
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(ethylene oxide)-based block copolymers with very high molecular weights for biomimetic calcium phosphate mineralization†
Tobias Maia,
Susanne Boyeb,
Jiayin Yuanc,
Antje Völkelc,
Marlies Gräwertc,
Christina Günterd,
Albena Ledererb and
Andreas Taubert*a
aInstitute of Chemistry, University of Potsdam, D-14476 Potsdam, Germany. E-mail: ataubert@uni-potsdam.de; Tel: +49 331 977 5773
bLeibniz Institut für Polymerforschung Dresden e.V., D-01069 Dresden, Germany
cMax Planck Institute of Colloids and Interfaces, D-14476 Potsdam, Germany
dInstitute of Earth and Environmental Sciences, University of Potsdam, D-14476 Potsdam, Germany
First published on 13th November 2015
Abstract
The present article is among the first reports on the effects of poly(ampholyte)s and poly(betaine)s on the biomimetic formation of calcium phosphate. We have synthesized a series of di- and triblock copolymers based on a non-ionic poly(ethylene oxide) block and several charged methacrylate monomers, 2-(trimethylammonium)ethyl methacrylate chloride, 2-((3-cyanopropyl)-dimethylammonium)ethyl methacrylate chloride, 3-sulfopropyl methacrylate potassium salt, and [2-(methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide. The resulting copolymers are either positively charged, ampholytic, or betaine block copolymers. All the polymers have very high molecular weights of over 106 g mol−1. All polymers are water-soluble and show a strong effect on the precipitation and dissolution of calcium phosphate. The strongest effects are observed with triblock copolymers based on a large poly(ethylene oxide) middle block (nominal Mn = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1). Surprisingly, the data show that there is a need for positive charges in the polymers to exert tight control over mineralization and dissolution, but that the exact position of the charge in the polymer is of minor importance for both calcium phosphate precipitation and dissolution.
000 g mol−1). Surprisingly, the data show that there is a need for positive charges in the polymers to exert tight control over mineralization and dissolution, but that the exact position of the charge in the polymer is of minor importance for both calcium phosphate precipitation and dissolution.
Introduction
Human enamel predominantly consists of calcium-deficient carbonated hydroxyapatite (HAP).1 Enamel is a robust material that is excellently adapted to the stresses exerted on teeth over the course of an animal's lifetime. Nonetheless, daily stresses like mechanical impact on chewing, biological (e.g. caries), or chemical attacks (e.g. erosion) will damage the enamel irretrievably. Unfortunately however, biological repair mechanisms of teeth are, in contrast to bone, virtually non-existent.2 There are only different exogenous strategies to minimize these everyday stresses and the resulting damage such as (i) adjustment of nutrition to minimize acid intake, which will lead to reduced chemical attack of the enamel surface3–6 (ii) improving the resistance of the enamel by (chemical) surface modification,3,7–13 (iii) counteracting the attacks of caries-inducing microorganisms, which will lead to a reduced biology-induced damage,14–20 or (iv) repair the damaged enamel by remineralization14–16,21 or other technologies.22,23There are several chemical strategies to stabilize the enamel. For example, the addition of fluoride7,13 to toothpastes or mouthwash facilitates the formation of resistant fluoride-substituted hydroxyapatite or fluorapatite (FAP) in the outermost enamel section. Alternatively, fluoride-containing tin compounds8–12 provide both a chemical stabilization via the fluoride and an antibacterial activity by way of the tin, which is located at the enamel surface and released over time. Other strategies to reduce biologically induced damage (i.e. caries) include the incorporation of antibacterial additives such as chlorhexidine14,20 or silver15,18 into dental care products and their regular application in dental hygiene. A further possibility to reduce adverse effects is to interrupt biofilm formation24 before caries bacteria can settle on the enamel.25,26
A rather new development is the interest in chemical strategies for remineralization of damaged enamel and dentin. Generally, these strategies involve the use of a synthetic material for filling existing defects in enamel and dentin. For example, HAP or amorphous calcium phosphate (ACP) nanoparticles provide some stabilization.14–16,21 Moreover, some of these systems combine remineralizing and antibacterial activities, for example by combination of calcium phosphate with silver species15 or chlorhexidine.14–16 One key issue here is that for a good functionality, the calcium phosphates must have a uniform size, shape, dimension, and chemical composition. This is often achieved via polymer additives that aid the calcium phosphate mineralization process. While there is a large body of work on polymer-controlled biomimetic calcium phosphate mineralization in general27–29 only a small fraction of the work focuses on dental applications.
One of these examples is the work of Kniep and coworkers,30–36 who described the formation of spherical FAP particles in gelatin hydrogels, mostly via double diffusion techniques. Initially, hexagonal rods form, which later on transform into fractal-like structures yielding FAP dumbbells and finally closed spheres. These materials fill dentin tubules and were later used as a functional component in the toothpaste Theramed S.O.S. sensitive.
In addition to inorganic compounds9,10,13,21,37–39 polymers like xanthan,40 pectin,41 casein,3 and others24,40–42 have also been used as additives in dental care products. Among others, these polymers are able to anchor on the surface of the teeth and act as barrier for protons or micro organisms.
Considering the chemical composition of the synthetic polymer additives used for calcium phosphate mineralization so far,27,28 the combination of poly(electrolyte)s with poly(ethylene oxide) (PEO) is one of the most popular choices.43–45 To a large extent, this is due the well-known biopassivation abilities of PEO46 and the strong interaction of poly(electrolyte)s with inorganic ions47–50 and surfaces.47,48,51–53 The combination of highly charged remineralizition-enhancing48,49,54–62 or dissolution-limiting63,64 polymer segments with the biopassivation of PEO is thus a viable strategy towards multifunctional polymers providing both anti-biofilm and remineralization-enhancing behavior in one single system.
In spite of this, there are only very few studies combining the two roles in one polymer. We have previously shown that negatively charged poly(sulfonate)-based block copolymers are efficient growth control agents for calcium phosphate and at the same time also reduce bacterial adhesion on the enamel surface.65
Clearly, an ideal additive should (i) adhere to the enamel surface without further damaging it, (ii) function as protective shield against incoming acids and bacteria, (iii) show bactericidal or bacteria-repellent properties, and (iv) initiate the remineralization at the damaged enamel surface sites.
Interestingly, positively charged,59,66–69 ampholytic,70 or betainic60,61,71 additives for calcium phosphate mineralization are much less common, despite the fact that such polymers show bactericidal72–74 and antifouling75–77 properties that should also be interesting for dental applications. Additionally, these polymers are able to interact with solids.78,79 The lack of studies on poly(cationic) additives is all the more surprising because the positive charge could also be interesting for phosphate enrichment and for improving the contact to the enamel surface, which is slightly negatively charged80 at ca. −0.02 C m−2. Accordingly, positively charged polymers could interact with the enamel surface and therefore offer a means of modifying the enamel surface for protection against biofilm formation and remineralization control at the same time. The current study therefore focuses on the role of PEO-based block copolymers, where the charged block is cationic, ampholytic, or betainic in nature, for biomimetic calcium phosphate mineralization.
Experimental part
Materials
Copper(I) chloride (technical grade, Carl Roth and Merck); ethylene diamine tetraacetic acid disodium salt (EDTA/Na2, Roth), [2-(methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (MEDSAH, 97%, Merck), 2-(dimethylamino)ethyl methacrylate (DMAEMA, ≥99%, containing monomethyl ether hydroquinone as inhibitor, Merck), 4-bromobutyronitrile (97%, abcr), poly(ethylene oxide) (PEO100k, nominal Mn = 100000 g mol−1, abcr), ammonium sulfate (≥99.0%, Sigma-Aldrich), α-bromoisobutyryl bromide (98%, Sigma-Aldrich), poly(ethylene oxide) (PEO4600, nominal Mn = 4600 g mol−1, Sigma-Aldrich), poly(ethylene oxide monomethylether) (MPEO5000, nominal Mn = 5000 g mol−1, Sigma-Aldrich), potassium peroxodisulfate (KPDS, ≥99%, Sigma-Aldrich), 3-sulfopropylmethacrylate potassium salt, (SPM, 98%, Sigma-Aldrich), 2-(trimethylammonium)ethyl methacrylate chloride (TMAEMA, 80 wt% in water, Sigma-Aldrich), and synthetic hydroxyapatite powder (Sigma-Aldrich, 98%) were used as received. N,N,N′,N′-tetramethyl ethylenediamine (TMEDA, ≈99%, Sigma-Aldrich) and triethylamine (≥99%, Sigma-Aldrich) were dried with phosphorous pentoxide. Solvents and polytetrafluoroethylene (PTFE) syringe filters were purchased from VWR international and used as received.
Monomer synthesis
The non-commercial monomer 2-((3-cyanopropyl)-dimethylammonium)ethyl methacrylate chloride (CPDMAEMA/Cl) was made by modification of DMAEMA. In a two neck flask 70 mmol (10.95 mL, 1 equivalent) of DMAEMA were dissolved in 20 mL of CHCl3. Subsequently, 70 mmol (6.45 mL, 1.0 equivalent) of 4-bromobutyronitrile dissolved in 30 mL of CHCl3 were added. The mixture was stirred at 50 °C for 1 day. After the addition of 50 mL of hexane, the CHCl3/hexane phase was removed. The colorless highly viscous residue was washed three times with 100 mL of hexane and two times with 50 mL of dioxane. The white viscous product was suspended in 50 mL of dioxane and freeze-dried to yield a white solid with 55–65% yield. The resulting monomer CPDMAEMA/Br is fairly stable but should be stored under argon because of its highly hygroscopic character.The chloride form CPDMAEMA/Cl was made by ion exchange chromatography using an aqueous, slightly yellow, solution of CPDMAEMA/Br on a DOWEX 1× 4-50 ion exchange resin. Completion of the exchange was verified with an 0.2 M silver nitrate solution. After removing the main part of the water under reduced pressure at 30 °C, the remaining water was removed by freeze-drying yielding a white solid. The product could only be stored under argon in the refrigerator for a few weeks as it tends to self-polymerize.
FTIR (ATR, 298 K): 2924 cm−1, C–H asymmetric stretching vibration; 2247 cm−1, CN stretching vibration; 1716 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration; 1634 cm−1, CO stretching; 1453 cm−1, C–H scissor vibration; 1294 cm−1, CH2 in-plane deformation vibration; 1156 cm−1, C–N stretching vibrations. 1H NMR (300 MHz, D2O, 298 K) d ppm 1.94 (t, J = 1.32 Hz, 3H) 2.19–2.31 (m, 2H) 2.66 (t, J = 7.06 Hz, 2H) 3.23 (s, 6H) 3.56 (dquin, J = 7.50, 4.00, 4.00, 4.00, 4.00 Hz, 2H) 3.81–3.87 (m, 2H) 4.64 (tt, J = 4.70, 2.30 Hz, 2H) 5.78 (quin, J = 1.50 Hz, 1H) 6.16 (quin, J = 0.90 Hz, 1H). EA experiment (calculated): C 43.3% (47.1%), H 7.1% (7.2%), N 8.9% (9.2%). HRMS (ESI-Q-TOF) m/z: [M − Br−]+ calcd for C12H21N2O2 225.1603; found 225.1598. [M − Br− + 1H]+ calcd for C12H21N2O2 226.1676; found 226.1624.
O stretching vibration; 1634 cm−1, CO stretching; 1453 cm−1, C–H scissor vibration; 1294 cm−1, CH2 in-plane deformation vibration; 1156 cm−1, C–N stretching vibrations. 1H NMR (300 MHz, D2O, 298 K) d ppm 1.94 (t, J = 1.32 Hz, 3H) 2.19–2.31 (m, 2H) 2.66 (t, J = 7.06 Hz, 2H) 3.23 (s, 6H) 3.56 (dquin, J = 7.50, 4.00, 4.00, 4.00, 4.00 Hz, 2H) 3.81–3.87 (m, 2H) 4.64 (tt, J = 4.70, 2.30 Hz, 2H) 5.78 (quin, J = 1.50 Hz, 1H) 6.16 (quin, J = 0.90 Hz, 1H). EA experiment (calculated): C 43.3% (47.1%), H 7.1% (7.2%), N 8.9% (9.2%). HRMS (ESI-Q-TOF) m/z: [M − Br−]+ calcd for C12H21N2O2 225.1603; found 225.1598. [M − Br− + 1H]+ calcd for C12H21N2O2 226.1676; found 226.1624.
Macroinitiators
Macroinitiator synthesis was done as described previously65 but using α-bromoisobutyryl bromide instead of bromoacetyl bromide to increase the rate of initiation with the TMAEMA and CPDMAEMA monomers. Fig. 1 shows the macroinitiators used in this study.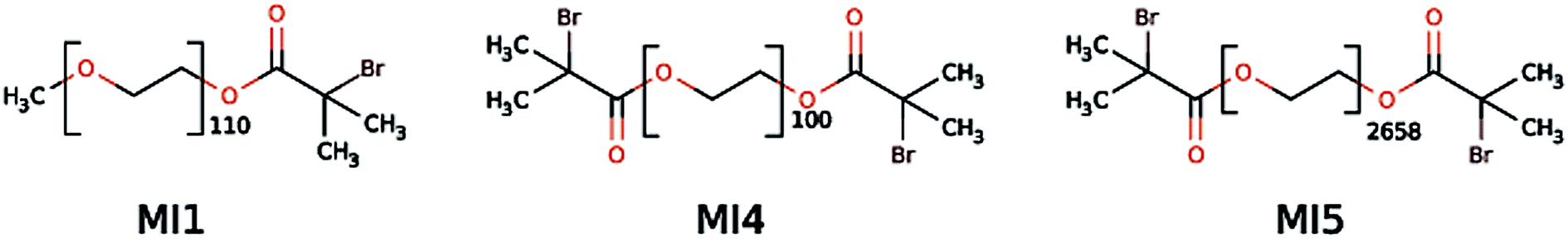 | ||
| Fig. 1 Macroinitiators used in the present study.81 MI is macroinitiator, MI2 and MI3 (not shown here) have been used in the previous study65 but have been replaced by MI4 and MI5, respectively, because MI4 and MI5 are more reactive than MI2 and MI3. Based on PEO these starters will lead to a biopassive block46 in the later block copolymers. | ||
Polymerization
Prior to polymerization, CuCl was cleaned and activated with glacial acetic acid followed by washing with ethanol and methyl tert-butyl ether (MTBE).50 Alternatively, CuCl was dissolved in aqueous HCl (25%) and reprecipitated by addition of water followed by washing with ethanol and MTBE.82,83 After both procedures the copper(I) chloride was dried at reduced pressure and stored under Ar.General polymerization procedure – 1. homopolymers
In a two neck flask the monomers were dissolved in water. The solution was flushed with argon and degassed five times. Then the initiator was added and the flask was sealed with a septum. The reaction mixture was stirred at 60 °C for 90 min. Then the flask was opened and air was bubbled through the reaction mixture to terminate the reaction. In the cases, where TMAEMA was used the liquid volume was reduced to ca. 50% by rotary evaporation. Purification was done by repeated precipitation. Sample nomenclature and analytical data of all polymers are given in Table S1.†General polymerization procedure – 2. block copolymers
In a two neck flask, the macroinitiator MI1 and the respective monomer were dissolved in water. The mixture was degassed and flushed 5 times with argon. Then CuCl and TMEDA were added, the flask was sealed with a septum, and the reaction mixture was stirred for 40–150 minutes at room temperature. To stop the reaction air was bubbled through the solution for 1 min. The product was precipitated from 300 mL of isopropyl alcohol and washed with MTBE.82,83 Purification was done by repeated precipitation. The final product was obtained by freeze-drying. Sample nomenclature and analytical data are given in Table S1† and Table 1. Chemical structures of all polymers are given in Fig. 2.| Polymer | GPC | 1HNMR | AF4 | Pn(mon) | Pn(PEO) | |||
|---|---|---|---|---|---|---|---|---|
| Mn [kg mol−1] | PDI | Mn [kg mol−1] | dn/dc [mL g−1] | Mn [kg mol−1] | PDI | |||
| C1 | 40.7 | 4.15 | 0.143 | 134.5 | 1.67 | |||
| C2 | 136.5 | 6.01 | 98.8 | 0.145 | 182.9 | 3.50 | 452 | 110 |
| C3 | 61.7 | 5.56 | 300.0 | 0.132 | 228.7 | 1.75 | 1422 | 100 |
| C1CN | 90.2 | 3.70 | 0.149 | |||||
| C2CN | 82.2 | 5.47 | 67.3 | 0.158 | 279.4 | 3.62 | 239 | 110 |
| C3CN | 119.0 | 4.02 | 85.8 | 0.159 | 253.0 | 4.22 | 311 | 100 |
| C4CN | 111.3 | 4.84 | 1293.2 | 4510 | 2658 | |||
| A1 | 0.150 | |||||||
| A2 | 764.0 | 0.134 | 2000 | 110 | ||||
| A3 | 443.3 | 0.137 | 1156 | 100 | ||||
| A4 | 2429.4 | 0.165 | 6093 | 2658 | ||||
| A1CN | 0.144 | |||||||
| A2CN | 179.1 | 0.146 | 403 | 110 | ||||
| A3CN | 123.6 | 0.146 | 275 | 100 | ||||
| A4CN | 397.2 | 0.152 | 648 | 2658 | ||||
| B1 | 0.143 | 1053 | 1.80 | |||||
| B2 | 630.8 | 0.085 | 1746 | 1.52 | 2240 | 110 | ||
| B3 | 555.9 | 0.144 | 1973 | 100 | ||||
| B4 | 11760.4 |
0.138 | 1133 | 1.28 | 41680 |
2658 | ||
Mineralization
Mineralization was done as described previously.65 In a typical experiment, 150 mg of a polymer were dissolved in 30 mL of doubly concentrated simulated body fluid (2SBF). 2SBF is a well-established system for artificial saliva first introduced by Kokubo et al.84 Then 15 mL of aqueous 0.1 M CaCl2 were added in three 5 mL steps interrupted by vortexing for 30 s. This mixture was stirred for 5 days at 500 rpm at room temperature. The product was purified by centrifugation and washing five times with pure water (ρ = 18.2 MΩ cm) and ethanol.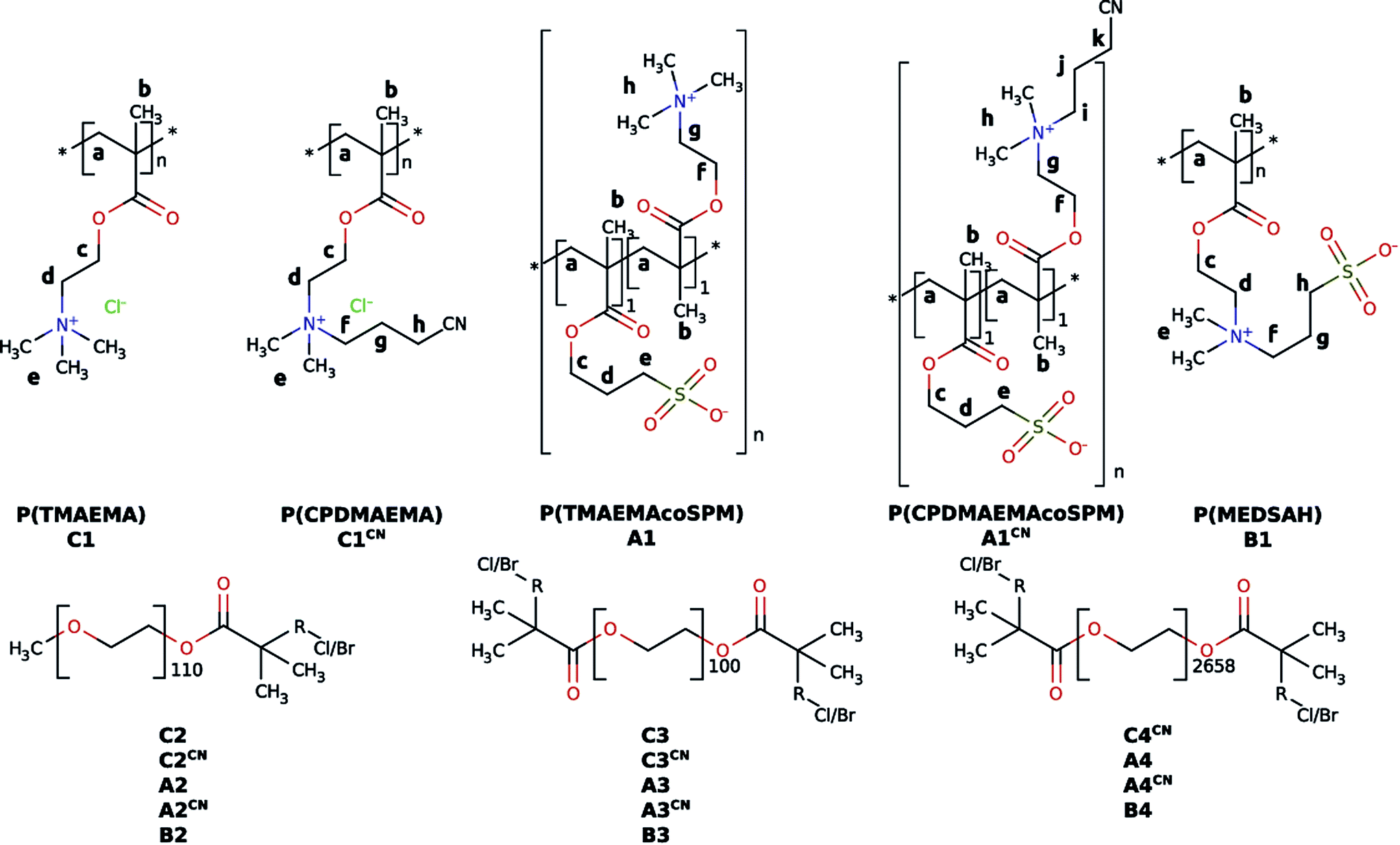 | ||
| Fig. 2 Chemical structures of the polymers synthesized in the present study.81 The lower case labels refer to 1H NMR assignments shown in Fig. 3 below. | ||
Polymer-induced calcium phosphate dissolution
Calcium phosphate dissolution was measured as described earlier.65 In 10 mL of millipore water (ρ = 18.2 MΩ cm) 100 mg of synthetic HAP powder were suspended and 2 to 50 mg of a polymer were dissolved. The mixtures were shaken for 10 days at room temperature. After centrifugation and filtration through a 0.2 μm PTFE syringe filter the concentration of Ca2+ in the aqueous phase was determined via ICP-OES.Asymmetric flow field-flow fraction
AF4 measurements were performed on a Eclipse 3 system (Wyatt) with a regenerated cellulose membrane (10 kDa cutoff) and a channel height of 350 μm using water with 0.2% sodium azide and 50 mmol L−1 NaNO3 as eluent. For detection, an Agilent Technologies UV-detector, a DAWN EOS multi-angle scattering detector (Wyatt Technologies), and an Optilab T-rEX RI-detector (Wyatt) at 690 nm were used. A flow rate of 1 mL min−1 at 25 °C was used with an exponential cross flow gradient from 3 to 0 mL min−1 for 15 min using a curve multiplier of 2.5. The sample size was 100 μL for C1, C2CN, C3CN and 50 μL for C2, C3, B1, B3, B4. Polymer concentration was 1.000 mg mL−1.Elemental analysis
EA was done on a Vario EL III (elementar).Gel permeation chromatography
Cationic polymers: GPC with simultaneous UV, RI, and MALLS (multi-angle laser light scattering, Wyatt Dawn Eos) detection was performed at room temperature in acetate buffer with 20% of methanol, a flow rate of 1.0 mL min−1, and using a PSS Novema MAX analytical linear S 10 column with porosities of 10 μm. Solutions containing ∼0.15 wt% polymer were filtered through 0.45 μm filters; the injected volume was 100 μL. Data were recorded and evaluated with the PSS-WinGPC Unichrom software package.Inductively coupled plasma-optical emission spectroscopy
ICP-OES experiments were performed on a VARIAN Vista MPX with axial plasma by dilution with water.Infrared spectroscopy
ATR-IR-spectra were recorded on a Thermo Nicolet 6700 from 500 to 4000 cm−1 at 1 cm−1 resolution and at least 32 scans per measurement. Data were evaluated with the Omnic 6.2 software.NMR spectroscopy
NMR spectra were recorded on a Bruker Avance 300 at room temperature.Mass spectrometry
Mass spectra were measured on an ESI-Q-TOF micro (Quadrupole-Time of Flight).Scanning electron microscopy & energy-dispersive X-ray spectroscopy
SEM and EDXS experiments were done on a JEOL JSM-6510 with an INCA-x-act detector. Acceleration voltage was 15 kV for EDXS and 8 to 15 kV for SEM. All samples were carbon-coated for 5 seconds using an EMITECH SC7620 sputter coater. For preparation the samples were dispersed in 5 mL ethanol, a few drops of the dispersion were deposited on the SEM sample holders and the ethanol was allowed to dry prior to inserting the samples into the microscope.Turbidimetry
The turbidity evolution during calcium phosphate precipitation reactions was quantified with a Shimadzu UV mini 1240 at 746 nm following a previously published protocol.65 The critical Ca2+ for precipitation concentration [Ca]P was obtained from x0 of the equation y = A1 + (A2 − A1)/(1 + e(x−x0)/dx), with A1 as the initial value, A2 as the final value, x0 as the function center and dx as the steepness of the fit. All measurements were reproduced in triplicate.X-ray diffraction
XRD experiments were done on a Siemens D5005 X-ray diffractometer with Cu anode and vertical circle goniometer with an optional sample holder for small amounts of sample. For preparation all samples were dispersed in 5 mL ethanol.Results
Polymer synthesis and composition
Polymers were characterized with 1H NMR spectroscopy, gel permeation chromatography (GPC), and asymmetric field-flow-field fractionation (AF4), Table 1, proving that in all cases polymers with very high molecular weights are obtained. Consistent with literature, AF4 detects higher molecular weights than 1H NMR spectroscopy.85,86 1H NMR spectroscopy further confirms that the copolymers made from either TMAEMA or CPDMAEMA/Cl (the cationic monomers) and the anionic monomer SPM have a 1:1 monomer ratio (Fig. 3 and S7–S11†). Further experiments using inductively coupled plasma-optical emission spectroscopy (ICP-OES) confirm that all polymers contain low fractions of Cu from the initiator and a slightly higher fraction of K from the SPM monomer in case of A-type (ampholyte) or from purification in the case of B-type (betaine) polymers (Table S12†). In spite of the fact that for all polymers analytical data can be provided, there are significant differences between the ampholytic A, betainic B, and cationic C-type polymers. These will now be discussed in more detail, starting with the cationic C polymers.
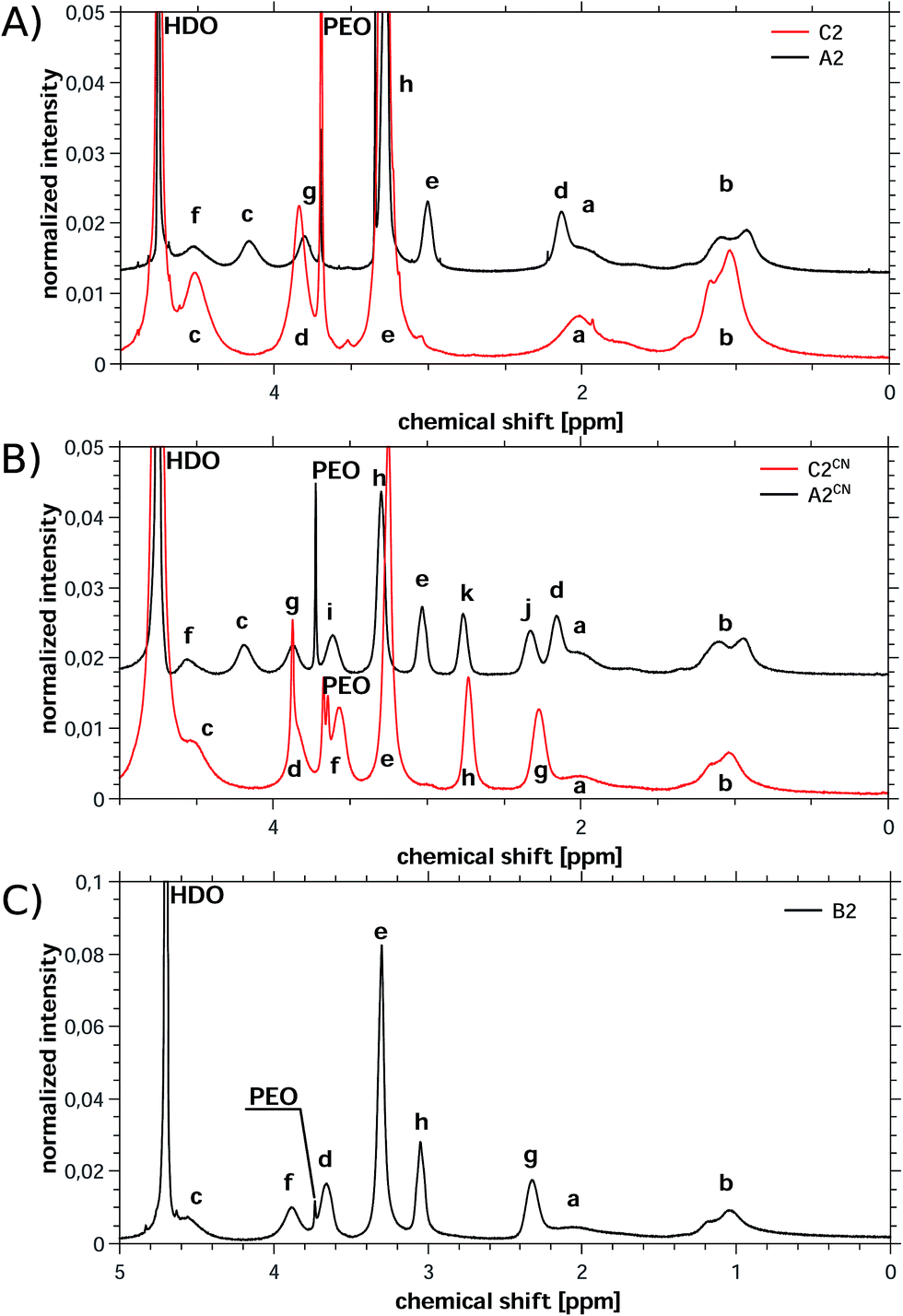 | ||
| Fig. 3 Representative 1H NMR spectra of (A) C2 and A2, (B) C2CN and A2CN and (C) B2. A2, A2CN and B2 were measured in the presence of KCl to improve solubility. Labels for peak assignments are given in Fig. 2. | ||
Under the experimental conditions chosen here, the cationic monomers TMAEMA and CPDMAEMA/Cl polymerize very slowly. Moreover, the high-molecular-weight macroinitiator MI5 shows poor initiation, which overall leads to very low conversions and yields below 9% for the combination of MI5 and TMAEMA or CPDMAEMA/Cl. The polymerization with the macroinitiators MI1 and MI4 is much more efficient (likely due to the lower molecular weight of MI1 and MI4) and the respective polymers can be isolated in reasonable yields (21% for C3 and over 55% for all other C-type polymers).
1H NMR proves the successful polymer synthesis for all cationic polymers enabling the determination of the molecular weight of all individual blocks from NMR data. The infrared spectra on the other hand confirm the presence of the cationic blocks, but do not show any signal that can be assigned to the PEO-based macroinitiators. This indicates, consistent with NMR spectroscopy, that the PEO fraction in the copolymers is rather low. However for the cationic polymers based on CPDMAEMA/Cl the infrared spectra also prove the stability of the cyanide group by the presence of the signal at 2248 cm−1.
All polyampholytes were synthesized using a molar monomer ratio (cation:anion) of 1:1. Indeed, 1H NMR spectroscopy confirms a 1:1 monomer ratio in the final polymers after purification. This is consistent with the work of Salamone87 and likely has its cause in a preorganization of the monomers in solution where a negatively charged SPM monomer is always nearby a positively charged ammonium monomer, TMAEMA or CPDMAEMA/Cl in our case.
Additionally the presence of both monomers is also confirmed by IR spectroscopy, which detects bands at 1037, 1041, and 1110 cm−1 indicative of the symmetric and asymmetric SO3 vibrations (SPM monomer) and bands at 1153 and 3046 cm−1 indicative of the C–N stretching vibrations in the TMAEMA and CPDMAEMA/Cl monomers, respectively. In contrast, no signal from the PEO blocks could be observed. This again indicates a relatively low mass fraction of the PEO blocks in the final polymers, consistent with NMR data.
From all polymers studied here, the betaine-based polymers could best be purified resulting in very low fractions of remaining copper and potassium (Table S12†). 1H NMR again shows the successful formation of the copolymers. The presence of signals from the PEO block and the betaine block enables the determination of the molecular weight from NMR spectroscopy (Table 1). B4 is the polymer with the highest molecular weight of the present study (Table 1) which is consistent with our previous study65 in the sense that the large PEO block again leads to the largest overall molecular weight. Consistent with the IR spectroscopy data on the polymers discussed above, also here, IR spectra only show signals characteristic of the betaine block, but not the PEO block (Fig. S2–S6†). Overall, these data show that the polymers are clean and have a well-defined composition, thus making them suitable additives for mineralization.
Calcium phosphate mineralization
We have previously shown that block copolymers made from essentially the same macroinitiators and SPM significantly delay the precipitation of calcium phosphate.65 To evaluate the efficiency of the current copolymers in calcium phosphate mineralization or inhibition we have again used the precipitation titration method introduced in our previous study.65 In short, calcium chloride was added to a calcium and phosphate-containing stock solution containing the polymer; by further addition of calcium chloride it is possible to determine, how effectively a polymer delays or accelerates precipitation of the mineral in relation to control reactions in the absence of polymer.Fig. 4 shows the calcium concentrations where precipitation was observed vs. the polymer composition. Somewhat surprisingly, the polymer chemistry appears to be of minor importance, as in all cases (cationic vs. ampholytic vs. betainic vs. anionic65) the calcium concentration at which precipitation is observed is in the same range of around 0.7 mg mL−1. Only C2CN (ca. 0.9 mg mL−1), C3CN (ca. 0.9 mg mL−1), and PSPM-b-PEO100k-b-PSPM (ca. 1.1 mg mL−1) stabilize solutions with much higher supersaturation. In absence of polymer-additives precipitation occurs at 0.542 mg mL−1.
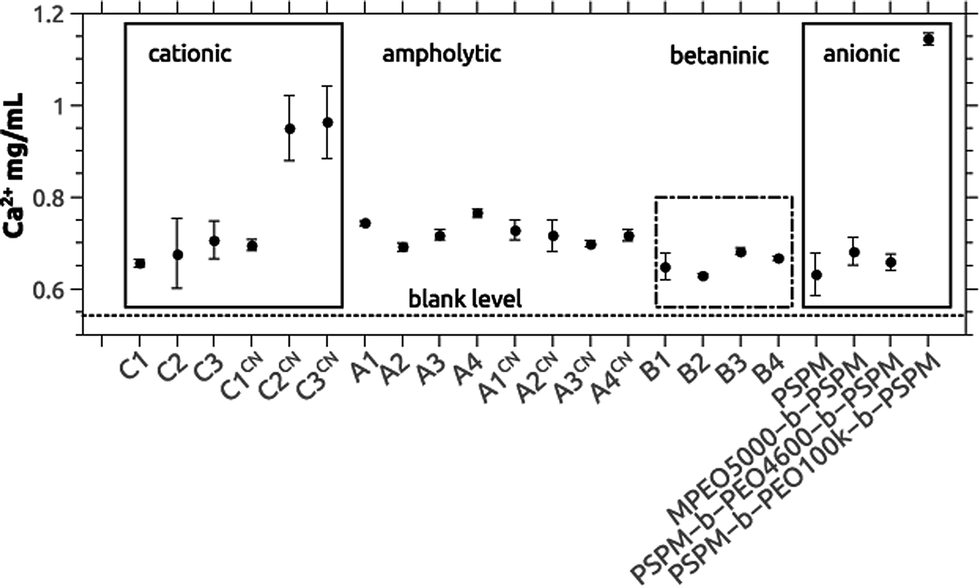 | ||
| Fig. 4 Precipitation concentrations [Ca]P determined by turbidity measurements using the titration method described in ref. 65. Data on the anionic block copolymers are from ref. 65. The dashed line at the bottom of the graph represents [Ca]P determined for samples without polymer additives (control sample). Absolute values are given in Table S13.†88 A single factor ANOVA analysis reveals that the average is the same. | ||
Although the precipitation concentration [Ca]P is roughly identical for all chemical groups present in the polymers, the particle morphologies of the precipitates obtained here differ significantly from products precipitated with the anionic block copolymers studied previously. While in the earlier case65 using 3-sulfopropyl methacrylate-based copolymer additives, relatively uniform spherical particles with diameters between 1 and a few μm were obtained, the cationic, ampholytic, and betainic additives studied here yield products that appear to be composed of much smaller nanoparticles. The particles are densely aggregated, which makes the determination of individual particle sizes and shapes more difficult than in the previous case. Nevertheless, the samples are homogeneous in themselves and do not contain two or more different particle types, Fig. 5.
 | ||
| Fig. 5 SEM images of precipitate obtained in presence of (A) MPEO5000-b-PSPM, (B) B3, (C) C3CN and (D) A1CN. SEM images of all polymers present in this study are shown in S17.† | ||
The samples were further studied with energy dispersive X-ray spectroscopy (EDXS). In all cases, Ca, P, O, S, C, Na, and Cl were found. The presence of Na and Cl is due to the fact that the SBF used for calcium phosphate precipitation contains NaCl. Some of the Cl may also have been introduced by the cationic polymers which have a chloride counterion. The presence of O is due to the polymer, the phosphate ions, and possibly the tape used to hold the samples on the SEM sample holder. The S and C is due to the polymer and the tape. Ca and P are due to the Ca and phosphate ions in the calcium phosphate precipitate.
The Ca/P ratios obtained from the EDX experiments (Fig. 6A, Table S16†) range from 1.33 to 1.52. The only exception are the samples precipitated with C1CN where Ca/P = 1.15 is observed. This is even below the Ca/P ratios for amorphous calcium phosphate (ACP, Ca/P = 1.5) or octacalcium phosphate (OCP, Ca/P = 1.33). The Ca/P ratio of ca. 1.15 in the samples grown with C1CN of 1.15 could indicate a mixture of phases, possibly containing brushite, a highly calcium-deficient form of hydroxyapatite, or an OCP-like phase.89,90
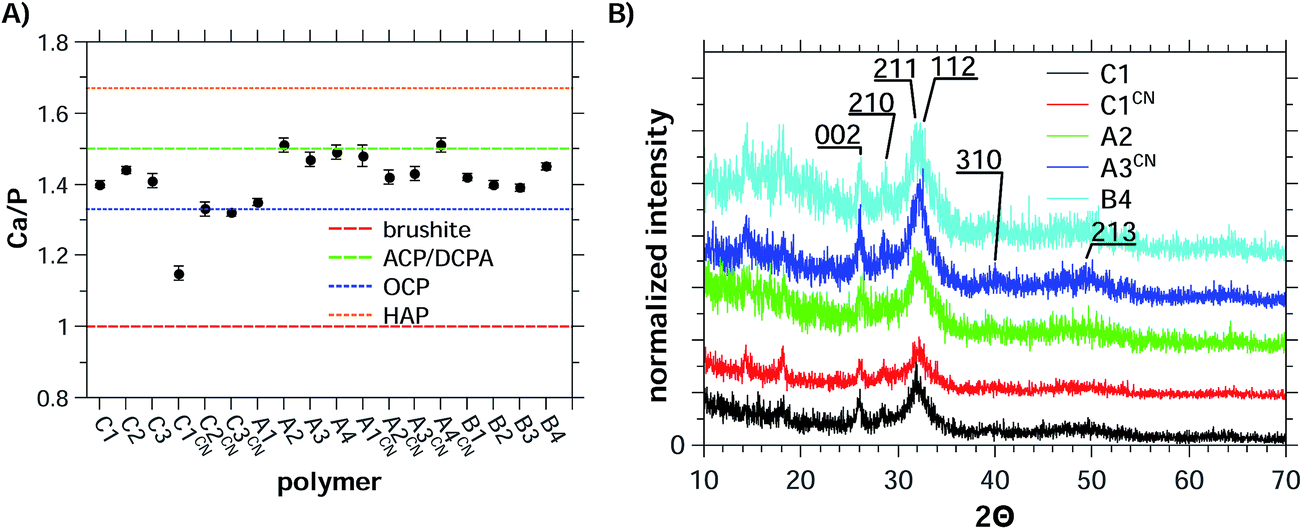 | ||
| Fig. 6 (A) Ca/P ratios obtained from EDX data of precipitates and (B) XRD patterns of precipitates.86 The dashed lines in (A) represent the Ca/P ratios of the different calcium phosphate phases28 OCP is octacalcium phosphate, ACP is amorphous calcium phosphate, HAP is hydroxyapatite, and DCPA is dicalcium phosphate anhydrate (monetite). | ||
Indeed, X-ray diffraction (Fig. 6B and S18†) shows that all samples are composed of HAP (JCPDS 03-0747, Ca10(PO4)6(OH)2). Although the patterns are noisy and exhibit low count rates (approximately 100–300) the main reflections could be assigned to HAP. This also applies to the samples obtained with C1CN, the sample exhibiting the low Ca/P ratio of only 1.15.
Calcium phosphate dissolution
We have previously shown that the polymers based on the SPM monomer have a beneficial effect on bacterial colonization of human enamel.65 An application in dental care can thus also be envisioned for the polymers studied here. An important factor to consider is the effect the polymers have on enamel stability, that is, whether or not the polymers dissolve the enamel of a tooth. Only polymers that do not significantly damage the enamel are candidates for application in dental care.To evaluate whether the current polymers are able to dissolve HAP (and hence damage enamel) we have studied the dissolution of synthetic HAP vs. polymer chemistry and polymer concentration. Dispersions of synthetic HAP in water with and without polymer present were used to evaluate the HAP dissolution efficiency. The amounts of Ca2+ released from the HAP powder vs. polymer chemistry and concentration were quantified using ICP-OES.
Fig. 7, 8, S14 and S15† summarize the results of the dissolution studies. Fig. 8 shows that the betaine-based copolymers are most attractive in that here, an increasing polymer concentration does not lead to an increased calcium phosphate dissolution. In contrast, the poly(ampholyte)s show a slight increase of the calcium concentration with polymer concentration and the poly(cation)s show an even stronger increase in calcium phosphate dissolution vs. polymer concentration.
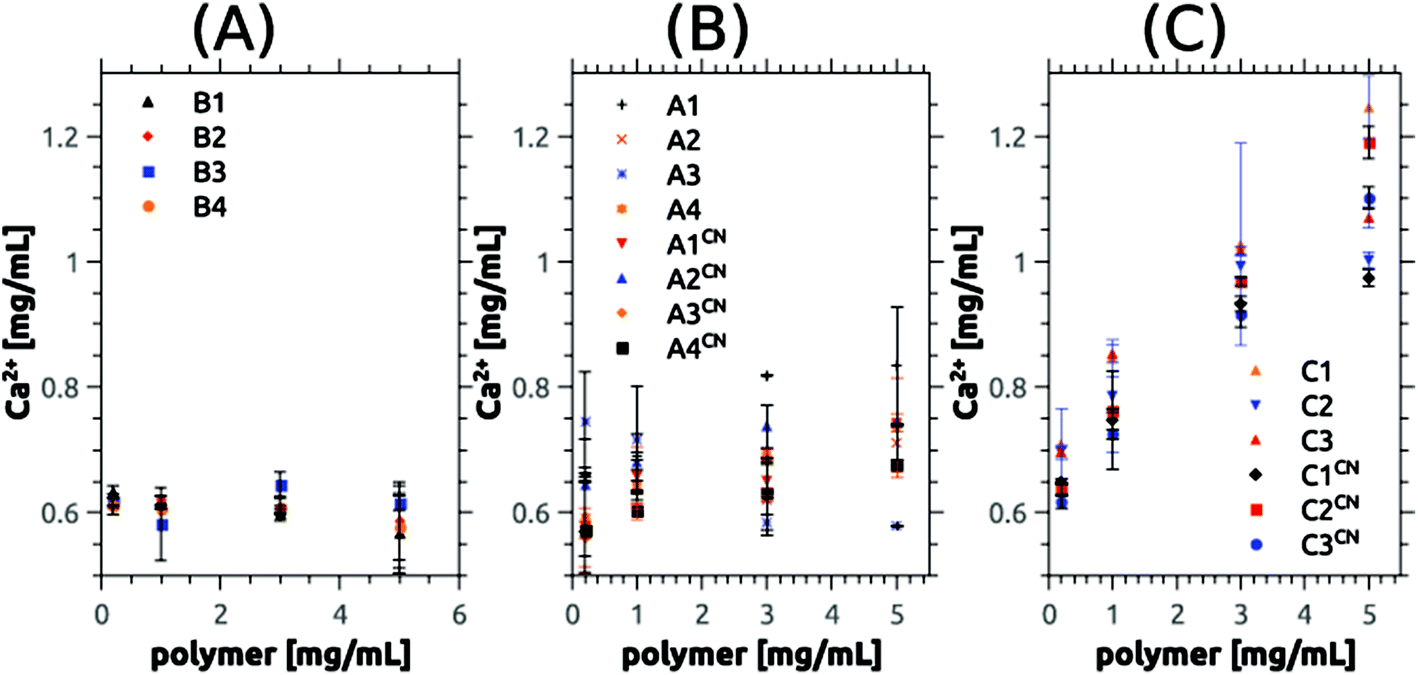 | ||
| Fig. 7 Absolute data of dissolution experiments showing concentration of calcium removed from the synthetic HAP powder vs. polymer chemistry and concentration. Panels show data from (A) betainic, (B) ampholytic, and (C) cationic copolymers. | ||
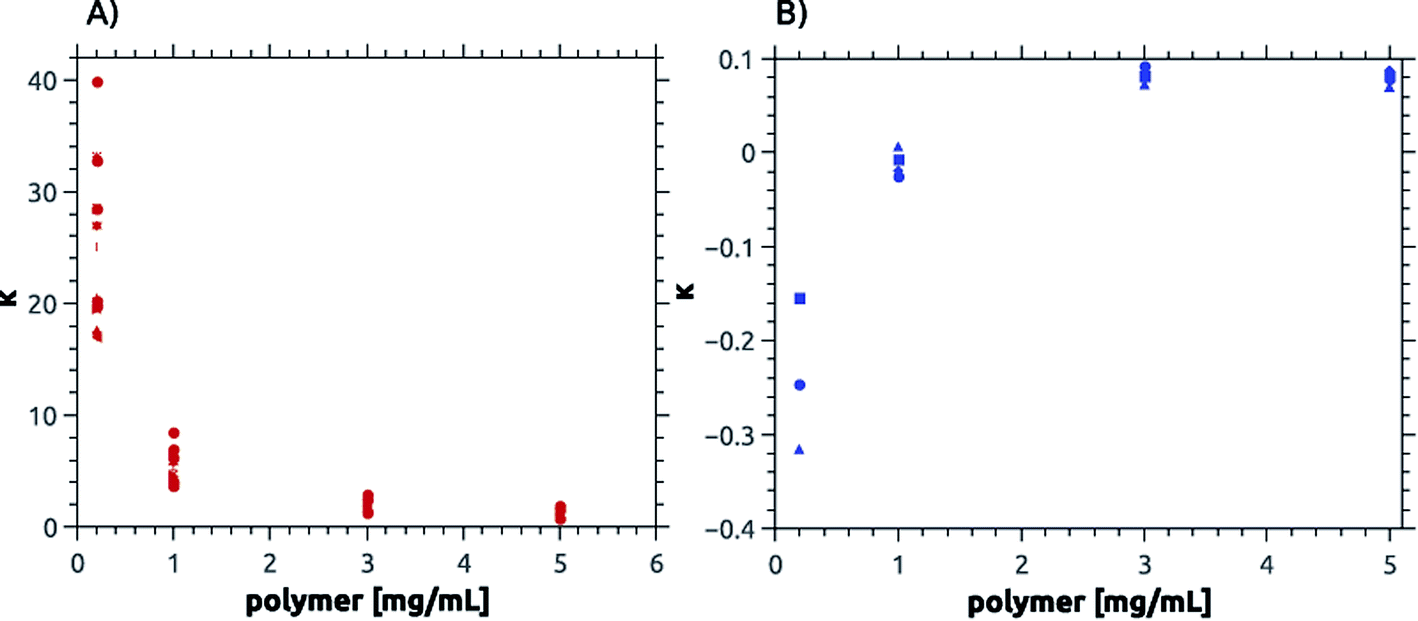 | ||
| Fig. 8 Calcium dissolution per virtual monomer unit, κ. Panel (A) shows values for cationic, ampholytic and betainic polymers and (B) for anionic polymers.65,88 Note the differences in the y-axis between (A) and (B). | ||
To better compare the effects, we have further used the dissolution parameter κ (eqn (1)) where n(monomer) is calculated from the respective molecular mass Mn and degree of polymerization P of the polymers used in this study. κ enables the comparison of the dissolution efficiency vs. numbers of functional monomers instead of polymer concentration and hence allows for the determination of relative dissolution effectiveness Fig. 7, 8, S14 and S15† summarize the results of the dissolution studies. Fig. 7 shows that the betaine-based copolymers are most attractive in that here, an increasing polymer concentration does not lead to an increased calcium phosphate dissolution. In contrast, the poly(ampholyte)s show a slight increase of the calcium concentration with polymer concentration and the poly(cation)s show an even stronger increase in calcium phosphate dissolution vs. polymer concentration.
To better compare the effects, we have further used the dissolution parameter κ (eqn (1)) where n(monomer) is calculated from the respective molecular mass Mn and degree of polymerization P of the polymers used in this study. κ enables the comparison of the dissolution efficiency vs. numbers of functional monomers instead of polymer concentration and hence allows for the determination of relative dissolution effectiveness.65
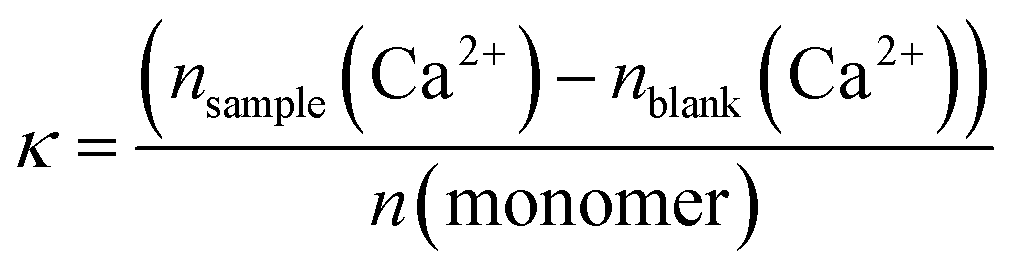 | (1) |
However, as Mn and P are obtained from 1H NMR measurements it is not possible to use eqn (1) for the polymers C1, C1CN, A1, A1CN and B1 because for these polymers, no signal of the PEO blocks could be obtained. Conversion of eqn (1) to eqn (2), where Mn(polymer)/P(polymer, ionic block) is the molar mass of a virtual monomer derived from the contribution of both the charged and the uncharged monomers. As the charged blocks are much larger than the PEO blocks, the contribution to this virtual molar mass is essentially caused by the molar mass of the charged monomers. The use of this virtual monomer molar mass enables the calculation of κ without knowledge of Mn or P. In the case of the charged homopolymers, the virtual molar mass of the monomers calculated in eqn (2) is equal to the real molar mass of the respective monomers.
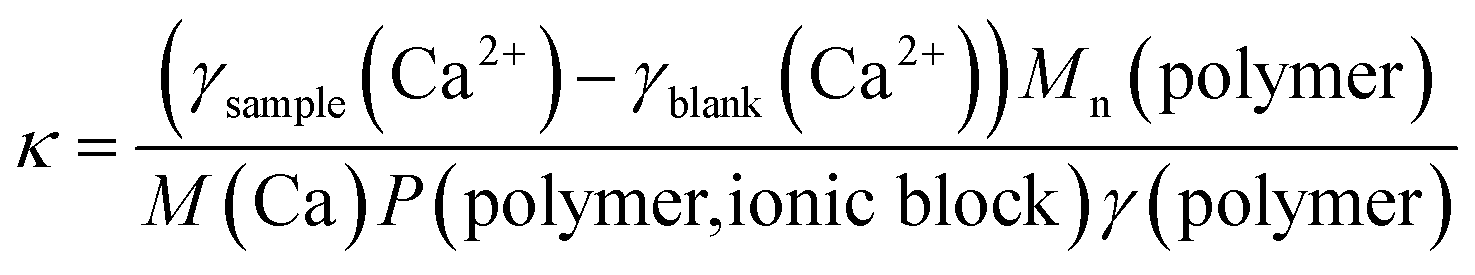 | (2) |
S14 and S15† and Fig. 8 summarize the change of κ vs. polymer concentration. Fig. 8 shows that κ is large for low polymer concentrations but decreases with increasing concentration of poly(cation)s, poly(ampholyte)s, and poly(betaine)s and reaches a value of 0.1 at a polymer concentration of 3 mg mL−1 and higher. In contrast, the poly(sulfonate)s studied earlier65 have slightly negative κ values at low polymer concentrations but also reach a value of ca. 0.1 at mg mL−1 and higher.
Discussion
Polymers are important additives for bioinspired mineralization and demineralization processes of calcium phosphate. The main focus has so far been on negatively charged (co)polymers and the amount of data on the effects of positively charged polymers is much lower.27,28 Data on the effects of zwitterionic, ampholytic, or betainic polymers are virtually non-existent. In the current article we describe the synthesis of a set of water-soluble cationic, ampholytic and betainic polymers and their role in calcium phosphate mineralization and dissolution. The effects of ampholytic and betainic polymers are described here for the first time.Polymerization was achieved via controlled radical polymerization. In all cases polymers were obtained, but the yields differ between ca. 9 and 90%. The presence of only positively charged monomers (TMAEMA or CPDMAEMA) dramatically reduces the polymerization efficiency. We currently speculate that the concentration of chloride ions (the counterions in the TMAEMA and CPDMAEMA monomers) in these systems is high enough to partly deactivate the polymerization catalyst,91,92 but this hypothesis will need to be investigated in the future. Moreover, the low activity of the high molecular weight initiator MI5 with these two monomers may also be due to the fact that the few available starting groups could not be effectively initiated by the inhibited catalyst.
In contrast, the presence of monomers carrying a negative charge (sulfonate or betaine monomers) dramatically improves the reactivity and higher yields were obtained. This is possibly due to the fact that less chloride is present in these systems leading to less deactivation of the polymerization catalyst.
Polymer characterization has turned out to be a challenge, mostly due to the somewhat limited solubility of the polymers with mixed charges. Although a number of experimental methods has been used for characterization (GPC, AF4, 1H NMR, Table 1) only 1H NMR provided information on a large fraction of the polymers. NMR spectroscopy confirms that in all cases polymers with very high molecular weights are obtained. GPC only provided reliable results for the positively charged samples, similar to other reports.85,93 The other polymers could not be investigated due to strong column–sample interactions.
Polymer analysis using AF4 was only possible for eight polymers (Table 1). The other polymers led to a strong membrane contamination due to strong interactions of the polymers with the membrane used in these experiments. Static light scattering (SLS) provided inconclusive results; this is mainly due to low dn/dc values and to the presence of at least two different species in the scattering data, indicating at least partial aggregation of the polymers. This is not unexpected because even polymers containing only hydrophilic blocks such as those studied here have been shown to aggregate in aqueous solution.94–98
One of the main goals of this study is the evaluation of the polymers as additives for calcium phosphate dissolution and precipitation. Precipitation experiments were done via an established method65 and revealed a consistent delay of the precipitation with polymer addition when compared to polymer-free control reactions. The differences between the different polymers are small. Moreover and somewhat surprisingly the efficiency of the polymers in delaying calcium phosphate precipitation is comparable to the anionic polymers studied earlier65 (Fig. 4). These data suggest that the type of charge is not the key effect here. Rather the high molecular weight and the accordingly high number of (positively and/or negatively) charged groups in the polymers may be sufficient to effectively trap very small aggregates, clusters or tiny nanoparticles. Likely, this is achieved by a combination of classical colloidal forces such as electrostatic, steric and electrosteric stabilization combined with a high affinity of the polyelectrolyte blocks to the first (small) inorganic precipitates. This hypothesis is further supported by the observation that the largest block copolymers delay calcium phosphate precipitation most effectively (Fig. 4) and by the fact that in all cases rounded and highly aggregated particles are observed in the SEM (Fig. 5).
Similar to the particle morphology, the crystal phase of the precipitates is indifferent to the exact chemical composition of the polymers as XRD (Fig. 6) always finds HAP as the product. The formation of HAP is further supported by EDXS (Fig. 6). With one exception, the Ca/P ratios determined by EDXS are between 1.33 and 1.53; in combination with XRD this suggests the formation of calcium deficient HAP. The formation of calcium deficient HAP in biomimetic syntheses is common, but it is again interesting to note that the polymer charge appears to be of minor importance in the mineralization reaction. This suggests that, consistent with literature,99,100 the pH during precipitation dominates the selection of the calcium phosphate phase. In contrast the charged groups of the polymers are mostly responsible for polymer–inorganic interaction and for trapping small clusters or particles; this in turn thus mostly affects particle sizes and morphologies rather than the crystal structure.
So far, the data of the current and the previous study65 therefore suggest that there is only a general and relatively unspecific effect of the charged polymers, but the type of charge (positive, negative, or mixed) appears less important, at least in the current case. This applies to both the morphology (spherical) and the composition (calcium deficient HAP). The sole exception can be observed when purely negatively charged polymers are used, as here the particles are still spherical and still consist of calcium deficient HAP, but the particles are larger and less aggregated than in all cases described here.65
As the surface of HAP is slightly negatively charged,80 polymers containing positively charged groups such as those investigated here, could favorably interact with HAP-like clusters or particles via electrostatic interaction. This interaction could lead to a strong growth inhibition and it could also be responsible for the much stronger aggregation of the particles than observed with the purely anionic additives observed before.65 Overall, these data show that betaines or ampholytes are interesting polymer additives for controlling and optimizing calcium phosphate mineralization.
Finally, as dental care is a potential field of application of these polymers, we have also investigated the resistance of synthetic HAP (as a simple model for enamel HAP) towards exposure of the polymers (Fig. 7, 8, S14 and S15†). The “enamel” loss here is presented by the calcium concentration in the solution after the experiment. Regardless of whether the calcium is free ionic or bound in clusters or nanoparticles it stems from damage of the enamel.
Consistent with the precipitation reactions described above, the effects of the negatively charged sulfonate copolymers differ from the effects observed here. Cationic, ampholytic, and betainic polymers show the same calcium phosphate dissolution capacity per virtual repeating unit, κ. As shown in Fig. 8, a plot of κ vs. polymer concentration shows that all polymers containing positively charged moieties exhibit a decreasing dissolution efficiency (i.e. a higher degree of protection or stabilization of the “enamel”) with increasing polymer concentration. In contrast, the poly(sulfonate)s show an increased dissolution efficiency vs. the polymer concentration65 resulting in a stronger destabilization of the “enamel” with increasing polymer concentration. Surprisingly all polymers approach a comparable limit of about κ = 0.1 but for the anionic polymers there is a stabilizing effect for HAP at low polymer concentration while there is a clear destabilizing effect for all polymers of the present study at low concentrations.
We currently speculate that this is again caused by the negatively charged HAP surface,80 similar to the delayed precipitation discussed above. Electrostatic interactions between the negatively charged HAP surface and the positively charged groups of the polymer will lead to an enrichment of the polymer at the HAP surface. In the case of ampholytic or betainic polymers, this will also lead to the effective interaction of calcium ions and the sulfonate groups present in the polymer. As a result, these polymers will have an enhanced calcium–sulfonate contact, which will result in an enhanced HAP dissolution. For the polymers containing cationic moieties there is an analogous interaction present between the ammonium groups and the phosphate on the surface the HAP. The reduced dissolution efficiency at higher polymer concentrations is likely due to the fact that the HAP surfaces are more densely covered with polymer and the removal of calcium from the HAP is therefore more difficult.
The different effects observed with the negatively charged polymers may be due to a weaker interaction of these polymers with the negatively charged HAP surface due to electrostatic repulsion. As a result, there are less calcium–sulfonate contacts, resulting in a reduced HAP dissolution at low polymer concentrations. Increasing polymer concentration may again lead to a more pronounced interaction of the polymer with the HAP surface and the formation of a polymer layer on the surface. This interpretation is consistent with several other studies56,65–67,101 where highly negatively charged polymers have been shown to interact quite differently with calcium phosphate than highly positively charged additives.
Conclusion
The current study shows that high molecular weight block copolymers with cationic, ampholytic, and betainic blocks can effectively be synthesized by controlled radical polymerization. The polymers have two common properties when compared to one another and to poly(sulfonate) copolymers: (1) all polymers delay the nucleation and growth of calcium phosphate by about the same factor and (2) there is no influence of the polymers on the crystal phase of the precipitates. There are, however, also two significant differences between the purely anionic (co)polymers and all other polymers: (1) only the poly(sulfonate) copolymers yield individual, large micrometer-sized spherical particles, while in all other cases, nanometer-sized and highly aggregated particles are observed and (2) the poly(sulfonate)s show a distinctly different behavior in calcium phosphate dissolution experiments. The reasons for these similarities and differences are likely to be found in electrostatic interactions of the polymers and the surface of HAP or one of its precursors.Acknowledgements
We thank Dr M. Heydenreich, A. Krtitschka, Y. Linde, A. Musiol, and A. Städtke for help with NMR spectroscopy, EA, and ICP-OES, respectively. The Max Planck Institute of Colloids & Interfaces and the University of Potsdam are thanked for financial support.References
- J. D. B. Featherstone and A. Lussi, Monogr. Oral Sci., 2006, 20, 66–76 CrossRef CAS PubMed.
- T. D. White, M. T. Black and P. A. Folkens, Human Osteology, Elsevier Science, 2011 Search PubMed.
- C. A. Hemaingway, A. J. White, R. P. Shellis, M. Addy, D. M. Parker and M. E. Barbour, Caries Res., 2010, 44, 525–530 CrossRef PubMed.
- J. O. Grippo, M. Simring and T. a. Coleman, J. Esthet. Restor. Dent., 2012, 24, 10–23 CrossRef PubMed.
- S. Kashket, J. Zhang and J. van Houte, J. Dent. Res., 1996, 75, 1885–1891 CrossRef CAS PubMed.
- A. Lussi, T. Jaeggi and D. Zero, Caries Res., 2004, 38, 34–44 CrossRef PubMed Suppl 1.
- A. C. Magalhães, A. Wiegand, D. Rios, M. A. R. Buzalaf and A. Lussi, Monogr. Oral Sci., 2011, 22, 158–170 Search PubMed.
- N. Schlueter, J. Klimek and C. Ganss, Clin. Oral Investig., 2011, 15, 361–367 CrossRef PubMed.
- N. Schlueter, M. Hardt, A. Lussi, F. Engelmann, J. Klimek and C. Ganss, Eur. J. Oral Sci., 2009, 117, 427–434 CrossRef CAS PubMed.
- N. Schlueter, L. Neutard, J. von Hinckeldey, J. Klimek and C. Ganss, Acta Odontol. Scand., 2010, 68, 180–184 CrossRef CAS PubMed.
- S. Miller, T. Truong, R. Heu, M. Stranick, D. Bouchard and A. Gaffar, Int. Dent. J., 1994, 44, 83–98 CAS.
- W. Wade, M. Addy, J. Hughes, S. Milsom and F. Doherty, J. Clin. Periodontol., 1997, 24, 81–85 CrossRef CAS PubMed.
- G. S. Ingram, E. A. Agalamanyi and S. M. Higham, J. Dent., 2005, 33, 187–191 CrossRef CAS PubMed.
- A. Kovtun, D. Kozlova, K. Ganesan, C. Biewald, N. Seipold, P. Gaengler, W. H. Arnold and M. Epple, RSC Adv., 2012, 2, 870 RSC.
- M. A. S. Melo, L. Cheng, M. D. Weir, R.-C. Hsia, L. K. a. Rodrigues and H. H. K. Xu, J. Biomed. Mater. Res., Part B, 2013, 101, 620–629 CrossRef PubMed.
- C. Chen, M. D. Weir, L. Cheng, N. J. Lin, S. Lin-Gibson, L. C. Chow, X. Zhou and H. H. K. Xu, Dent. Mater., 2014, 30, 891–901 CrossRef CAS PubMed.
- M. Sin, Y. Sun and Y. Chang, ACS Appl. Mater. Interfaces, 2014, 6, 861–873 CAS.
- S.-J. Ahn, S.-J. Lee, J.-K. Kook and B.-S. Lim, Dent. Mater., 2009, 25, 206–213 CrossRef CAS PubMed.
- N. B. Arweiler, T. M. Auschill, N. Donos and A. Sculean, Clin. Oral Investig., 2002, 6, 205–209 CrossRef PubMed.
- S. Twetman, A. Hallgren and L. G. Petersson, Caries Res., 1995, 29, 188–191 CrossRef CAS PubMed.
- P. Tschoppe, D. L. Zandim, P. Martus and A. M. Kielbassa, J. Dent., 2011, 39, 430–437 CrossRef CAS PubMed.
- A. Lussi, Monogr. Oral Sci., 2006, 20, 1–8 CAS.
- E. Combe, F. J. T. Burke and D. W. Bernard, Dental Biomaterials, Springer, US, 2013 Search PubMed.
- A. Shimotoyodome, T. Koudate, H. Kobayashi, J. Nakamura, I. Tokimitsu, T. Hase, T. Inoue, T. Matsukubo and Y. Takaesu, Antimicrob. Agents Chemother., 2007, 51, 3634–3641 CrossRef CAS PubMed.
- J. D. Rogers, R. J. Palmer, P. E. Kolenbrander and F. A. Scannapieco, Infect. Immun., 2001, 69, 7046–7056 CrossRef CAS PubMed.
- J. Li, E. J. Helmerhorst, C. W. Leone, R. F. Troxler, T. Yaskell, a. D. Haffajee, S. S. Socransky and F. G. Oppenheim, J. Appl. Microbiol., 2004, 97, 1311–1318 CrossRef CAS PubMed.
- K. Bleek and A. Taubert, Acta Biomater., 2013, 9, 6283–6321 CrossRef CAS PubMed.
- S. Schweizer and A. Taubert, Macromol. Biosci., 2007, 7, 1085–1099 CrossRef CAS PubMed.
- S. V. Dorozhkin and M. Epple, Angew. Chem., 2002, 114, 3260–3277 CrossRef.
- R. Kniep and S. Busch, Angew. Chem., Int. Ed. Engl., 1996, 35, 2624–2626 CrossRef CAS.
- S. Busch, H. Dolhaine, A. DuChesne, S. Heinz, O. Hochrein, F. Laeri, O. Podebrad, U. Vietze, T. Weiland and R. Kniep, Eur. J. Inorg. Chem., 1999, 1999, 1643–1653 CrossRef.
- P. Simon, W. Carrillo-Cabrera, P. Formánek, C. Göbel, D. Geiger, R. Ramlau, H. Tlatlik, J. Buder and R. Kniep, J. Mater. Chem., 2004, 14, 2218 RSC.
- R. Kniep and P. Simon, Biomineralization, 2007, 270, 73–125 CAS.
- H. Tlatlik, P. Simon, A. Kawska, D. Zahn and R. Kniep, Angew. Chem., Int. Ed. Engl., 2006, 45, 1905–1910 CrossRef CAS PubMed.
- P. Simon, U. Schwarz and R. Kniep, J. Mater. Chem., 2005, 15, 4992 RSC.
- S. Busch, U. Schwarz and R. Kniep, Adv. Funct. Mater., 2003, 13, 189–198 CrossRef CAS.
- S. B. Huang, S. S. Gao and H. Y. Yu, Biomed. Mater., 2009, 4, 034104 CrossRef CAS PubMed.
- F. D. Babcock, J. C. King and T. H. Jordan, J. Dent. Res., 2015, 57, 933–938 CrossRef.
- C. Ganss, N. Schlueter, M. Hardt, P. Schattenberg and J. Klimek, Caries Res., 2008, 42, 2–7 CrossRef CAS PubMed.
- M. E. Barbour, R. P. Shellis, D. M. Parker, G. C. Allen and M. Addy, Eur. J. Oral Sci., 2005, 113, 457–461 CrossRef CAS PubMed.
- M. Beyer, J. Reichert, E. Heurich, K. D. Jandt and B. W. Sigusch, Dent. Mater. Off. Publ. Acad. Dent. Mater., 2010, 26, 831–839 CAS.
- W. A. van der Reijden, M. J. Buijs, J. J. Damen, E. C. Veerman, J. M. ten Cate and A. V. Nieuw Amerongen, Caries Res., 1997, 31, 216–223 CrossRef CAS PubMed.
- E. Zhulina, A. V. Dobrynin and M. Rubinstein, Eur. Phys. J. E, 2001, 5, 41–49 CrossRef CAS.
- M. Zhang, A. Ishii, N. Nishiyama, S. Matsumoto, T. Ishii, Y. Yamasaki and K. Kataoka, Adv. Mater., 2009, 21, 3520–3525 CrossRef CAS.
- Y.-Y. Hu, X. P. Liu, X. Ma, A. Rawal, T. Prozorov, M. Akinc, S. K. Mallapragada and K. Schmidt-Rohr, Chem. Mater., 2011, 23, 2481–2490 CrossRef CAS.
- M. J. Joralemon, S. McRae and T. Emrick, Chem. Commun., 2010, 46, 1377–1393 RSC.
- M. J. Roman, E. a. Decker and J. M. Goddard, ACS Appl. Mater. Interfaces, 2014, 6, 5383–5387 CAS.
- A. Tsortos and G. H. Nancollas, J. Colloid Interface Sci., 2002, 250, 159–167 CrossRef CAS PubMed.
- P. Bar-Yosef Ofir, R. Govrin-Lippman, N. Garti and H. Füredi-Milhofer, Cryst. Growth Des., 2004, 4, 177–183 Search PubMed.
- Y. Xu, A. Walther and A. H. E. Müller, Macromol. Rapid Commun., 2010, 31, 1462–1466 CrossRef CAS PubMed.
- A. V. Dobrynin, R. H. Colby and M. Rubinstein, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3513–3538 CrossRef CAS.
- J. Plank, A. Brandl and N. R. Lummer, J. Appl. Polym. Sci., 2007, 106, 3889–3894 CrossRef CAS.
- A. Habbaba and J. Plank, J. Am. Ceram. Soc., 2010, 93, 2857–2863 CrossRef CAS.
- O. Casse, O. Colombani, K. Kita-Tokarczyk, A. H. E. Müller, W. Meier and A. Taubert, Faraday Discuss., 2008, 139, 179 RSC.
- J. Ye, D. Wang, D. N. Zeiger, W. C. Miles and S. Lin-gibson, Biomacromolecules, 2013, 14, 3417–3422 CrossRef CAS PubMed.
- R. J. Coleman, K. S. Jack, S. Perrier and L. Grøndahl, Cryst. Growth Des., 2013, 13, 4252–4259 CAS.
- T. Iwatsubo, S. P. Kusumocahyo, T. Kanamori and T. Shinbo, J. Appl. Polym. Sci., 2006, 100, 1465–1470 CrossRef CAS.
- T. Iwatsubo, K. Sumaru, T. Kanamori, T. Shinbo and T. Yamaguchi, Biomacromolecules, 2006, 7, 95–100 CrossRef CAS PubMed.
- L.-J. Zhang, H.-G. Liu, X.-S. Feng, R.-J. Zhang, L. Zhang, Y.-D. Mu, J.-C. Hao, D.-J. Qian and Y.-F. Lou, Langmuir, 2004, 20, 2243–2249 CrossRef CAS PubMed.
- M. Kanapathipillai, Y. Yusufoglu, A. Rawal, Y.-Y. Hu, C.-T. Lo, P. Thiyagarajan, Y. E. Kalay, M. Akinc, S. Mallapragada and K. Schmidt-Rohr, Chem. Mater., 2008, 20, 5922–5932 CrossRef CAS.
- Y. Yusufoglu, Y. Hu, M. Kanapathipillai, M. Kramer, Y. E. Kalay, P. Thiyagarajan, M. Akinc, K. Schmidt-Rohr and S. Mallapragada, J. Mater. Res., 2011, 23, 3196–3212 CrossRef.
- L. Chen, K. Liang, J. Li, D. Wu, X. Zhou and J. Li, Arch. Oral Biol., 2013, 58, 975–980 CrossRef CAS PubMed.
- P. Schaad, J.-M. Thomann, J.-C. Voegel and P. Gramain, Colloids Surf., A, 1994, 83, 285–292 CrossRef CAS.
- P. Schaad, J.-M. Thomann, J.-C. Voegel and P. Gramain, J. Colloid Interface Sci., 1994, 164, 291–295 CrossRef CAS.
- T. Mai, E. Rakhmatullina, K. Bleek, S. Boye, J. Yuan, A. Völkel, M. Gräwert, Z. Cheaib, S. Eick, C. Günter, A. Lederer, A. Lussi and A. Taubert, Biomacromolecules, 2014, 15, 3901–3914 CrossRef CAS PubMed.
- M. Junginger, K. Kita-Tokarczyk, T. Schuster, J. Reiche, F. Schacher, A. H. E. Müller, H. Cölfen and A. Taubert, Macromol. Biosci., 2010, 10, 1084–1092 CrossRef CAS PubMed.
- M. Junginger, K. Bleek, K. Kita-Tokarczyk, J. Reiche, A. Shkilnyy, F. Schacher, A. H. E. Müller and A. Taubert, Nanoscale, 2010, 2, 2440–2446 RSC.
- A. Shkilnyy, J. Brandt, A. Mantion, O. Paris, H. Schlaad and A. Taubert, Chem. Mater., 2009, 21, 1572–1578 CrossRef CAS.
- S. Grohmann, H. Rothe and K. Liefeith, Biointerphases, 2012, 7, 1–13 CrossRef PubMed.
- S. Segman-Magidovich and H. Rapaport, J. Phys. Chem. B, 2012, 116, 11197–11204 CrossRef CAS PubMed.
- P. Liu and J. Song, Biomaterials, 2013, 34, 2442–2454 CrossRef CAS PubMed.
- S. Lin, J. Wu, H. Jia, L. Hao, R. Wang and J. Qi, RSC Adv., 2013, 3, 20758–20764 RSC.
- B. B. Hsu and A. M. Klibanov, Biomacromolecules, 2011, 12, 6–9 CrossRef CAS PubMed.
- D. Park, J. Wang and A. M. Klibanov, Biotechnol. Prog., 2006, 22, 584–589 CrossRef CAS PubMed.
- R. Quintana, M. Gosa, D. Jańczewski, E. Kutnyanszky and G. J. Vancso, Langmuir, 2013, 29, 10859–10867 CrossRef CAS PubMed.
- Y. Chang, W.-J. Chang, Y.-J. Shih, T.-C. Wei and G.-H. Hsiue, ACS Appl. Mater. Interfaces, 2011, 3, 1228–1237 CAS.
- J. B. Schlenoff, Langmuir, 2014, 30, 9625–9636 CrossRef CAS PubMed.
- S. Kudaibergenov, W. Jaeger and A. Laschewsky, in Adv. Polym. Sci., 2006, vol. 201, pp. 157–224 Search PubMed.
- S. E. Kudaibergenov, in Polymer Latexes – Epoxide Resins – Polyampholytes, Springer Berlin Heidelberg, Berlin, Heidelberg, 1999, pp. 115–197 Search PubMed.
- J. Vandiver, D. Dean, N. Patel, W. Bonfield and C. Ortiz, Biomaterials, 2005, 26, 271–283 CrossRef CAS PubMed.
- Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions, Marvin 15.11.16.0, ChemAxon, 2015, http://www.chemaxon.com Search PubMed.
- E. W. A. F. Holleman, Lehrbuch der anorganischen Chemie, deGruyter, 101. Aufl., 1995 Search PubMed.
- W. L. F. Armarego and C. Chai, Purification of Laboratory Chemicals, Elsevier Science, Sixth edn, 2009 Search PubMed.
- T. Kokubo, S. Ito, Z. T. Huang, T. Hayashi, S. Sakka, T. Kitsugi and T. Yamamuro, J. Biomed. Mater. Res., 1990, 24, 331–343 CrossRef CAS PubMed.
- K. E. S. Locock, L. Meagher and M. Haeussler, Anal. Chem., 2014, 86, 2131–2137 CrossRef CAS PubMed.
- M. Wagner, C. Pietsch, L. Tauhardt, A. Schallon and U. S. Schubert, J. Chromatogr. A, 2014, 1325, 195–203 CrossRef CAS PubMed.
- J. C. Salamone, L. Quach, A. C. Watterson, S. Krauser and M. U. Mahmud, J. Macromol. Sci., Part A: Pure Appl. Chem., 1985, 22, 653–664 CrossRef.
- I. Vasilef, QTIPLOT, Data Analysis and Scientific Visualisation, Universiteit Utrecht, Utrecht, Niederlande, 2011 Search PubMed.
- E. D. Eanes, in Octacalcium Phosphate, Karger, Basel, 2001, vol. 18, pp. 130–147 Search PubMed.
- W. J. E. M. Habraken, J. Tao, L. J. Brylka, H. Friedrich, L. Bertinetti, A. S. Schenk, A. Verch, V. Dmitrovic, P. H. H. Bomans, P. M. Frederik, J. Laven, P. van der Schoot, B. Aichmayer, G. de With, J. J. deYoreo and N. a. J. M. Sommerdijk, Nat. Commun., 2013, 4, 1507 CrossRef PubMed.
- N. V. Tsarevsky, T. Pintauer and K. Matyjaszewski, Macromolecules, 2004, 37, 9768–9778 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 1858–1863 CrossRef CAS.
- T. D. Michl, K. E. S. Locock, N. E. Stevens, J. D. Hayball, K. Vasilev, A. Postma, Y. Qu, A. Traven, M. Haeussler, L. Meagher and H. J. Griesser, Polym. Chem., 2014, 5, 5813 RSC.
- A. Blanazs, N. J. Warren, A. L. Lewis, S. P. Armes and A. J. Ryan, Soft Matter, 2011, 7, 6399–6403 RSC.
- O. Casse, A. Shkilnyy, J. Linders, C. Mayer, D. Häussinger, A. Völkel, A. F. Thünemann, R. Dimova, H. Cölfen, W. Meier, H. Schlaad and A. Taubert, Macromolecules, 2012, 45, 4772–4777 CrossRef CAS.
- W. Agut, A. Brûlet, D. Taton and S. Lecommandoux, Langmuir, 2007, 23, 11526–11533 CrossRef CAS PubMed.
- M. Arotçaréna, B. Heise, S. Ishaya and A. Laschewsky, J. Am. Chem. Soc., 2002, 124, 3787–3793 CrossRef.
- J. H. Lee and M. L. Shofner, Langmuir, 2013, 29, 10940–10944 CrossRef CAS PubMed.
- R. Palanivelu, A. Mary Saral and A. Ruban Kumar, Spectrochim. Acta, Part A, 2014, 131, 37–41 CrossRef CAS PubMed.
- X. Lu and Y. Leng, Biomaterials, 2005, 26, 1097–1108 CrossRef CAS PubMed.
- V. Ball, M. Michel, F. Boulmedais, J. Hemmerle, Y. Haikel, P. Schaaf and J. C. Voegel, Cryst. Growth Des., 2006, 6, 327–334 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data of all polymers, IR-spectra and 1H NMR spectra of polymers, Cu and K contents from ICP-OES, absolute data for calcium precipitation titration, κ values for all polymers, EDXS data of precipitates, SEM images of all precipitates, XRD data of all precipitates, from HAP dissolution experiments. See DOI: 10.1039/c5ra20035k |
| This journal is © The Royal Society of Chemistry 2015 |