
Stereoselective synthesis of alkyl styryl selenides in one-pot: a straightforward approach by in situ dialkyl diselenide formation under transition metal-free conditions†
Abstract
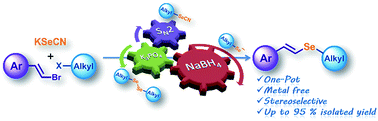
A simple procedure for the synthesis of alkyl styryl selenides was developed. This methodology involves the in situ generation of selenolate anions by a one-pot and three-step protocol where a nucleophilic substitution between KSeCN and alkyl halides affords the corresponding alkyl selenocyanate in the first step, giving dialkyl diselenide by a further K3PO4-assisted reaction. Subsequent reduction yields the alkyl selenolate anions which react with substituted (E,Z)-β-styryl halides to afford the corresponding vinyl selenides with retention of stereochemistry and from moderate to excellent yields. This procedure does not require a catalyst, proceeds under an air atmosphere and within a short reaction time, and is free from diselenide and malodorous and air-sensitive alkyl selenols as starting materials.
 Please wait while we load your content...
Please wait while we load your content...

