 Indeed this is the case for the Fe2O3/GA electrode which shows a much increased IPCE value under back side illumination. It is worth mentioning that the highest IPCE value achieved is 8.5% for Fe2O3/GA at 470 nm which is twice the value obtained for Fe2O3 showing the superior activity of GA modified hematite electrodes.
Mott–Schottky (MS) and electrochemical impedance spectroscopy (EIS) analysis
To gain further insights into the effect of organic additives and doping on the photoactivity of electrodeposited hematite films we conducted EIS and MS analysis. The donor densities and the flat band potentials of the semiconductor electrodes can be accurately estimated from MS plots using eqn (2) if the electrode is planar and no surface states are involved. Considering the morphologies and the sizes of the hematite grains, both criteria cannot be met with any of the electrodes under investigation. The situation is directly manifested by a strong frequency dependence of the complex impedance and the curved nature of the MS plots (Fig. 9a–d). Therefore the determination of the donor densities from the slope of these curved MS plots is very unreliable. A similar type of behaviour was reported for sputter deposited and powder pressed hematite photoanodes.66,67 However, the intercepts are fairly similar for the measured frequencies and can be used to estimate the flat band potentials. The plots are fitted in the range of 0.4–0.7 V vs. RHE and the values obtained are 0.53 ± 0.02 V, 0.61 ± 0.05 V, 0.49 ± 0.02 V, 0.54 ± 0.05 V for Fe2O3, Fe2O3/Sds, Fe2O3/C343, and Fe2O3/GA, respectively. The values obtained are close to the onset of the photocurrent in the H2O2 solution measured under chopped light illumination which is 0.53–0.55 V (Fig. 4Sb†). Reported hematite flat band potentials range between 0.4 and 0.6 V vs. RHE and thus our values coincide with these.54,56,68 After the Ti treatment the MS plots become more flat (Fig. 6Sb†) indicating an increase in donor density, however, extracting the flat band potentials and the donor densities quantitatively is not possible.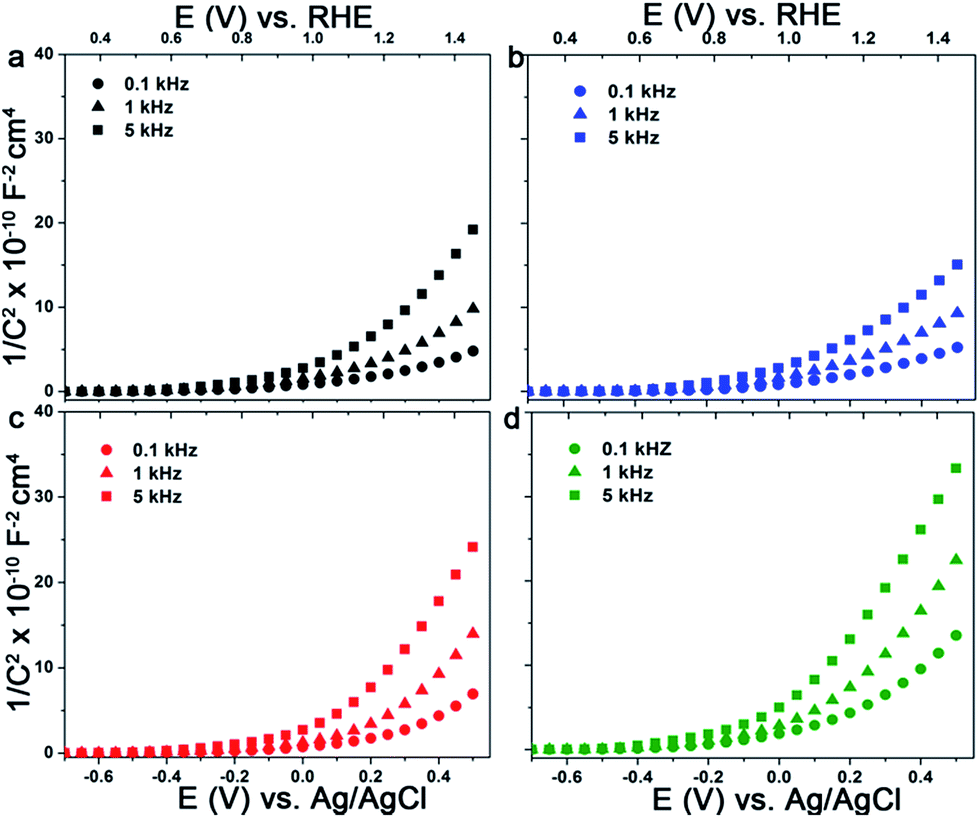 | ||
| Fig. 9 MS plots of electrodeposited Fe2O3 films with and without organic additives in 1 M NaOH at the indicated frequencies: (a) Fe2O3, (b) Fe2O3/Sds, (c) Fe2O3/GA and (d) Fe2O3/C343. | ||
Fig. 10a and b show the Nyquist plots for Fe2O3 and Fe2O3/GA obtained from measurements under illumination at 1.23 V vs. RHE for the electrodes before and after the loading of Ti and Co-Pi. The plots consist of two semi-circles at bias potentials <1.4 V vs. RHE which is consistent with the behaviour of hematite photoanodes under illumination.24 The experimental data were fitted to an equivalent circuit model consisting of two RC units each containing a resistance in parallel with a constant phase element (CPE) (Fig. 10a inset).68,69 In this model the CPE replaces the pure capacitive elements to account for the non-ideal behaviour originating from the non-homogeneity of the films and roughness. The model consists of the space charge capacitance for bulk hematite (CPE1) and the surface state capacitance (CPE2). R1 and R2 represent the resistance of the bulk hematite and the charge transfer resistance from the surface states to the solution, respectively. The series resistance Rs accounts for the resistance of the TCO and/or the electrolyte. The fitted data are collected and summarized in Table 1.
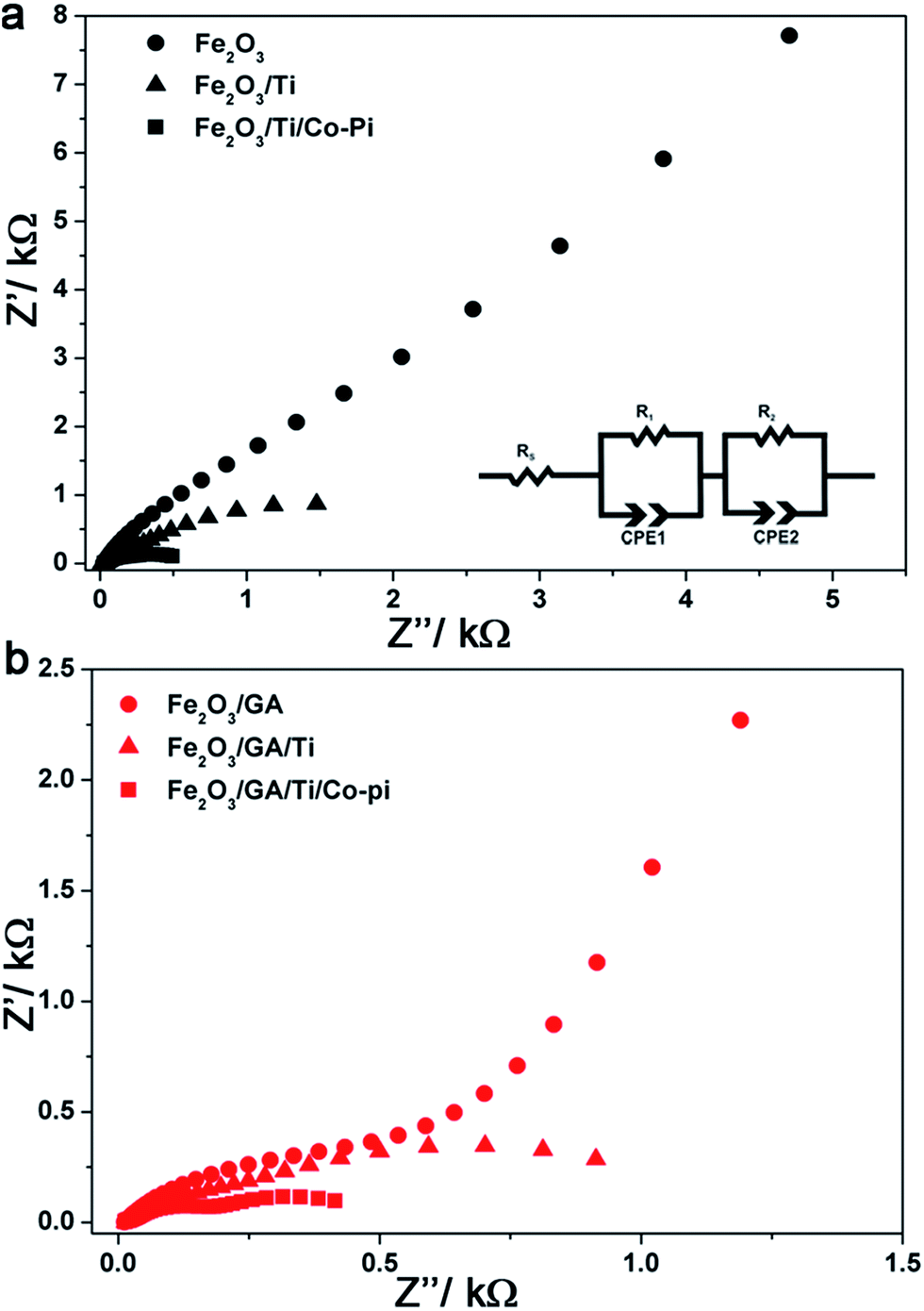 | ||
| Fig. 10 EIS results and analysis: effect of Ti and Co-Pi loading on the complex impedance plots of (a) Fe2O3 and (b) Fe2O3/GA. The inset shows the equivalent circuit model used for fitting the data. | ||
| Electrodes | R s/Ω | R 1/Ω | R 2/Ω | CPE1/μF | CPE2/μF |
|---|---|---|---|---|---|
| Fe2O3 | 17.7 | 825 | 98533 |
18.1 | 26.7 |
| Fe2O3/GA | 13.9 | 730 | 35946 |
27.4 | 82.7 |
| Fe2O3/Ti | 19.2 | 200 | 2493 | 31.8 | 58.8 |
| Fe2O3/GA/Ti | 20.9 | 155 | 1235 | 28.3 | 175.1 |
| Fe2O3/Ti/Co-Pi | 32.5 | 145.2 | 430 | 58.1 | 370.3 |
| Fe2O3/GA/Ti/Co-Pi | 22.9 | 150 | 303 | 41.5 | 408.2 |
The organic additive GA greatly reduces the charge transfer resistance (R2) responsible for the water oxidation reaction due to a favourable morphology which allows the easy penetration of the electrolyte. The larger grain sizes due to the fused nanoparticles lead to fewer grain boundaries. Doping with Ti further reduces R2 indicating a facilitated charge transfer from the hematite surface to the solution. Notably, the resistance of the bulk hematite (R1) is affected and after the Ti treatment the value is reduced by a factor of about four for both electrodes which further supports the Ti incorporation into the bulk of the film during the drop cast procedure. The Co-Pi treatment slightly decreases the bulk resistance, greatly reduces the charge transfer resistance and increases the surface state capacitance resulting in high photocurrent densities.
Conclusion
In summary, we have shown that the use of small organic molecules tunes the size and morphology of electrodeposited hematite thin films inducing enhanced photoactivity. The improved photoelectrochemical activity of the organically modified hematite films can be attributed to a favorable film morphology which allows easy penetration of the electrolyte and better charge separation. Better accessibility of the electrolyte is demonstrated by a dependence of the photocurrent and IPCE on the direction of the illumination. However, the functionality of the organic additives govern the type and strength of the interactions as sulfonates impart no unique morphology as opposed to the carboxylate ligands. Comparison of the photocurrent density at 1.4 V vs. RHE shows that the Fe2O3/GA electrode displays a photocurrent density 5–6 times higher than that of Fe2O3. The photocurrent density can be further enhanced by loading with a Ti dopant and a Co-Pi OER co-catalyst, observable by a cathodic shift of the onset of the photocurrent. The main role of the organic additive, specifically GA, is the reduction of the charge transfer resistance from the hematite surface to the solution which is the kinetic bottleneck for the overall water splitting reaction, as evidenced by EIS. The organic additives particularly GA imparts moderate preferential crystal orientation but there is no ordering in the resulting films.Acknowledgements
This work is supported by the Deutsche Forschungsgemeinschaft (DFG) (WA 1116/23-1) within the priority program SPP1613. We are very grateful to Dr Carsten Dosche for his help during the XPS experiments.References
- A. Maczulka, Renewable energy Sources: Sources and Methods, Library of Congress Cataloging, 2010, ch. 5 Search PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. Graetzel, Nature, 2001, 414, 338–343 CrossRef PubMed.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347–370 CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- K. Sivula, F. le Formal and M. Graetzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- M. J. Katz, S. C. Riha, N. C. Jeong, A. B. F. Martinson, O. K. Farha and J. T. Hupp, Coord. Chem. Rev., 2012, 256, 2521–2529 CrossRef CAS.
- D. K. Bora, A. Braun and E. C. Constable, Energy Environ. Sci., 2013, 6, 407–425 CAS.
- U. Bjorksten, J. Moser and M. Graetzel, Chem. Mater., 1994, 6, 858–863 CrossRef.
- A. B. Murphy, P. R. F. Barnes, L. K. Randeniya, I. C. Plumb, I. E. Greyb, M. D. Horne and J. A. Glawsscock, Int. J. Hydrogen Energy, 2006, 31, 1999–2017 CrossRef CAS.
- J. H. Kennedy and K. W. Frese Jr, J. Electrochem. Soc., 1978, 125, 709–714 CrossRef CAS.
- L. Vayssieres, N. Beermann, S.-E. Lindquist and A. Hagfeldt, Chem. Mater., 2001, 13, 233–235 CrossRef CAS.
- A. Kay, I. Cesar and M. Graetzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS PubMed.
- Y. Lin, S. Zhou, S. W. Sheehan and D. Wang, J. Am. Chem. Soc., 2011, 133, 2398–2401 CrossRef CAS PubMed.
- S. C. Warren, K. Voïtchovsky, H. Dotan, C. M. Leroy, M. Cornuzl, F. Stellacci, C. Hebert, A. Rothschild and M. Graetzel, Nat. Mater., 2013, 12, 812–849 CrossRef PubMed.
- R. Franking, L. Li, M. A. Lukowski, F. Meng, Y. Tan, R. J. Hamers and S. Jin, Energy Environ. Sci., 2013, 6, 500–512 CAS.
- J. A. Glasscock, P. R. F. Barnes, I. C. Plumb and N. Savvides, J. Phys. Chem. C, 2007, 111, 16477–16488 CAS.
- S. S. Yarahmadi, K. G. Upul Wijayantha, A. A. Tahir and B. Vaidhyanathan, J. Phys. Chem. C, 2009, 113, 4768–4778 Search PubMed.
- A. K. Shwarsctein, Y. S. Hu, A. J. Forman, G. D. Stucky and E. W. McFarland, J. Phys. Chem. C, 2008, 112, 15900–15907 Search PubMed.
- D. K. Zhong and D. R. Gamelin, J. Am. Chem. Soc., 2010, 132, 4201–4207 Search PubMed.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS PubMed.
- F. le Formal, N. Tétreault, M. Cornuz, T. Moehl, M. Graetzel and K. Sivula, Chem. Sci., 2011, 2, 737–743 RSC.
- T. Hisatomi, F. le Formal, M. Cornuz, J. Brillet, N. Tétreault, K. Sivula and M. Graetzel, Energy Environ. Sci., 2011, 4, 2512–2515 CAS.
- C. D. Park, D. Magana and A. E. Stiegman, Chem. Mater., 2007, 19, 677–683 CrossRef CAS.
- S. U. M. Khan and J. Akikusa, J. Phys. Chem. B, 1999, 103, 7184–7189 CrossRef CAS.
- C. J. Sartoretti, B. D. Alexander, R. Solarska, I. A. Rutkowska and J. Augustynski, J. Phys. Chem. B, 2005, 109, 13685–13692 CrossRef CAS PubMed.
- A. A. Tahir, K. G. U. Wijayantha, S. S. Yarahmadi, M. Mazhar and V. McKee, Chem. Mater., 2009, 21, 3763–3772 CrossRef CAS.
- M. Lie, H. Fjellvåg and A. Kjekshus, Thin Solid Films, 2005, 488, 74–81 CrossRef CAS.
- S. C. Riha, B. M. Klahr, E. C. Tyo, S. Seifert, S. Vajda, M. J. Pellin, T. W. Hamann and A. B. F. Martinson, ACS Nano, 2013, 7, 2396–2405 CrossRef CAS PubMed.
- Y. S. Hu, A. K. Shwarsctein, A. J. Forman, D. Hazen, J. N. Park and E. W. McFarland, Chem. Mater., 2008, 20, 3803–3805 CrossRef CAS.
- P. S. Shinde, G. H. Go and W. J. Lee, J. Mater. Chem., 2012, 22, 10469–10471 RSC.
- R. S. Schrebler, L. Ballesteros, A. Burgos, E. C. Munoz, P. Grez, D. Leinen, F. Martin, J. R. Ramos-Barrado and E. A. Dalchiele, J. Electrochem. Soc., 2011, 158, D500–D505 CrossRef CAS.
- R. Schrebler, K. Belloa, F. Veraa, P. Curya, E. Munoza, R. del Ríoa, H. G. Meiera, R. Córdovaa and E. A. Dalchiele, J. Electrochem. Soc., 2006, 9, C110–C113 CAS.
- R. L. Spray and K. S. Choi, Chem. Mater., 2009, 21, 3701–3709 CrossRef CAS.
- A. K. Shwarsctein, Y.-S. Hu, A. J. Forman, G. D. Stucky and E. W. McFarland, J. Phys. Chem. C, 2008, 112, 15900–15907 Search PubMed.
- P. Kumar, P. Sharma, R. Shrivastav, S. Dass and V. R. Satsangi, Int. J. Hydrogen Energy, 2011, 36, 2777–2784 CrossRef CAS.
- Y. Liu, Y. X. Yu and W. D. Zhang, Electrochim. Acta, 2012, 59, 121–127 CrossRef.
- X. Shi, K. Zhang, K. Shin, J. H. Moon, T. W. Lee and J. H. Park, Phys. Chem. Chem. Phys., 2013, 15, 11717–11722 RSC.
- C. Boeckler, T. Oekermann, A. Feldhoff and M. Wark, Langmuir, 2006, 22, 9427 CrossRef CAS PubMed.
- H. Graaf, F. Lüttich, C. Dunkel, M. Wark and T. Oekermann, J. Phys. Chem. C, 2012, 116, 5610–5613 CAS.
- M. J. Siegfried and K.-S. Choi, J. Am. Chem. Soc., 2006, 128, 10356–10357 CrossRef CAS PubMed.
- L. F. Xu, Y. Guo, Q. Liao, J. P. Zhang and D. S. Xu, J. Phys. Chem. B, 2005, 109, 13519–15522 CrossRef CAS PubMed.
- S. Jiao, L. Xu, K. Hu, J. Li, S. Gao and D. Xu, J. Phys. Chem. C, 2010, 114, 269–273 CAS.
- G. A. Gamlen and D. O. Jordan, J. Chem. Soc., 1953, 435–1443 Search PubMed.
- L. A. Marusak, R. Messier and W. B. White, J. Phys. Chem. Solids, 1980, 41, 981–984 CrossRef CAS.
- K. M. Ross, D. M. A. Smith and M. Dupuis, J. Chem. Phys., 2003, 118, 6455–6466 CrossRef.
- D. Delichere, S. Joiret, C. Pallotta and A. Hugot-Le Goff, J. Electrochem. Soc., 1988, 135, 305–310 CrossRef.
- D. Bersani, P. P. Lottici and A. Montenero, J. Raman Spectrosc., 1999, 30, 355–360 CrossRef CAS.
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Graetzel, J. Phys. Chem. C, 2009, 113, 772–782 CAS.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529 CrossRef CAS.
- C. Toussaint, H. L. le Tran, P. Colson, J. Dewalque, Bénédicte Vertruyen, B. Gilbert, N. D. Nguyen, R. Cloots and C. Henrist, J. Phys. Chem. C, 2015, 119, 1642–1650 CAS.
- L. Fu, H. Yu, Y. Li, C. Zhang, X. Wang, Z. Shao and B. Yia, Phys. Chem. Chem. Phys., 2014, 16, 4284–4290 RSC.
- H. Dotan, K. Sivula, M. Graetzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 CAS.
- I. S. Cho, M. Logar, C. H. Lee, L. Cai, F. B. Prinz and X. Zheng, Nano Lett., 2014, 14, 24–31 CrossRef CAS PubMed.
- G. Wang, Y. Ling, D. A. Wheeler, K. E. N. George, K. Horsley, C. Heske, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3503–3509 CrossRef CAS PubMed.
- T. Hisatomi, H. Dotan, M. Stefik, K. Sivula, A. Rothschild, M. Grätzel and N. Mathews, Adv. Mater., 2012, 24, 2699–2702 CrossRef CAS PubMed.
- F. le Formal, K. Sivula and M. Graetzel, J. Phys. Chem. C, 2012, 116, 26707–26720 CAS.
- T. Hisatomi, F. le Formal, M. Cornuz, J. Brillet, N. Tetreault, K. Sivula and M. Graetzel, Energy Environ. Sci., 2011, 4, 2512–2515 CAS.
- L. Bertoluzzi, L. Badia-Bou, F. Fabregat-Santiago, S. Gimenez and J. Bisquert, J. Phys. Chem. Lett., 2013, 4, 1334–1339 CrossRef CAS PubMed.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109–114 RSC.
- D. K. Zhong, M. Cornuz, K. Sivula, M. Graetzel and D. R. Gamelin, Energy Environ. Sci., 2011, 4, 1759–1764 CAS.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS PubMed.
- L. M. Peter, K. G. U. Wijayantha and A. A. Tahir, FaradayDiscuss., 2012, 155, 309–322 RSC.
- F. A. Benko, J. Longo and F. P. Koffyberg, J. Electrochem. Soc., 1985, 132, 609–613 CrossRef CAS.
- C. D. Bohn, A. K. Agrawal, E. C. Walter, M. D. Vaudin, A. A. Herzing, P. M. Haney, A. A. Talin and V. A. Szalai, J. Phys. Chem. C, 2012, 116, 15290–15296 CAS.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Energy Environ. Sci., 2012, 5, 7626–7636 CAS.
- J. Y. Kim, J.-W. Jang, D. H. Youn, J. Y. Kim, E. S. Kim and J. S. Lee, RSC Adv., 2012, 2, 9415–9422 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images, UV-vis spectra, I–V curves, XPS and EDX data. See DOI: 10.1039/c5ra21290a |
| This journal is © The Royal Society of Chemistry 2015 |