
Fabrication of a non-enzymatic Ni(II) loaded ZSM-5 nanozeolite and multi-walled carbon nanotubes paste electrode as a glucose electrochemical sensor†
Seyed Karim Hassaninejad-Darzi*
Research Laboratory of Analytical & Organic Chemistry, Department of Chemistry, Faculty of Science, Babol University of Technology, Shariati Av., P.O. Box: 484, Babol, Iran 47148-71167. E-mail: hassaninejad@nit.ac.ir; Fax: +98 1132334203; Tel: +98 1132334203
First published on 9th December 2015
Abstract
Effective electro-oxidation of glucose is critically important in developing analytical sensors and carbohydrate-based fuel cells. In this study, a template-free ZSM-5 nanozeolite was synthesized hydrothermally with spherical particle diameters of 40–60 nm, as characterized by scanning electron microscopy. Then, a carbon paste electrode (CPE) was modified by multi-walled carbon nanotubes (MWCNTs), ZSM-5 nanozeolite and Ni2+ ions (Ni–MW–ZSM-5/CPE). Electrochemical studies of this electrode were performed using cyclic voltammetry which exhibits the redox behavior of the Ni(III)/Ni(II) couple in alkaline medium. This modified electrode was used as an anode for the electrocatalytic oxidation of glucose in 0.1 mol L−1 NaOH solution. The results confirmed that ZSM-5 nanozeolite at the surface of the CPE improved the catalytic efficiency of the dispersed nickel ions toward glucose oxidation. The values of electron transfer coefficient, electrode surface coverage and charge-transfer rate constant for Ni(III)/Ni(II) redox couple were found to be 0.65, 4.04 × 10−8 mol cm−2 and 0.184 s−1, respectively. Also, the diffusion coefficient and the mean value of the catalytic rate constant for glucose and redox sites of the electrode were found to be 1.66 × 10−4 cm2 s−1 and 1.136 × 108 cm3 mol−1 s−1, respectively. The sensor showed an acceptable linear range of 0.5–6.1 mM with a detection limit of 0.14 mM (S/N = 3) by cyclic voltammetry technique. Moreover, differential pulsed voltammetry method revealed a linear range of 0.0001–0.01 mM with a detection limit of 3.5 × 10−5 mM. Based on the results, the fabricated electrode (Ni–MW–ZSM-5/CPE) showed good catalytic activity, good stability, high sensitivity and reproducibility.
1. Introduction
Glucose is the primary energy source of the body. The level of glucose in the blood has been applied for diagnosis of diabetes. Besides the need for monitoring and determining of glucose in the case of diabetes patients, it is also necessary for non-diabetic critical care patients in order to control glucose levels.1 Oxidation of most carbohydrates can be easily detected by electrochemical methods. However, their electrochemical oxidation is not easy, as it requires a large overpotential on conventional electrodes.2 Glucose is a keen metabolite for living organisms, especially in the case of patients suffering from diabetes. Since Clark and Lyons3 reported the first enzyme electrode in 1962, and other electrochemical biosensors have later been employed successfully for the determination of glucose. Glucose oxidase (GOx) is one of the famous enzymes for detection of glucose in solution due to its being inexpensive, stable and of practical use.4 Application of GOx in biosensors has also brought about several problems. For example, activity of GOx can be easily affected by temperature, pH, humidity and toxic chemicals.5 Also, a complicated procedure, including adsorption, cross-linking, entrapment, and electropolymerization, is required for the immobilization of the enzyme on the solid electrode and this may decrease the activity of the GOx.5,6Non-enzyme electrochemistry-based method has several advantages over enzyme-based methods for glucose detection in terms of stability, simplicity, reproducibility, cost, and the fact that it is free of oxygen limitation.7 Considering these respects, the enzymeless glucose sensor is an attractive alternative technique. Actually, many efforts have been tried for the determination of glucose without enzyme, gold nanoparticles onto modified glassy carbon electrode,8 CuO nanosheets electrode,9 CuO/ZnO composite nanoarrays,10 magnetic copper ferrite onto multiwalled carbon nanotubes,11 nickel electrode,12,13 hierarchical Cu–Co–Ni electrode,14 and Pt nanoparticles encapsulated in carbon microspheres.15 Also, modified carbon paste electrodes16,17 and palladium nanoparticles deposited on graphene18 were proposed for determination of glucose without enzyme. Velarde et al.19 studied oxidation of D-glucose with hydrogen peroxide as oxidant over several titanium-containing NaY zeolites. It is important to develop a nonenzymatic sensor that has high sensitivity and stability, and is interference free, for the determination of glucose by the method of electrochemical oxidation.
Production of modified electrodes for electrocatalysis has been extensively developed and investigated by researchers.20–22 Modified electrodes provides an excellent way to accelerate charge transfer processes, decrease the over potentials as well as to increase the intensity of the corresponding voltammetric responses.20,23,24 Zeolites and nanozeolites are ordered porous crystalline materials with wide practical applications.25 They have high surface areas with strongly organized microporous channel systems which is an advantage compared to other classical support materials of interest for fuel cell technology.25 One of the most representative artificial zeolites, ZSM-5 zeolite, has been widely used in industry as catalysts, adsorbents, ion exchangers and zeolite membranes.26 Also, zeolites and nanozeolites have been so far utilized in zeolite modified electrodes (ZMEs) and applied in electrocatalysis reaction.27
In the recent years, multi-walled carbon nanotubes (MWCNTs) have received great attention as nanomaterials for the fabrication of electronic devices because of its surprising physical and electrical properties such as highly elastic modules, high tensile strength, good chemical stability, large surface-to-volume ratio, and high thermal and electrical conductivity.28,29 As an electrode material, MWCNTs provide a novel platform for fabricating chemical sensors, and can be utilized to help electron-transfer between the fabricated electrode and the electroactive species.30,31 The surprising electrochemical features of MWCNTs make them appropriate for usage in faradaic processes (e.g., the fast electron transfer kinetics of MWCNTs, due to the presence of edge plane graphite sites within the walls and at the ends of MWCNTs) or in non-faradaic processes such as the large changes in conductance due to the presence of the cloud of electrons surrounding their walls, can accept or withdraw charges from or to molecules in their near chemical environment.32
Nickel is a relatively abundant and low cost material that is utilized widely in numerous industrial applications. It is well established that Ni can be used as a catalyst based on its surface oxidation properties and has long-term stability in alkaline solutions.25,26 According to the literatures,33,34 the oxidation of glucose to glucolactone (two hydrogen are liberated in this process) can be catalyzed by the NiOOH/Ni(OH)2 redox couple at Ni electrode or modified electrode with nickel in the alkaline medium.
In respect of literature survey, no ZSM-5 nanozeolites and MWCNTs were employed for the modification of CPE for electrocatalytic oxidation of glucose. In this study, ZSM-5 nanozeolite was synthesized and characterized by different techniques. Then, this nanozeolite was utilized for modification of CPE and applied for electrocatalytic oxidation of glucose in the alkaline medium.
2. Experimental
2.1. Reagents and materials
Tetraethylorthosilicate (TEOS), sodium aluminate (NaAlO2), sodium hydroxide, glucose and NiCl2·6H2O were of analytical reagent grade and were purchased from Merck Company and used without further purification. Graphite powder and paraffin oil (density 0.88 g cm−3) as the binding agent (both from Daejung company) were used for preparing the pastes. Multi-walled carbon nanotubes (MWCNTs) more than 95 wt% purity was purchased from Iranian Nanomaterials Pioneers Company. Also double distilled water was used throughout the experiment.2.2. Preparation of organic template free ZSM-5 nanozeolite
ZSM-5 nanozeolite has been synthesized using tetraethylorthosilicate (TEOS) and sodium aluminate (NaAlO2) as a silica and aluminum sources, respectively. In a typical synthesis, sodium aluminate was dissolved in NaOH solution under stirring, followed by successive addition of double distilled water. Then, TEOS was added to the above mixture and stirred until a homogeneous gel was obtained. The gel was transferred into a Teflon lined stainless steel autoclave and heated at 180 °C for 24 h under static condition. After this procedure, the product was separated by centrifugation (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm), washed several times with double distilled water, dried overnight at 90 °C and calcinated at 550 °C for 5 h in air. The molar ratio of the above reactants was as follows: 1.0Al2O3:30SiO2:3.3Na2O:1350H2O.35
000 rpm), washed several times with double distilled water, dried overnight at 90 °C and calcinated at 550 °C for 5 h in air. The molar ratio of the above reactants was as follows: 1.0Al2O3:30SiO2:3.3Na2O:1350H2O.35
2.3. Apparatus and characterization
XRD pattern was recorded by X-ray diffractometer instrument (XRD, GBC-MMA) with Be filtered Cu Kα radiation (1.5418 Å) operating at 35.4 kV and 28 mA. The scanning range of 2θ was set between 5° and 50° with scan rate of 0.05 degree per second. The morphology of the synthesized zeolite was investigated with scanning electron microscopy (SEM, VEGA2-TESCAN) and transmission electron microscopy (TEM, JSM-1400 transmission electron microscope, JEOL Japan, with an accelerating voltage of 100 kV). The Bruker FT-IR spectrometer (Vector 22) was utilized for recording of Fourier transform infrared (FT-IR) spectrum at room temperature. The particle size distributions of synthesized zeolite was determined using laser particle sizer Analysette 22 NanoTec plus (Fritsch GmbH, Germany) caused by the solid material suspended in distilled water. Disintegration of the solid powders was supported by an ultrasound bath with incorporated stirrer. The electrochemical experiments were performed using a potentiostat/galvanostat (SAMA500 electroanalyzer system, Isfahan, Iran) with a voltammetry cell in a three electrodes configuration at 25 ± 1 °C. The Ag|AgCl|KCl (3 M) and platinum wire (Azar Electrode Co., Iran) were used as reference and auxiliary electrodes, respectively. The CPE, modified CPE with ZSM-5 nanozeolites and modified CPE with ZSM-5 nanozeolites and MWCNTs were used as the working electrode in the electrochemical experiments.2.4. Preparation of the working electrodes
Typically to prepare MW–ZSM-5/CPE, 0.05 g of ZSM-5 nanozeolite (25 wt%) and 0.02 g of MWCNTs (10 wt%) were mixed with 130 mg of graphite powder and then, paraffin oil was blended with the mixture in a mortar by hand mixing for 30 min until a uniformly wetted paste was obtained. This paste was packed into the end of a glass tube (ca. 0.35 cm i.d. and 10 cm long) and the copper wire was used for electrical contact. A new surface was achieved by pushing an excess of the paste out of the tube and polishing with a weighing paper. For comparison, unmodified CPE (bare CPE), MW/CPE and ZSM-5/CPE were also prepared in the same mentioned method.3. Results and discussion
3.1. Characterization of ZSM-5 nanozeolite
XRD powder pattern of synthesized ZSM-5 nanozeolite is presented in Fig. 1. The crystallization products matched the characteristic peaks of ZSM-5 nanozeolite at 2θ values of 7.9, 8.9, 23.2 and 24.5 degrees with the reference sample26,36 indicated that pure phase of ZSM-5 nanozeolite was prepared. It must be emphasized that when TEOS was used as the silicon source, it can be hydrolyzed and produce alcohol which was proved to have structure directing effect on the formation of ZSM-5 nanozeolite.35 The crystallite size (Dc) of synthesized ZSM-5 nanozeolite was also calculated using Debye–Scherrer equation:37
 | (1) |
 | ||
| Fig. 1 The representation XRD pattern of synthesized ZSM-5 nanozeolite. | ||
The SEM image of the crystalline phase provides useful approach to the determination of size and morphology of the obtained crystals. SEM image of synthesized ZSM-5 nanozeolite is illustrated in Fig. 2, which indicates the formation of spherical nanosized particle with diameter between 60–90 nm. Similar result was obtained from TEM image. Fig. 3 displays histograms of particle size distributions of synthesized ZSM-5 nanozeolite according to the Mie theory. The average particle sizes of ZSM-5 nanozeolites are between 45 and 130 nm and the mean particle size is about 87 nm, corresponding to cumulative volume frequency of 84%.
 | ||
| Fig. 2 The representation (a) SEM and (b) TEM images of synthesized ZSM-5 nanozeolite. | ||
 | ||
| Fig. 3 Particle size distribution of ZSM-5 nanozeolite. | ||
FTIR spectroscopy is a very useful technique for obtaining vibrational information about the species in materials. Fig. 4 illustrates FT-IR spectrum of as-synthesized ZSM-5 nanozeolite. The bands located at 1070–1230 cm−1 are characteristic of SiO4 tetrahedron units. The absorption bands at 1230 and 550 cm−1 supply information on the difference between ZSM-5 nanozeolite and other types of zeolites. The external asymmetric stretching vibration near 1230 cm−1 is due to the presence of structures containing four chains of four-member rings between SiO4 and AlO4 tetrahedral of ZSM-5 structure and is a structure sensitive IR band of ZSM-5 zeolite.38 The bands near 1100 cm−1 and 800 cm−1 are due to the internal asymmetric stretching and external symmetric stretching of external linkages, respectively. Also, the band at about 550 cm−1 is attributed to a structure-sensitive vibration caused by the double four-member rings of the external linkages and the band near 450 cm−1 is ascribed to the Si–O and Al–O bending vibration due to the formation of only ZSM-5 crystal.39 The broad bands at 2800–3700 cm−1 (centered at 3450 cm−1) and band at about 1630 cm−1 are attributed to the stretching and bending vibration of water molecules adsorbed on hydroxyl groups, respectively.26
 | ||
| Fig. 4 The FT-IR spectrum of synthesized ZSM-5 nanozeolite. | ||
3.2. Electrochemistry of fabricated electrodes
Potassium ferricyanide (K4Fe(CN)6) was selected as a probe to evaluate the performance of the fabricated bare CPE, MW/CPE, ZSM-5/CPE and MW–ZSM-5/CPE electrodes using CV technique. Fig. 5 illustrates the typical CVs of the electrochemical oxidation of K4Fe(CN)6 at the surface of these electrodes in the 10 mM of K4Fe(CN)6 and 0.1 M KCl solution. It is obvious that the electron transfer rate was slow, with a peak-to-peak separation (ΔEp) of 240 mV at the CPE indicating an irreversible electron transfer process. Meanwhile, the MW/CPE, ZSM-5/CPE and MW–ZSM-5/CPE displayed a well-shaped cyclic response for the Fe(CN)63−/Fe(CN)64− redox couple with a ΔEp of 130, 170 and 150 mV, respectively. These are a quasi-reversible system because ΔEp is greater than 59 mV that expected for a reversible system. As can be seen, the oxidation and reduction currents for the Fe(CN)63−/Fe(CN)64− redox couple is higher than that at the surface of MW/CPE and ZSM-5/CPE electrodes. Therefore, the fabricated MW–ZSM-5/CPE electrodes was selected for further experiments. The obtained result from CV of MW–ZSM-5/CPE in various buffered solutions with different pH value does not show any shift in the anodic peak potentials for oxidation of K4Fe(CN)6. It can be concluded that the electrochemical behavior of the Fe(CN)63−/Fe(CN)64− redox couple in the MW–ZSM-5/CPE electrode is not dependent on the solution pH.40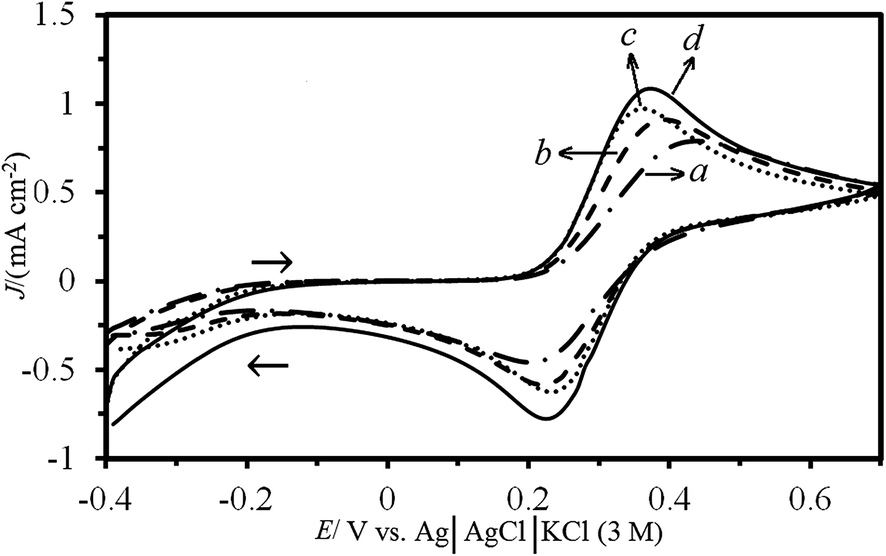 | ||
| Fig. 5 The cyclic voltammograms of (a) bare CPE, (b) ZSM-5/CPE, (c) MW/CPE and (d) MW–ZSM-5/CPE in the presence of 10 mM K4Fe(CN)6 solution at a scan rate of 20 mV s−1 and pH of 7.0 in 0.1 M KCl as supporting electrolyte before immersion of these electrode in 0.5 M NiCl2 solution. | ||
3.3. Electrochemical behavior of fabricated electrodes in alkaline solution
Inset (a) in Fig. 6 shows CVs of bare CPE and MW–ZSM-5/CPE in 0.1 M NaOH solution at the potential range from 0.2 to 0.7 V vs. Ag|AgCl|KCl (3 M) and potential sweep rate of 20 mV s−1. As can be seen, no current was obtained with these electrodes and the background current for MW–ZSM-5/CPE is larger than that at bare CPE due to high surface area. Also, Fig. S1 (in the ESI†) shows CVs of MW/CPE and ZSM-5/CPE in 0.1 M NaOH solution. No current was obtained with these electrodes and the background current for ZSM-5/CPE is larger than that at MW/CPE. The inclination of the base line for the voltammogram of ZSM-5/CPE electrode can be attributed to the additional resistance obtained from the presence of ZSM-5 nanozeolite and the oxidation–reduction process of oxygen species in the molecular sieve layer.41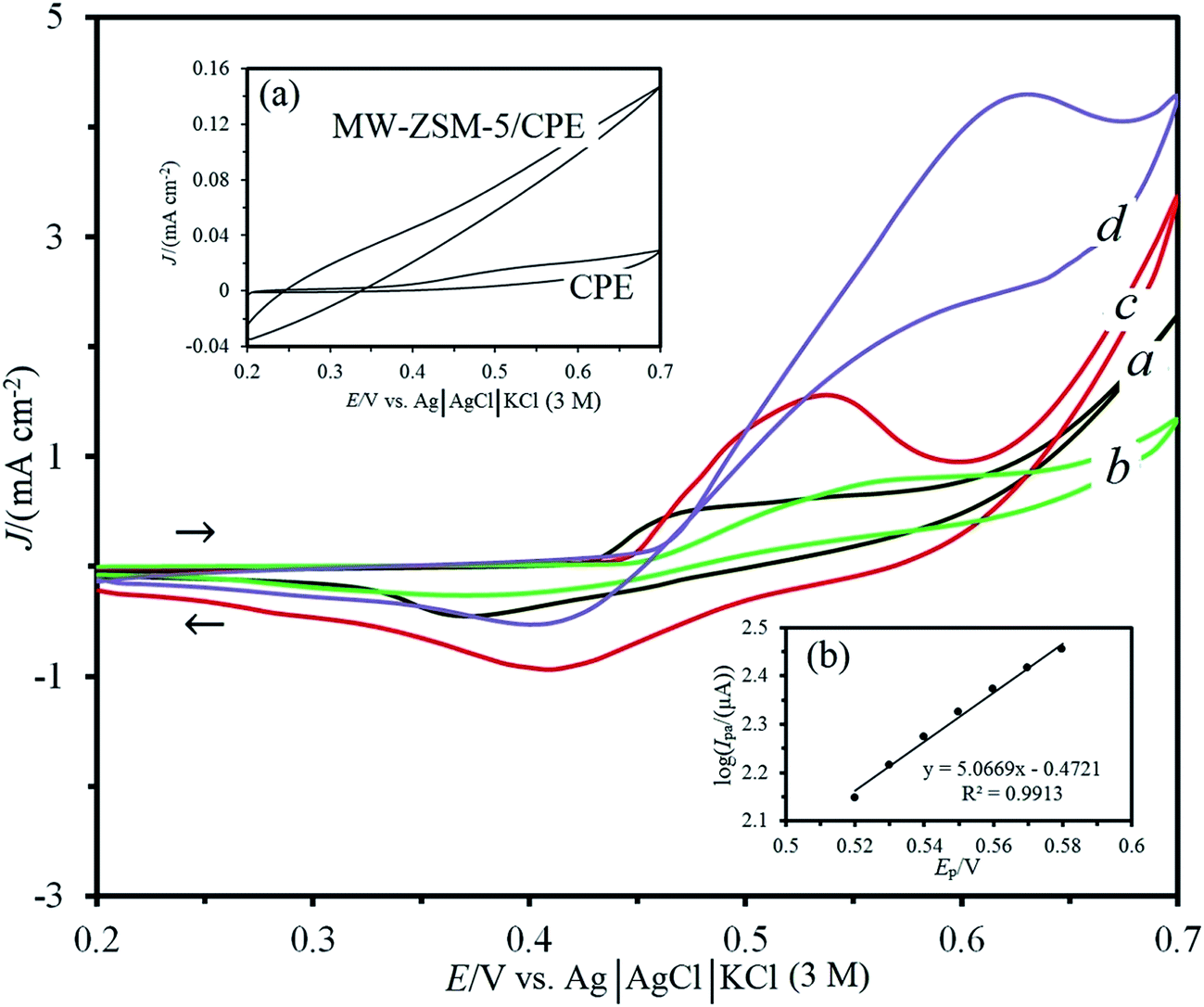 | ||
| Fig. 6 The cyclic voltammograms of the Ni/CPE and Ni–MW–ZSM-5/CPE in the absence (a and c) and presence (b and d) of 0.005 M glucose in 0.1 M NaOH at scan rate of 20 mV s−1, respectively. The inset (a) shows cyclic voltammograms of the bare CPE and MW–ZSM-5/CPE in 0.1 M NaOH before immersion in 0.5 M NiCl2 solution. The inset (b) displays Tafel plot derived from the rising part of the curve d. | ||
In order to incorporate Ni2+ ions at the surface of the CPE and MW–ZSM-5/CPE electrodes, these electrodes were immersed in a well-stirred aqueous solution of 0.5 M NiCl2 for 10 min at 150 rpm and then washed completely with distilled water to remove the surface adsorbed species. Fig. 6c demonstrates CV of Ni–MW–ZSM-5/CPE (MW–ZSM-5/CPE that immersed in the 0.5 M NiCl2) in 0.1 M NaOH solution and scan rate of 20 mV s−1. It can be inferred that the electrochemical behavior of Ni–MW–ZSM-5/CPE modified electrode in alkaline solution is similar to that of Ni anode.40,42,43 These redox waves are ascribed to the oxidation of Ni(OH)2 at interface of the MW–ZSM-5/electrolyte to NiOOH and reduction of NiOOH to Ni(OH)2 at scan rate of 20 mV s−1 with a peak potential of 0.54 and 0.41 V vs. Ag|AgCl|KCl (3 M), respectively.42,43 The broad cathodic peak can be ascribed to the phase transformation of β-NiOOH to γ-NiOOH due to slow, irreversible overcharging during cycling, and the corresponding reduction to α-Ni(OH)2. This phenomenon originally reported by Bode et al.44
After stabilization of nickel species on the surface of the electrodes, the anodic and cathodic peak currents for Ni–MW–ZSM-5/CPE were much greater than those at Ni/CPE (see Fig. 6a and c) and were greater than those at Ni–MW/CPE and Ni–ZSM-5/CPE (see curves a in Fig. S2 and S3†). It can be realized that the peak current of Ni(OH)2 oxidation at the surface of Ni–MW–ZSM-5/CPE is about 2.8, 2.3 and 1.5-fold greater than that at the surfaces of Ni/CPE, Ni–MW/CPE and Ni–ZSM-5/CPE, respectively. Also, the Ni/CPE, Ni–MW/CPE, Ni–ZSM-5/CPE and Ni–MW–ZSM-5/CPE displayed a well-shaped cyclic response for the Ni(OH)2/NiOOH redox couple with ΔEp of 150, 120, 140 and 130 mV, respectively. As can be seen in Fig. S2,† the presence of the MWCNTs in CPE could enhance the peak currents and decrease the oxidation potential by decreasing the overpotential. The advantages of Ni–MW/CPE had been explained by the good electrical conductivity, high chemical stability and high surface area and fast electron transfer rate of MWCNTs.45 Also, the peak current of Ni(OH)2 oxidation at the surface of Ni–ZSM-5/CPE is much greater than that at bare CPE (see curves a in Fig. S3† and 6). It can be expected that diffusion of Ni2+ ions in ZSM-5 nanozeolite is much faster owing to their coordination to the nanozeolite framework, the bigger cages and channels and fast migration of Ni2+ ions from the cages.23,46 These results confirmed that the presence of both MWCNTs and ZSM-5 on the fabrication of Ni–MW–ZSM-5/CPE had a great influence on improving the oxidation currents and decreasing the overpotential.
The electrocatalytic oxidation of the glucose was investigated at the surface of Ni/CPE and Ni–MW–ZSM-5/CPE in 0.1 M NaOH solution. Curves b and d in Fig. 6 display the CVs for electrocatalytic oxidation of the glucose in the surfaces of Ni/CPE and Ni–MW–ZSM-5/CPE electrodes in 0.1 M NaOH solution and 0.005 M glucose at the scan rate of 20 mV s−1, respectively. In the presence of glucose, an increase in current was observed at the surface of Ni–MW–ZSM-5/CPE but no significant variation in the current intensity was observed at the surface of Ni/CPE. For comparison, the Ni–MW/CPE and Ni–ZSM-5/CPE modified electrodes were utilized to 0.005 M glucose oxidation in 0.1 M NaOH solution at the scan rate of 20 mV s−1 (see curves b in Fig. S2 and S3†). The anodic current densities for electrooxidation of glucose at the surface of Ni–MW/CPE and Ni–ZSM-5/CPE were obtained to be 0.98 and 1.2 mA cm−2, respectively. The Ni–ZSM-5/CPE displayed a similar oxidation peak for glucose to the Ni–MW–ZSM-5/CPE, meanwhile the catalytic oxidation current for the Ni–ZSM-5/CPE modified electrode was much smaller than that at the surface of the Ni–MW–ZSM-5/CPE. For example, the anodic current densities at 0.005 M glucose for Ni–MW–ZSM-5/CPE and Ni–ZSM-5/CPE are 4.3 and 1.2 mA cm−2, respectively. This may be due to the unique structure and good electrical properties of MWCNTs, which could help to increase the electrical conductivity of catalysts materials.28,29 As a result, this improvement in the current densities demonstrates that incorporation of both MWCNTs and ZSM-5 onto a carbon paste electrode enhances the electrochemical signal of glucose oxidation.
It can be observed clearly in Fig. 6d that the oxidation of glucose gives rise to a typical electrocatalytic response, with an increase in the anodic peak current and together with a decrease in the cathodic peak current. The oxidation potential of glucose is observed at 0.63 V vs. Ag|AgCl|KCl (3 M) that is positive than the potential observed for Ni2+ to Ni3+ transition at the Ni–MW–ZSM-5/CPE in the absence of glucose (i.e. 0.54 V). This recommends an interaction between the glucose and the film redox sites confined at the electrode surface.
In order to obtain information about the rate determining step, a Tafel plot was illustrated for Ni–MW–ZSM-5/CPE using the data derived from the raising part of the current–voltage of curve d in Fig. 6. This part of voltammogram, known as Tafel region, is affected by electron transfer kinetics between the glucose and the modified electrode.47 Inset (b) in Fig. 6 illustrates the plot of logIp vs. Ep in the presence of 0.005 M glucose at scan rate of 20 mV s−1 in 0.1 M NaOH solution on the surface of Ni–MW–ZSM-5/CPE. The Tafel slope is equal to n(1 − α)F/2.303RT which comes up to 5.0669 V per decade, which indicates that transfer coefficient (α) for electrooxidation of glucose is about 0.85.
The electrocatalytic behavior of the modified electrodes with ZSM-5 nanozeolite ratio of 5, 10, 15, 20, 25 and 30 wt% together with 10 wt% of MWCNTs with respect to the graphite powder were studied by CV technique in 0.005 M glucose at 0.1 M of NaOH solution (see Fig. S4 in ESI†). It was experimentally determined that higher anodic current of glucose oxidation is observed when 25% of nanozeolite was used. It is suggested that with increasing the nanozeolite over than 25% in the modified electrode, the resistance of the modified electrode may increase due to low conductivity of ZSM-5 nanozeolite and a decrease in glucose oxidation current is distinguished. Also, at lower ratios of nanozeolite in Ni–MW–ZSM-5/CPE, the amount of available pores for Ni2+ insertion decrease and available NiOOH active site is reduced for glucose oxidation which ultimately cause a low current density.25,48 The amount of MWCNTs in Ni–MW–ZSM-5/CPE had a significant effect on the anodic oxidation current of glucose. Four different modified carbon paste electrodes (5, 7.5, 10 and 12.5 wt% of MWCNTs together with 25 wt% of ZSM-5 nanozeolite with respect to the graphite powder) were prepared and studied for their voltammetric signal under identical conditions in 0.005 M glucose at 0.1 M NaOH solution (see Fig. S5 in ESI†). The result showed that with increasing the amount of MWCNTs 10 wt%, the oxidation peak current for glucose was increased and then began to level off. Therefore, we selected these conditions (MWCNTs 10 wt% and ZSM-5 nanozeolite 25 wt% with respect to the graphite powder) for preparation of modified electrode.
3.4. Effect of scan rate on the NiOOH to Ni(OH)2 conversion
The electrochemical behavior of Ni–MW–ZSM-5/CPE was studied in 0.1 M NaOH at various scan rates. Fig. 7 displays the CVs of the Ni–MW–ZSM-5/CPE electrode in 0.1 M NaOH at various scan rates from 5 to 500 mV s−1. With increasing of scan rate, the anodic and cathodic currents were enhanced and the anodic peaks moved to more positive potentials and the cathodic peaks shifted to more negative potential values. This result indicates a limitation in the charge-transfer kinetics which is due to chemical interactions between the electrolyte ions and the modified electrode.49,50 This observation is possible according to a theory defined by Laviron for the linear potential sweep voltammetric response in the case of surface confined electroactive species at the small concentrations.51 The expressions for peak to peak separation of ΔEp > 0.2/n V, where n is the number of exchanged electrons, can be written as the following equations:
 | (2) |
 | (3) |
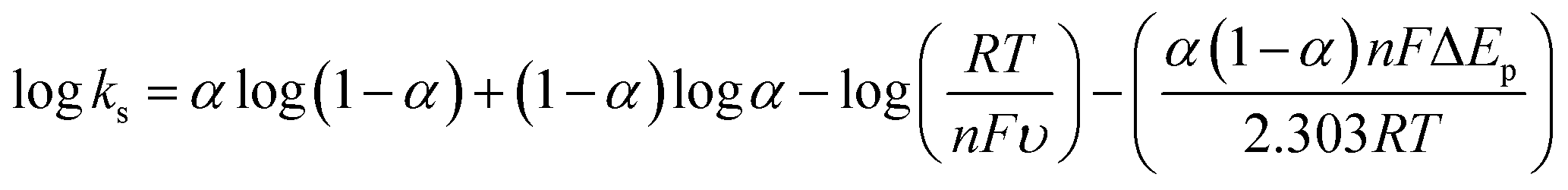 | (4) |
 | ||
| Fig. 7 The cyclic voltammograms of the Ni–MW–ZSM-5/CPE in 0.1 M NaOH at various scan rates from inner to outer: 0.005, 0.010, 0.015, 0.025, 0.040, 0.060, 0.080, 0.100, 0.150, 0.200, 0.250, 0.300, 0.350, 0.400, 0.450 and 0.500 V s−1. The inset (a) shows the plot of Ep vs. logυ for CVs at scan rates 0.100–0.500 for anodic peaks a and cathodic peaks b. The inset (b) shows the dependency of Ipa and Ipc on lower values of υ (0.005–0.080 V s−1) for anodic peaks a and cathodic peaks b. | ||
The inset (a) in Fig. 7 displays the plots of the variation of Ep with respect to the logarithm υ in the ranges of 0.100–0.500 V s−1 for both anodic and cathodic peaks in 0.1 M NaOH onto Ni–MW–ZSM-5/CPE. It can be seen that Ep is proportional to logυ at υ > 0.100 V s−1 as demonstrated by Laviron.51 From inset (a) in Fig. 7 and eqn (2) and (3), the value of anodic electron transfer coefficient (α) is found to be 0.65. This results show that the rate limiting steps for cathodic and anodic might not be the same step.48 According to the eqn (4), the mean value of charge-transfer rate constant (ks) is calculated to be 0.184 s−1.
The inset (b) in Fig. 7 shows the plots of anodic and cathodic peak currents for oxidation–reduction of the NiOOH/Ni(OH)2 redox couple versus scan rate on the surface of Ni–MW–ZSM-5/CPE at low values from 0.005 to 0.008 V s−1 in 0.1 M NaOH solution. This dependence is probably due to electrochemical activity of immobilized redox species at the surface of modified electrode. The electrode surface coverage (Γ*) can be calculated from the linear part of the plot and using the following equation which corresponds to reversible process with adsorbed species.47
 | (5) |
3.5. Electrocatalytic oxidation of glucose at the surface of Ni–MW–ZSM-5/CPE
As can be seen in Fig. 6c and d, the enhanced anodic peak was appeared around 0.63 V vs. Ag/AgCl/KCl (3 M); meanwhile, cathodic peak current around 0.41 V was decreased after addition of glucose. It can be deduced that the over potential for glucose electrooxidation was decreased in the surface of modified electrode. Also, the anodic current for glucose oxidation is higher than that for Ni(OH)2/NiOOH conversion in the same scan rate. It can be concluded that the applied modifier in this process contributed directly to the electrocatalytic oxidation of glucose with electrochemical catalytic (EC′) mechanism. According to the literature,33 the oxidation of glucose to gluconolactone (two hydrogen are liberated in this process) can be catalyzed by the NiOOH/Ni(OH)2 redox couple in the alkaline medium. When glucose diffuses from the bulk solution to the surface of the electrode, it is quickly oxidized to gluconolactone by the NiOOH species on the surface of modified electrode. Therefore, the amount of NiOOH species decreases due to its chemical reaction with glucose. Simply, the electrocatalytic oxidation mechanism of glucose at Ni–MW–ZSM-5/CPE can be described by the following equations that is the basis for the fabrication of a nonenzymatic sensor for electrochemical detection of glucose:| [(Ni(OH)2–MW–ZSM-5/CPE)] + OH− ⇄ [(NiOOH–MW–ZSM-5/CPE)] + H2O + e− (E) |
| [(NiOOH–MW–ZSM-5/CPE)] + glucose → [(Ni(OH)2–MW–ZSM-5/CPE)] + glucolactone (C′) |
3.6. Effect of scan rate on the oxidation of glucose
The dependence of the glucose electrocatalytic oxidation current on the scan rate of the potential (υ) under the optimal conditions was investigated in the range of 10–300 mV s−1 at the surface of Ni–MW–ZSM-5/CPE. Fig. 8a displays CVs of Ni–MW–ZSM-5/CPE in the presence of 0.005 M glucose in 0.1 M NaOH. The anodic peak currents (Ipa) increased and the anodic peak potentials shifted to more positive directions by increasing the scan rate. It can be proposed that a kinetic limitation was occurred in the reaction between the redox sites of the glucose and Ni–MW–ZSM-5/CPE at high scan rate.49,50 The increase in the peak current with the υ can be considered for adsorption or diffusion control of the process. Fig. 8b shows the plot of anodic current density (Jpa) versus square root of scan rate (υ1/2) that obtained from CVs of Ni–MW–ZSM-5/CPE in 0.005 M glucose and 0.1 M NaOH solution. As can be observed, the plot of Jpa versus υ1/2 was found to be linear with equation of Jpa (μA) = 0.4542υ1/2 (mV s−1)1/2 + 2.0091 and R2 = 0.9976, meanwhile, the plot of Jpa against υ did not show a linear curve (see Fig. S6 in ESI†). From this observation, it can be realized that this process is diffusion-controlled process rather than surface-controlled process.47,48 | ||
| Fig. 8 (a) The cyclic voltammograms of the Ni–MW–ZSM-5/CPE in the presence of 0.005 M glucose in 0.1 M NaOH at various scan rates from inner to outer: 0.010, 0.025, 0.050, 0.075, 0.100, 0.150, 0.200 and 0.300 V s−1. (b) Variation of Jpa vs. υ1/2. (c) The plot of logIpa vs. logυ and (d) the plot of Ipa/υ1/2 vs. υ. | ||
From the theoretical point of view, a slope of 0.5 or 1.0 is expected for the plot of logIpa vs. logυ under diffusion- or adsorption-controlled process, respectively.42 A linear dependence is observed between logIpa and logυ at the surface of Ni–MW–ZSM-5/CPE in the glucose oxidation (see Fig. 8c). From linear section, the slope of 0.3099 is found close to the theoretically predicted value of 0.5 for a purely diffusion-controlled current. However, the contribution of a kinetic limitation to the overall process cause to the small alteration with theoretical value.48,52 Fig. 8d displays a plot of scan rate normalized current (Ipa/υ1/2) vs. logarithm scan rate (logυ). The characteristic shape of an electrochemical catalytic (EC′) mechanism is detected which express that the electrode reaction is coupled with an irreversible follow up chemical step.53 Also, at higher potential scan rates, the cathodic peak current relating to Ni(III) reduction to Ni(II) appears which confirms EC′ mechanism.50
3.7. Chronoamperometric studies
The electrocatalytic oxidation of the glucose at the surface of modified electrode was also considered by chronoamperometry method. Fig. 9 illustrates chronoamperometric measurements for glucose at the Ni–MW–ZSM-5/CPE. It shows the current–time profiles obtained at a working electrode potential of 650 mV vs. Ag|AgCl|KCl (3 M) for various concentrations of glucose (0, 0.147, 0.476 and 1.67 mM). It was found that the observed current from chronoamperograms was in good agreement with the detected current from cyclic voltammetry experiment and the current increases as the glucose concentration increases (curves b–d). This result supports our conclusion about the catalytic role of NiOOH for oxidation of glucose that glucose oxidation starts directly after the formation of the first amount of NiOOH on the surface of electrode.54 | ||
| Fig. 9 Main panel: chronoamperograms obtained at Ni–MW–ZSM-5/CPE in 0.1 M NaOH solution in the a absence and presence of b 0.147, c 0.476 and d 1.67 mM glucose, (the potential step was 0.65 V vs. Ag/AgCl/KCl (3 M)). The inset (a) displays plot of I vs. t−1/2, derived from the data of chronoamperogram b. The inset (b) illustrates plot of the slope of the straight lines against glucose concentration. The inset (c) shows plots of Icat/IL vs. t1/2, obtained from chronoamperograms b–d. | ||
An exponential behavior of achieved I–t curves shows that a diffusion-controlled process has occurred according to Cottrell equation. From the chronoamperometric study, the diffusion coefficient of glucose, D, was determined in aqueous solution by using Cottrell equation as below:55
| I = nFACD1/2π−1/2t−1/2 | (6) |
485 C mol−1 and A = 0.096 cm2).56
Chronoamperometry can be applied for the evaluation of the catalytic rate constant (kcat) of the electrocatalytic oxidation of glucose on the active sites of the modified electrode according by the following equation:49
 | (7) |
3.8. Effects of glucose concentration and calibration curves
Fig. 10 illustrates the effect of glucose concentration on its electrooxidation current onto Ni–MW–ZSM-5/CPE at scan rate of 25 mV s−1. It is clearly observed that the anodic peak current increased with increasing of glucose up to the concentration of 6.1 mM (see Fig. 10a and b). In the concentrations above 6.1 mM, no remarkable increase in the anodic peak current was observed. It can be specified that this effect may be due to the saturation of active sites and/or poisoning the electrode surface with adsorbed intermediates. Thus, 6.1 mM of glucose represented the optimum concentration after which the adsorption of the oxidation products at the surface of electrode may cause the stoppage of further oxidation. The dependence of the anodic peak current on the glucose concentration is shown in the Fig. 10c. As can be seen, the plot of peak current versus glucose concentration consists of two linear regions with different slopes, corresponding to two different substrate concentration ranges. The decrease in sensitivity (slope) in the second linear range is caused by kinetic limitations.58 From analysis of these data, we expected that the detection limit (S/N = 3) in the lower linear range was calculated to be 0.14 mM. | ||
| Fig. 10 The cyclic voltammograms of the Ni–MW–ZSM-5/CPE in 0.1 M NaOH solution with various concentrations of glucose: (a) 0.0–3.4 mM and (b) 3.8–6.1 mM at scan rate of 20 mV s−1. (c) The plot of electrocatalytic current density (J) vs. glucose concentration. | ||
Since differential pulsed voltammetry (DPV) has a much higher current sensitivity and better resolution than CV, DPV was investigated under the optimized conditions for determination of glucose. Fig. 11 displays typical DPVs with increasing glucose concentration leading to a linear increase in the peak current. Results indicated that this sensor gave a good linear response to the glucose concentration with two glucose concentration ranges: 1 × 10−4 to 1 × 10−3 mM which could be fitted to the regression equation of Jp = 1194.9Cglucose + 3.4944 (R2 = 0.9975, Cglucose is in mM), and 1 × 10−3 to 1 × 10−2 mM which could be fitted to the regression equation of Jp = 126.49Cglucose + 4.593 (R2 = 0.9979, Cglucose is in mM). The detection limit (S/N = 3) in the lower linear range was calculated to be 3.5 × 10−5 mM which may be attributed to the high efficiency of the electron transfer mediator MWCNTs and ZSM-5 nanozeolites. This value is much lower than those reported in enzymeless glucose sensor in the literature (see Table 2) and CV method in this work.
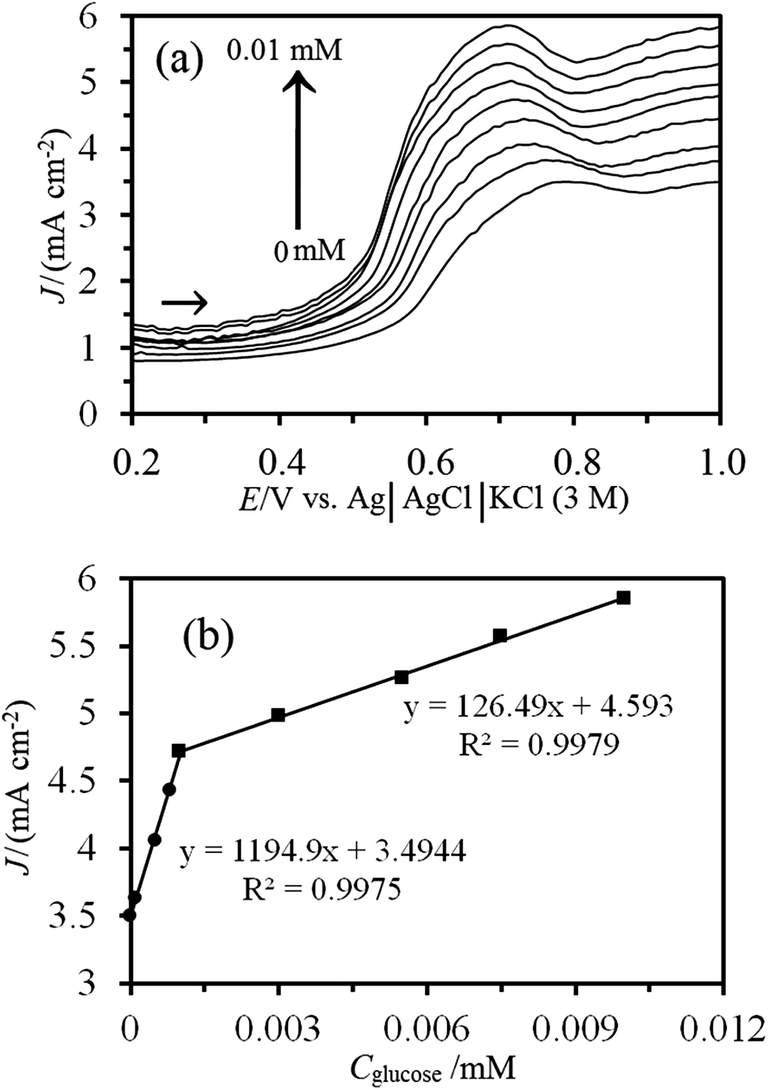 | ||
| Fig. 11 (a) Differential pulse voltammograms of Ni–MW–ZSM-5/CPE in 0.1 M NaOH solution containing different concentrations of glucose from inner to outer: 0, 0.0001, 0.0005, 0.0008, 0.001, 0.003, 0.0055, 0.0075 and 0.01 mM (the scan rate: 50 mV s−1, step height: 5 mV, step width: 1 s, pulse width: 200 ms, pulse height: 50 mV, and sample period: 50 ms). (b) The plot of electrocatalytic current density (J) as a function of glucose concentrations. | ||
| Glucose sensors | Applied potential (V) vs. Ag/AgCl/KCl (3 M) | Sensitivity (μA mM−1 cm−2) | Linear range | LOD (μM) | Ref. |
|---|---|---|---|---|---|
| Cu–Co–Ni/carbon nanofibers/GCE | 0.55 | 104.68 | 10 μM to 4.30 mM | 3.05 | 14 |
| Ni(II)–Qu–MWCNT–IL–PE | 0.48 | 36.9 (μA mM−1) | 5 μM to 2.8 mM | 1.0 | 15 |
| Ni/Al-LDH/chitosan/GCE | 0.48 | 0.182 (μA mM−1) | 10 μM to 10 mM | 10.0 | 24 |
| Cu-based metal–organic framework | 0.59 | — | 0.2–8.0 mM | 29.8 | 51 |
| Cu nanocluster/MWCNT/GCE | 0.65 | 251.38 | 0.7–3.5 mM | 0.21 | 54 |
| Dimethylglycoxime/Cu (DMG–CuNPs) | 0.69 | 700 | 1 μM to 5.0 mM | 0.5 | 55 |
| CuO/modified Cu electrode | 0.50 | 2792.64 | 0.8 μM to 2.2 mM | 0.8 | 56 |
| Ni/carbon nanofiber paste electrode | 0.60 | 420.4 | 2 μM to 2.5 mM | 1.0 | 57 |
| Ordered mesoporous CPE | 0.45 | 10.81 | 0.5–2.5 mM | 20.0 | 58 |
| 2.5–5 mM | |||||
| CuO nanorods/graphite | 0.60 | 371.43 | 4 μM to 8 mM | 4.0 | 59 |
| Nanoscale Ni(OH)2 modified CILE | 0.55 | 202 | 50 μM to 23 mM | 6.0 | 60 |
| Ni–MW–ZSM-5/CPE | 0.63 | 510.3 | 0.5–6.1 mM | 140 | This work |
| 1194.9 | 0.1–10 μM | 0.035 |
3.9. Reproducibility and stability of the Ni–MW–ZSM-5/CPE
The reproducibility and stability of the fabricated electrode were evaluated via the comparison of the currents of different electrodes using CV technique. The anodic current of five modified Ni–MW–ZSM-5/CPE electrodes to 0.005 M glucose was tested independently, and the relative standard deviation (RSD) was 3.26%. A reproducible current response with a RSD of 2.62% was observed for 5 successive assays of 0.005 M glucose. The long-term stability was explored by measuring a glucose solution intermittently, and the electrode was stored at room temperature when it is not in use. After 1 and 3 months, the electrode response to electrocatalytic oxidation of glucose retains 93% and 87% of initial value, respectively.Comparisons of fabricated sensor with other nonenzymatic glucose sensor are done and results are listed in Table 2. As can be seen in this table, the proposed glucose sensor shows good sensing performance in terms of LOD and sensitivity. In comparison with some previously reported works,10,14,16,17,34,56,59–63 it seems that Ni–MW–ZSM-5/CPE can act as an efficient electrocatalyst in glucose oxidation process. It must be noted that the sensitivity of the fabricated electrode is higher than that of previous works (see Table 2).
3.10. Interferences and blood serum sample measurement
Because the electrochemical oxidation of NiOOH to glucose has no special selective response, the effects of some interfering substances on the glucose biosensor were investigated in 0.1 M NaOH solution. The interference compounds were dopamine (DA), ascorbic acid (AA) and urea acid (UA) in view of a possible application of the developed biosensor to the analysis of glucose in human serum. Since the normal physiological level of glucose in the human blood is about 30 times of AA, DA and UA,64 the interference effect of 20 μM AA, DA and UA were investigated on the CV response of 600 μM of glucose. At this level, the current response for such electroactive interfering species to that of glucose by the sensor was below 5%. Therefore, good selectivity for glucose can be achieved with the fabricated sensor.In order to observe whether there is no matrix effect in blood, the fabricated sensor was used to determine glucose in blood serum samples with CV method. When considering the normal and clinical range of blood sugar (4–6 mM) in human blood serum, the analytical applicability of the Ni–MW–ZSM-5/CPE biosensor for analysis of glucose in human blood developed in this work proves promising. Therefore, the Ni–MW–ZSM-5/CPE shows a sufficient selectivity to be used for determination of glucose at high or low levels in blood.
4. Conclusions
In the present study, an organic template-free method was applied in the synthesis of ZSM-5 nanozeolite and the role of this nanozeolite and MWCNTs in the electrocatalytic process of glucose oxidation was investigated with fabrication of MW–ZSM-5/CPE electrode. This process was investigated using cyclic voltammetry, chronoamperometry and DPV techniques. It was seen that in the presence of glucose, the anodic oxidation current increases while the cathodic current decreases at the surface of Ni–MW–ZSM-5/CPE. The fabricated Ni–MW–ZSM-5/CPE electrode has large electrochemical surface area, and exhibits superior electrocatalytic performance for oxidation of glucose with decreasing over potential vs. bare CPE and shows good electrocatalytic activity toward glucose compared to many of the previously reported electrodes. The CV and DPV methods used for the determination of glucose exhibited good sensitivity, selectivity and reproducibility. This new non-noble catalyst has some advantages such as low cost and stability, ease of preparation and regeneration, stable response and very low ohmic resistance.References
- L. Liu, Q. Ma, Y. Li, Z. P. Liu and X. Su, Biosens. Bioelectron., 2015, 63, 519–524 CrossRef CAS PubMed.
- C. Barrera, I. Zhukov, E. Villagra, F. Bedioui, M. A. Paez, J. Costamagna and J. H. Zagal, J. Electroanal. Chem., 2006, 589, 212–218 CrossRef CAS.
- L. C. Clark and J. C. Lyons, Ann. N. Y. Acad. Sci., 1962, 102, 29–45 CrossRef PubMed.
- A. T. E. Vilian and S.-M. Chen, RSC Adv., 2014, 4, 50771–50781 RSC.
- J. Wang, D. Li, M. Yang and Y. Zhang, Anal. Methods, 2014, 6, 7161–7165 RSC.
- S. Palanisamy, C. Karuppiah and S. M. Chen, Colloids Surf., B, 2014, 114, 164–169 CrossRef CAS PubMed.
- S. Park, H. Boo and T. D. Chung, Anal. Chim. Acta, 2006, 556, 46–57 CrossRef CAS.
- G. Chang, H. Shu, K. Ji, M. Oyama, X. Liu and Y. He, Appl. Surf. Sci., 2014, 288, 524–529 CrossRef CAS.
- L. Tian and B. Liu, Appl. Surf. Sci., 2013, 283, 947–953 CrossRef CAS.
- T. Soejima, K. Takada and S. Ito, Appl. Surf. Sci., 2013, 277, 192–200 CrossRef CAS.
- Y. Zhang, E. Zhou, Y. Li and X. He, Anal. Methods, 2015, 7, 2360–2366 RSC.
- J. Chen, C. X. Zhao, M. M. Zhi, K. Wang, L. Deng and G. Xu, Electrochim. Acta, 2012, 66, 133–138 CrossRef CAS.
- R. Galindo, S. Gutierrez, N. Menendez and P. Herrasti, J. Alloys Compd., 2014, 586, S511–S515 CrossRef CAS.
- H. Liu, X. Lu, D. Xiao, M. Zhou, D. Xu, L. Sunb and Y. Song, Anal. Methods, 2013, 5, 6360–6367 RSC.
- X. Niu, H. Zhao, M. Lan and L. Zhou, Electrochim. Acta, 2015, 151, 326–331 CrossRef CAS.
- L. Zheng, J. Q. Zhang and J. F. Song, Electrochim. Acta, 2009, 54, 4559–4565 CrossRef CAS.
- S. Donmez, F. Arslan, N. Sarı, N. K. Yetim and H. Arslan, Biosens. Bioelectron., 2014, 54, 146–150 CrossRef CAS PubMed.
- E. Mijowska, M. Onyszko, K. Urbas, M. Aleksandrzak, X. Shi, D. Moszynski, K. Penkala, J. Podolski and M. E. Fray, Appl. Surf. Sci., 2015, 355, 587–592 CrossRef CAS.
- A. M. Velarde, P. Bartl, T. E. W. Nieen and W. F. Hoelderich, J. Mol. Catal. A: Chem., 2000, 157, 225–236 CrossRef CAS.
- A. Walcarius, Chem. Soc. Rev., 2013, 42, 4098–4140 RSC.
- A. Walcarius, C. R. Chim., 2005, 8, 693–712 CrossRef CAS.
- N. Atar, T. Eren, M. L. Yola, H. Karimi-Maleh and B. Demirdogena, RSC Adv., 2015, 5, 26402–26409 RSC.
- A. Samadi-Maybodi, S. K. Hassani Nejad-Darzi, M. R. Ganjali and H. Ilkhani, J. Solid State Electrochem., 2013, 17, 2043–2048 CrossRef CAS.
- H. Karimi-Maleh, P. Biparva and M. Hatami, Biosens. Bioelectron., 2013, 48, 270–275 CrossRef CAS PubMed.
- S. K. Hassaninejad-Darzi and M. Rahimnejad, J. Iran. Chem. Soc., 2014, 11, 1047–1056 CrossRef CAS.
- S. K. Hassaninejad-Darzi, A. Samadi-Maybodi and M. Ghobakhluo, J. Porous Mater., 2013, 20, 909–916 CrossRef CAS.
- B. Yuan, C. Xu, L. Liu, Q. Zhang, S. Ji, L. Pi, D. Zhang and Q. Huo, Electrochim. Acta, 2013, 104, 78–83 CrossRef CAS.
- C. Niu, E. K. Sichel, R. Hoch, D. Moy and H. Tennent, Appl. Phys. Lett., 1997, 70, 1480–1482 CrossRef CAS.
- Y. Zhang, E. Zhou, Y. Li and X. He, Anal. Methods, 2015, 7, 2360–2366 RSC.
- A. T. E. Vilian and S.-M. Chen, RSC Adv., 2014, 4, 50771–50781 RSC.
- J. Wang, Electroanalysis, 2005, 17, 7–14 CrossRef CAS.
- C. E. Banks and R. G. Compton, Analyst, 2006, 131, 15–21 RSC.
- L. M. Lu, L. Zhang, F. L. Qu, H. X. Lua, X. B. Zhang, Z. S. Wu, S. Y. Huan, Q. A. Wang, G. L. Shen and R. Q. Yu, Biosens. Bioelectron., 2009, 25, 218–223 CrossRef CAS PubMed.
- H. Ai, X. Huang, Z. Zhu, J. Liu, Q. Chi, Y. Li, Z. Li and X. Ji, Biosens. Bioelectron., 2008, 24, 1048–1052 CrossRef CAS PubMed.
- L. Zhang, S. Liu, S. Xie and L. Xu, Microporous Mesoporous Mater., 2012, 147, 117–126 CrossRef.
- A. Gurses, C. Dogar, M. Yalc, M. Akyldz, R. Bayrak and S. Karaca, J. Hazard. Mater., 2006, 131, 217–228 CrossRef CAS PubMed.
- H. P. Klug and L. E. Alexander, X-ray Diffraction Procedures, 2nd edn, Wiley, New York, 1964 Search PubMed.
- R. M. Mohamed, H. M. Aly, M. F. El-Shahat and I. A. Ibrahim, Microporous Mesoporous Mater., 2005, 79, 7–12 CrossRef CAS.
- Y. Li and J. N. Armor, Appl. Catal., B, 1992, 1, L31–L40 CrossRef CAS.
- S. K. Hassaninejad-Darzi, J. Electroceram., 2014, 33, 252–263 CrossRef CAS.
- A. A. El-Shafei, A. M. A. Elhafeez and H. A. Mostafa, J. Solid State Electrochem., 2010, 14, 185–190 CrossRef CAS.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem., 1971, 31, 39–49 CrossRef CAS.
- K. Nagashree and M. Ahmed, J. Solid State Electrochem., 2010, 14, 2307–2320 CrossRef CAS.
- H. Bode, K. Dehmelt and J. Witte, Electrochim. Acta, 1966, 11, 1079–1087 CrossRef CAS.
- S. Salmanpour, T. Tavana, A. Pahlavan, M. A. Khalilzadeh, A. A. Ensafi, H. Karimi-Maleh, H. Beitollahi, E. Kowsari and D. Zareyee, Mater. Sci. Eng., C, 2012, 32, 1912–1918 CrossRef CAS.
- J. W. Li and G. Calzaferri, J. Electroanal. Chem., 1994, 377, 163–175 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 2001 Search PubMed.
- S. N. Azizi, S. Ghasemi and H. Yazdani-Sheldarrei, Int. J. Hydrogen Energy, 2013, 38, 12774–12785 CrossRef CAS.
- M. Vilas-Boas, C. Freire, B. de Castro and A. R. Hillman, J. Phys. Chem. B, 1998, 102, 8533–8540 CrossRef CAS.
- R. Ojani, J. B. Raoof and S. Zamani, Bioelectrochemistry, 2012, 85, 44–49 CrossRef CAS PubMed.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- A. Velazquez-Palenzuela, F. Centellas, J. A. Garrido, C. Arias, R. M. Rodriguez, E. Brillas and P. L. Cabot, J. Power Sources, 2011, 196, 3503–3512 CrossRef CAS.
- D. K. J. Gosser, Cyclic voltammetry-simulation and analysis of reaction mechanism, Wiley, New York, 1993 Search PubMed.
- M. M. Dimos and G. J. Blanchard, J. Phys. Chem. C, 2010, 114, 6019–6026 CAS.
- R. Greef, R. Peat, L. M. Peter, D. Pletcher and J. Robinson, Instrumental methods in electrochemistry, Ellis Horwood, Chichster, 1990 Search PubMed.
- C. Wei, X. Li, F. Xu, H. Tan, Z. Li, L. Sun and Y. Song, Anal. Methods, 2014, 6, 1550–1557 RSC.
- R. Ojani, J. B. Raoof and S. Fathi, Electroanalysis, 2008, 20, 1825–1830 CrossRef CAS.
- M. Mazloum-Ardakani, M. Abolhasani, B. F. Mirjalili, M. A. Sheikh-Mohseni, A. Dehghani-Firouzabadi and A. Khoshroo, Chin. J. Catal., 2014, 35, 201–209 CrossRef CAS.
- X. Kang, Z. Mai, X. Zou, P. Cai and J. Mo, Anal. Biochem., 2007, 363, 143–150 CrossRef CAS PubMed.
- Q. Xu, Y. Zhao, J. Z. Xu and J. J. Zhu, Sens. Actuators, B, 2006, 114, 379–386 CrossRef CAS.
- J. C. Ndamanisha and L. Guo, Bioelectrochemistry, 2009, 77, 60–63 CrossRef CAS PubMed.
- X. Wang, C. Hua, H. Liu, G. Du, X. He and Y. Xi, Sens. Actuators, B, 2010, 144, 220–225 CrossRef CAS.
- A. Safavi, N. Maleki and E. Farjami, Biosens. Bioelectron., 2009, 24, 1655–1660 CrossRef CAS PubMed.
- J. Chen, W. Zhang and J. Ye, Electrochem. Commun., 2008, 10, 1268–1271 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20622g |
| This journal is © The Royal Society of Chemistry 2015 |