
A modification of a conventional technique for the synthesis of hydrazones of racemic carbonyls: prevention of spontaneous chiral inversion
Abstract
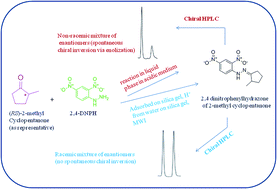
Conventionally hydrazones and other derivatives of carbonyls are synthesized under acidic conditions when spontaneous chiral inversion is a common problem if the carbonyl compound is chiral. A new method has been developed involving solid phase microwave-assisted conditions for the synthesis of 2,4-dinitrophenyl hydrazone(s) of chiral carbonyl compounds wherein there occurred no inversion of configuration. The method provided high yields (91–95%) in short reaction times (4–6 min). The method proposed clearly has synthetic advantages over current practices. The hydrazones were characterized by IR, 1H NMR and CHN analysis. The hydrazones represent enantiomeric pairs tagged with a strong chromophore rather than diastereomers. The enantiomeric pairs were separated by HPLC using an α1-acid glycoprotein column and the best resolution of all the analytes was achieved with a mobile phase containing 0.5% 2-propanol in 10 mM citrate phosphate buffer at pH 6.5. The chromatographic peaks clearly showed base line separation with comparable peak areas and thus the results confirmed that there was no spontaneous inversion of configuration during derivatization. The chromatograms corresponding to the products obtained by conventional procedure (from the racemic mixtures of analytes) showed peaks with unequal areas suggesting formation of enantiomers in unequal amounts (i.e., non racemic mixtures) because of spontaneous inversion of the configuration during derivatization.
 Please wait while we load your content...
Please wait while we load your content...

