
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic investigations of a bifunctional squaramide organocatalyst in asymmetric Michael reaction and observation of stereoselective retro-Michael reaction†
Eszter
Varga
a,
László Tamás
Mika
b,
Antal
Csámpai
c,
Tamás
Holczbauer
a,
György
Kardos
a and
Tibor
Soós
*a
aInstitute of Organic Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar Tudósok körútja 2A, Budapest, Hungary H-1117. E-mail: soos.tibor@ttk.mta.hu
bBudapest University of Technology and Economics, Faculty of Chemical Technology and Biotechnology, Department of Chemical and Environmental Process Engineering, Műegyetem rkp. 3, Budapest, H-1111, Hungary
cInstitute of Chemistry, Eötvös Loránd University, Pázmány Péter str. 1A, Budapest, H-1117, Hungary
First published on 27th October 2015
Abstract
The mechanism of the addition of acetylacetone to β-nitrostyrene catalyzed by a cinchona based squaramide catalyst was studied in detail under synthetically relevant conditions. The reaction was monitored by in situ IR and 1H-NMR spectroscopy and a reaction mechanism was proposed based on these kinetics experiments. It was found that the reaction shows nearly first order dependence on both substrates and catalyst. Our investigations also revealed that the catalyst was able to promote stereoselective retro-Michael reaction.
Introduction
Organocatalysis occupies a commanding position amongst the recent methodological advances owing to its broad utility, versatility and robustness.1 The ever-expanding list of applied catalysts2 and activation modes3 has markedly contributed to the recent expansion of synthetic chemists' toolkit. The practical advantages of these methodologies have been also valued which include lack of sensitivity toward moisture and oxygen and the ready availability of these catalysts. These features confer a direct synthetic benefit in the construction of highly elaborated chiral molecules.4 Consequently, organocatalysis has far-reaching implications that stretch beyond the current academic practice; it is becoming a broadly integrated and widely applied transformation in contemporary drug design and development.5Although numerous papers have been published concerning the application and development of organocatalysts, comparable few studies have been focused on the mechanism.6 The mechanistic understanding of a catalytic cycle is fundamentally important to design more active and selective catalytic systems. In addition, it helps bench chemists to make rational predictions as to how to modify experimental parameters to maximize product yields.
Albeit the bifunctional catalysis concept has been around for decades,7 the discovery of a bifunctional thiourea organocatalyst by Takemoto8 has had an enormous impact on the evolution of organocatalysis and also the field of asymmetric catalysis. This class of catalysts9 (Scheme 1) proved to be a highly efficient promoter for multitude of asymmetric transformations.10
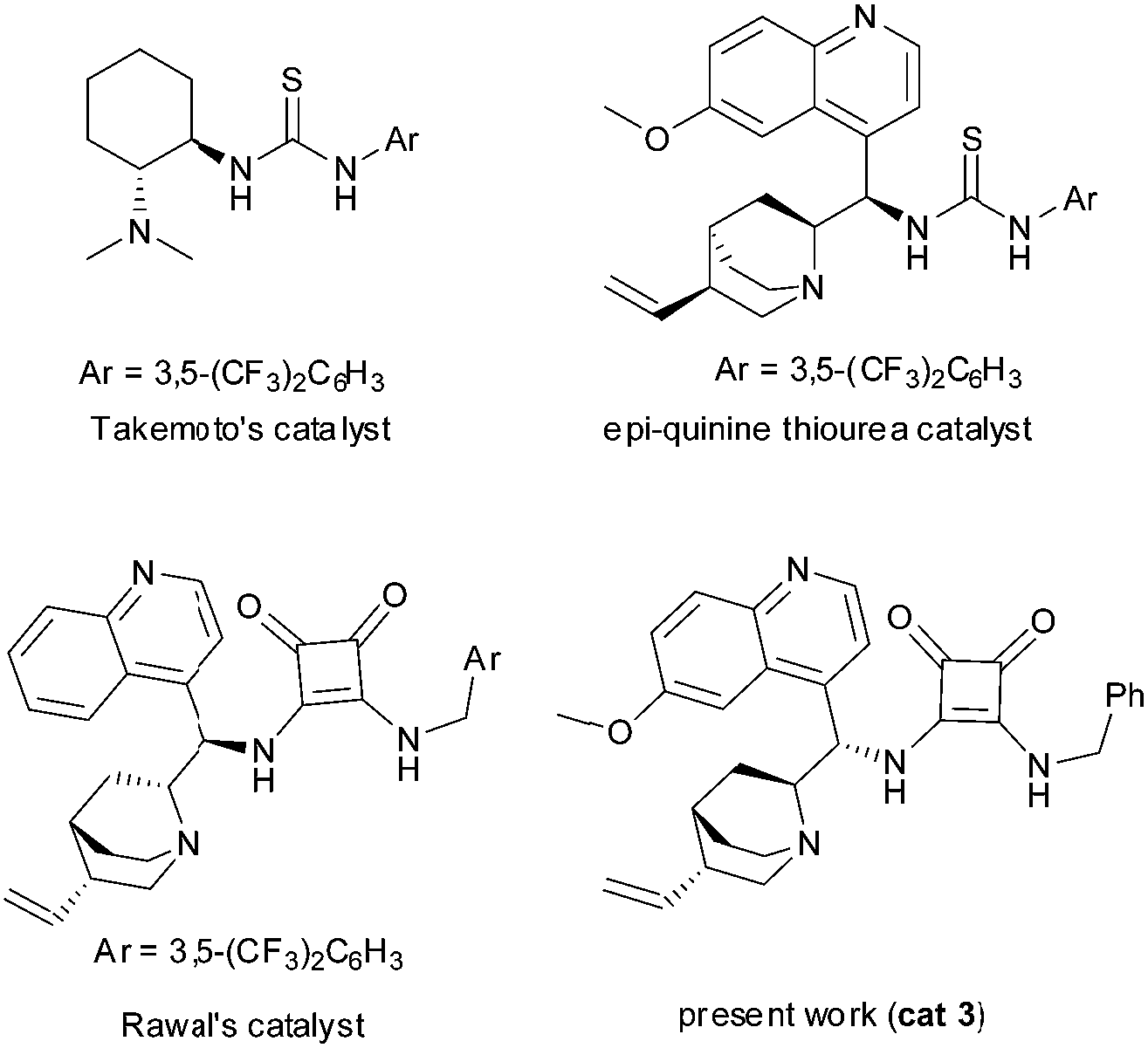 | ||
| Scheme 1 Bifunctional thiourea- and squaramide-based organocatalysts. | ||
Despite the apparent impact of these catalysts on the contemporary asymmetric catalysis, the mechanism of these transformations is still far from being well understood. Based on experimental and theoretical studies,11 the generally accepted mechanism of bifunctional organocatalyzed Michael addition involves the simultaneous and dual activation of the nucleophile and the electrophile through hydrogen bonds. Nevertheless, theoretical studies on the substrates–catalyst ternary complex have disclosed the subtleties of the bifunctional activation and three distinctly different pathways have been postulated (Scheme 2, routes A–C).11a,b,e While all of these routes involve the dual activation of reactants, these pathways are different in which hydrogen bond interacts with the incoming electrophile. To unify these alternative pathways, we have recently formulated the chiral oxyanion hole concept.12 Finally, our parallel solution phase NMR studies of bifunctional thiourea organocatalysts have revealed additional structural and dynamic consequence of bifunctionality, including catalyst dimerization and a possible self-activation mechanism.13
 | ||
| Scheme 2 Postulated transition state variants in bifunctional organocatalyzed reactions. | ||
As a continuation of our efforts to gain deep insight into non-covalent bifunctional organocatalysis, herein, we report the detailed kinetic investigation of a bifunctional squaramide organocatalyst promoted Michael addition, which study reinforces the deciding role of the ternary complex, suggests the possible sequence of steps and provide additional subtleties about the bifunctionality; the stereoablative retro-Michael reaction.
Results and discussion
Kinetic studies were performed on Michael addition between β-nitrostyrene (1) and acetylacetone (2) promoted by a cinchona based bifunctional organocatalyst (3) (Scheme 3). The applied catalyst was recently reported as an efficient and readily available organocatalyst (easily accessible even in multi-gram scale).14 This benzyl substituted catalyst (3) exhibits similar activity to its CF3 congener (Scheme 1), a catalyst that was developed earlier by Rawal.9c This model Michael reaction was selected because it was relatively fast in the presence of 1 mol% of catalyst (3) and resulted Michael adduct (4) with high enantioselectivity (>99%) and yield (98%).14 The kinetic parameters were obtained by in situ IR and NMR measurements and the limiting factor of the reaction, e.g. catalyst deactivation, substrate and product inhibitions were investigated as well. The application of in situ spectroscopy was also expected to provide molecular level information concerning possible key intermediates in real time.15,16 | ||
| Scheme 3 Application of cinchona–squaramide bifunctional organocatalyst (3) in the Michael addition between β-nitrostyrene (1) and acetylacetone (2). | ||
Characterization of the catalyst
At the outset, NMR spectroscopy and X-ray crystallography studies of the catalyst were performed to characterize the catalyst in solid and solution phase. The CDCl3 solution phase structure of the catalyst (3) was determined by 1H- and 13C-NMR spectroscopy at 310 and 320 K, respectively. Assignments were confirmed by 2D-COSY, 2D-HSQC, 2D-HMBC and NOESY methods.17 The catalyst has two antagonistic active sites, the double hydrogen bond donor squaramide moiety and the tertiary amine, and their spatial arrangement is critical for the dual activation of substrates. Based on the comparison of the 1H-NMR spectra of catalyst (3) at two different temperatures, it can be seen that the molecule can freely rotate around the critical C8–C9 bond. Consequently, dynamic equilibrium of multiple conformers exists at ambient temperature and the spatial orientation of the active sites can vary.The crystal structure of the catalyst (3) was then determined by X-ray crystallography (Fig. 1)18 and proved to be consistent with the solution phase structures. Nevertheless, owing to the influence of lattice forces, the solid phase conformer is not necessarily the most stable of the feasible molecular conformations.
 | ||
| Fig. 1 The crystallographically independent molecule in the asymmetric unit of the crystal (3) with the atomic labelling. Displacement ellipsoids are drawn at the 30% probability level. | ||
Kinetic studies
To determine the rate law and elucidate the composition of the rate-determining step, kinetic studies were conducted using the method of initial rates in synthetically relevant concentration range. The model reaction was monitored by in situ IR spectroscopy (Fig. 2) and the band of β-nitrosytrene (1) at 1345 cm−1, band of acetylacetone (2) at 1621 cm−1 and band of the product (3) at 1556 cm−1 were selected to determine the concentration of corresponding species in CHCl3 solution in real time. After calibration and testing the reproducibility of the measurement, the reaction orders in reactants and in the catalyst were determined.17 It is important to note that we were not able to detect any transient species during these studies.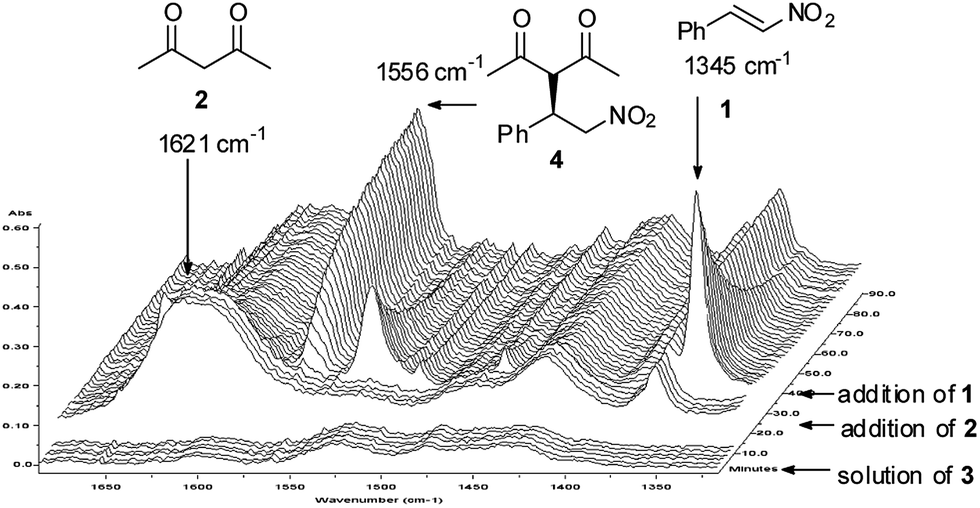 | ||
| Fig. 2 Representative 3D plot of IR spectra for the Michael addition of (1) and (2) using catalyst (3). Conditions: [1]0 = 0.3 M; [2]0 = 0.6 M; [3] = 0.006 M in CHCl3, T = 25 °C. | ||
The catalyst order in the rate law
The reaction was carried out at different catalyst loadings (1–7 mol%). The catalyst concentrations were varied between 0.003 M and 0.021 M in two series of experiments as follows: using twofold β-nitrostyrene (1) or twofold acetylacetone (2) excess. When [1]0 = 0.3 M and [2]0 = 0.6 M were applied, and the concentration of (3) was varied, an excellent linear correlation could be obtained (Fig. 3A). By applying [1]0 = 0.6 and [2]0 = 0.3 M, similar trend for the initial rates were achieved under identical conditions (Fig. 3B). Accordingly, the reaction rate shows first order dependence in the concentration of catalyst (3). While bifunctional organocatalysts tend to aggregate in solution,13 the reaction was still first order in catalyst (3).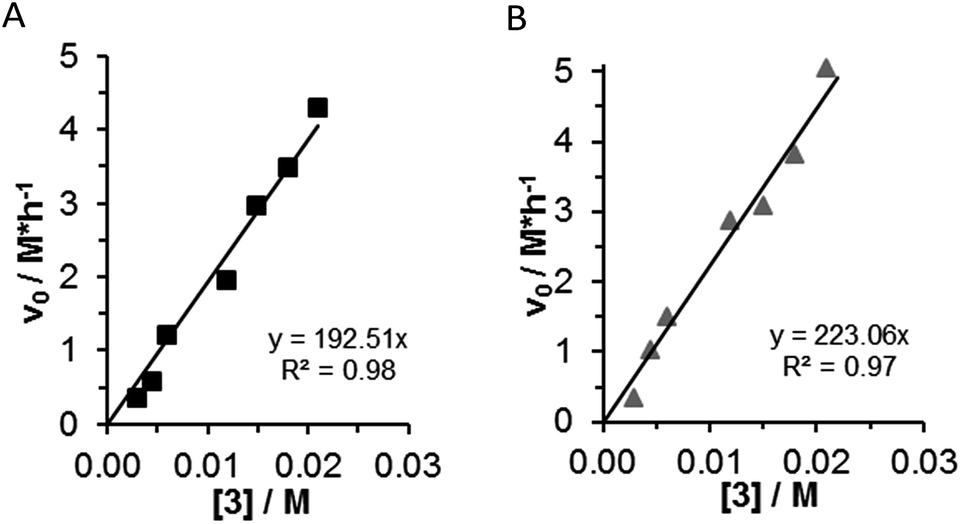 | ||
| Fig. 3 Initial reaction rate vs. catalyst (3) concentration by two sets of experiments. (A) In the case of nucleophile excess, square: [1]0 = 0.3 M; [2]0 = 0.6 M; [3] = 0.003–0.021 M; (B) in the case of electrophile excess, triangle [1]0 = 0.6 M; [2]0 = 0.3 M; [3] = 0.003–0.021 M. | ||
The nucleophilic substrate complexation and order in the rate law
Theoretical studies on bifunctional thiourea catalysis indicated that the nucleophilic substrate could be deprotonated by the catalyst.19 Therefore, this binary complex formation might be the preceding pre-equilibria step of the reaction sequence before the rate-determining step.In the case of the bifunctional squaramide catalyst 3, the immediate formation of acetylacetone/catalyst complex could be detected by NMR. The stoichiometry and the strength of this complex formation were then determined by Job's method.
The (2–3) complex formation was systematically studied by 1H-NMR spectroscopy using catalyst 3 concentrations between 0.02 and 0.2 M. The expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex was verified by the Job plot and the association constants were determined by fitting the data to a 1:1 complexation isotherm using a nonlinear curve fitting (Fig. 4).20 The constant proved to be Ka = 98.89 ± 1.02 M−1. Since the determined association constants (K) fall in the range of 10 to 1000 M−1, the applied NMR method is indeed adequate for accurate equilibrium constant determination in the system under investigation.21
:1 complex was verified by the Job plot and the association constants were determined by fitting the data to a 1:1 complexation isotherm using a nonlinear curve fitting (Fig. 4).20 The constant proved to be Ka = 98.89 ± 1.02 M−1. Since the determined association constants (K) fall in the range of 10 to 1000 M−1, the applied NMR method is indeed adequate for accurate equilibrium constant determination in the system under investigation.21
 | ||
| Fig. 4 (A) Curve-fitting on measured chemical shifts-difference. (B) Plot of weighted 1H-NMR shifts versus mole fraction and the calculated complex concentrations versus mole fraction. | ||
Next, the order of the acetylacetone (2) in the rate law was determined by kinetic measurements at two different β-nitrostyrene ([1]0 = 0.3 M; 0.6 M) initial concentrations. The concentration of acetylacetone (2) was varied between 0.15–1.5 M. The studied range can be separated into two regimes. At low initial concentrations ([2]0 = 0.15–0.75 M) of acetylacetone (2) the reaction has a non-saturation kinetic but less than first order in nucleophile. The order of 2 was determined by logarithmic transformation of initial reaction rates (Fig. 5B). At low β-nitrostyrene initial concentration ([1]0 = 0.3 M, □ on Fig. 5A and B) the order of the nucleophile (2) was found 0.9 with kobs,1 = 1.47 ± 0.27 h−1 observed rate constant, while at higher β-nitrostyrene concentration ([1]0 = 0.6 M, Δ on Fig. 5A and B) the order of nucleophile decreased; it became 0.5 with the observed rate constant of kobs,2 = 2.72 ± 0.49 h−1 (Fig. 5B). These results indicated that there was a competition between substrates for active sites of the catalyst and nitrostyrene 1 could form a non-productive complex with the catalyst 3.
 | ||
| Fig. 5 Determination of reaction order in acetylacetone (2), (A) initial reaction rate vs. initial concentration of (2), [2]0 = 0.15–1.5 M, [3] = 0.006 M, square [1]0 = 0.3 M, triangle [1]0 = 0.6 M, (B) natural logarithm of initial reaction rate. | ||
Above [2]0 = 0.75 M the reaction rate did not increase further and the reaction showed saturation kinetics. Interestingly, above a certain initial concentration, acetylacetone (2) could inhibit the reaction.22 So one can infer that the excess of the acetylacetone (2) encroached on the accessible binding site of acetylacetone/catalyst 1:1 pre-complex suggesting that the reaction was not a base catalysed (monofunctional) reaction, but followed a bifunctional mechanism.
The electrophilic substrate order in the rate law
First, the catalyst–electrophile interaction was investigated by NMR spectroscopy to gauge the complexation ability, but only a slight change of the NMR chemical shift of the catalyst was detected and these shifts did not correlate with the concentration change. Consequently, the parameters of complex formation could not be determined. These observations indicate that either no complex or weak non-specific complex formed between the catalyst 3 and β-nitrostyrene (1).23The reaction rate dependence on the concentration of β-nitrostyrene (1) was also studied at two different initial concentration of acetylacetone ([2]0 = 0.3 M; 0.6 M). The initial concentration of the electrophile (1) was varied from 0.3 to 1.1 M. By increasing the concentration of (1), increasing reaction rate could be observed achieving a maximum at [1]0 = 0.8 M (Fig. 6A). Excellent linear correlation was observed in the concentration range of 0.3–0.7 M of electrophile 1. Above 0.8 M β-nitrostyrene (1) concentration, a slight decrease in the reaction rate was detected. The first order dependence on (1) was found in 0.3–0.7 M electrophile initial concentration regime (Fig. 6B). Value of kobs,3 = 1.73 ± 0.31 h−1 was obtained as observed rate constant ([2]0 = 0.3 M, ■), while at high [2]0 concentration (▲) the observed rate constant was calculated as kobs,4 = 3.15 ± 0.57 h−1.
 | ||
| Fig. 6 Determination of order in β-nitrostyrene (1), (A) initial reaction rate vs. initial concentration of (1), [1]0 = 0.3–1.1 M, [3] = 0.006 M, square [2]0 = 0.3 M, triangle [2]0 = 0.6 M; (B) natural logarithm of initial reaction rate. | ||
Based on these findings, it seems reasonable to conclude that the acetylacetone (2) binding was the first step of the reaction sequence and because the high concentration of the electrophile 1 hindered the access of the nucleophile to the catalyst site causing the reaction-rate decrease. Additionally, these experiments could reinforce again that there was a competitive binding of substrates to the same active site of the catalyst 3.
The product inhibition
The saturation kinetics at high substrate initial concentration (both at [1]0, [2]0) can imply also product inhibition. In order to investigate the possible inhibition effect of the product, the reaction was monitored in the presence of different concentrations of the chiral product ((R)-3-(2-nitro-1-phenylethyl)pentane-2,4-dione, R-4) and its enantiomer (S-4).24 Different concentration of R- and S-Michael adduct 4 was added to the reaction mixture and strong product inhibition was observed (Fig. 7). The acetylacetone equivalent product ([R-4]0 = 0.3 M) caused 50% rate decrease. The determined order of the product in the rate law was −0.5. Interestingly, the enantiomer product (S-4) had practically the same inhibitory effect, so the inhibition had no enantiospecificity.17 | ||
| Fig. 7 (A) Initial reaction rate at different initial product (circle R-4, or × S-4) concentration, [1]0 = 0.6 M; [2]0 = 0.3 M, [3] = 0.006 M. (B) Natural logarithm of initial reaction rate in the presence R-4. | ||
This prompted us to study the Michael adducts–catalyst interaction via NMR spectroscopy. The concentrations of the applied Michael adducts (R-4, S-4, or the racemic RS-4) were in the range as the catalyst was used for the reaction (0.006 M). The measurements were carried out in CDCl3. Nearly all protons of catalyst 3 were shifted in the Michael adduct–catalyst mixtures (using R-4 or S-4) compare to the free catalyst solution (Fig. 8A–C). The slight change in the NMR spectra of catalyst (Δ = 0.01–0.1 ppm) indicates that the catalyst can form a complex or set of complexes with both Michael adducts but these complexes are weak. During these studies, an interesting observation was also made, in the case of (R-4) product–catalyst 1:1 mixture, about 25% of the product transformed back to starting materials within one hour (Fig. 8A). The chiral organocatalyst (3), however, could not promote this retro-Michael reaction on the enantiomer of the product (S-4). Furthermore, applying the racemic mixture of product (RS-4), only one of the enantiomers could undergo the reversible process affording an enantioriched Michael adduct in which the S-enantiomer was enriched (Table 1). These findings indicate that the bifunctional organocatalyst can control not only the stereoselective formation but also the stereoselective decomposition of a Michael adduct according to the principle of microscopic reversibility (the stereo-determining transition state for forward and back reaction is identical).25 Accordingly, the above capacity of 3 indicates that this type of catalyst might be utilized for stereoablative processes.26
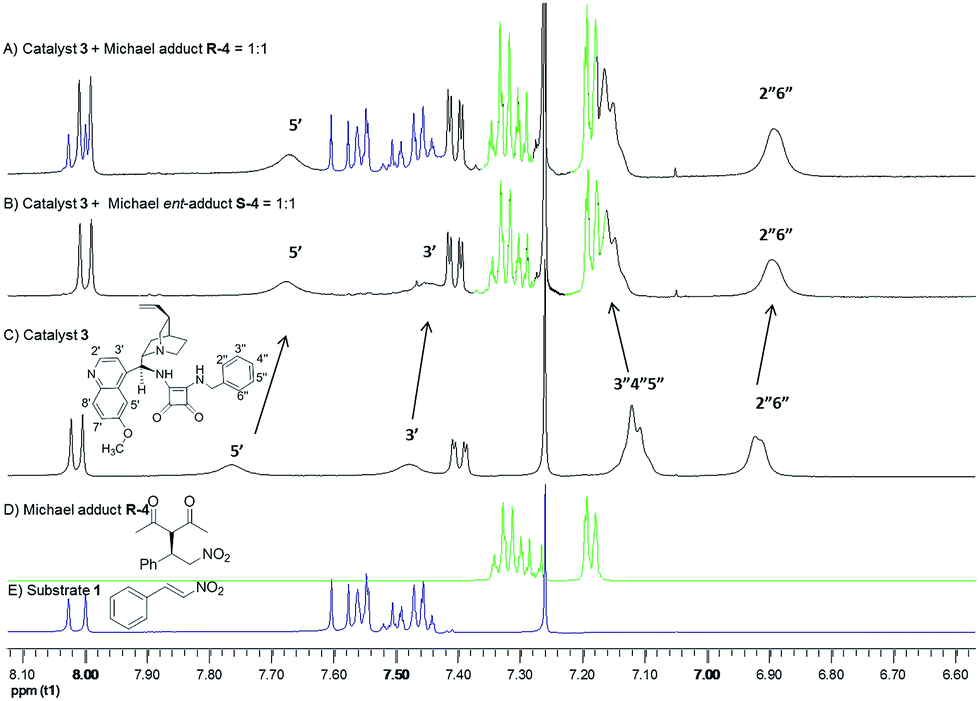 | ||
| Fig. 8 NMR complexation studies of the R-4 or S-4 Michael adducts–catalyst mixtures. | ||
| Substrate | Ratio | ee/% | Retro-Michael product/% |
|---|---|---|---|
| Michael adduct (R-4) | 1:1 |
95 | 25 |
| Michael ent-adduct (S-4) | 1:1 |
−97.4 | 0 |
| Michael rac-adduct (RS-4) | 1:1 |
−23.0 | 20 |
The activation energy
The activation energy was acquired by Arrhenius plot analysis with the measurement of reactions rates between 0–45 °C. The reaction rate markedly increased at higher temperature, the reaction time was only 20 minutes at 45 °C compared to the 2 h at 0 °C using 2 mol% catalyst (Fig. 9A). The apparent activation energy of the reaction was deduced to be 20.8 ± 3.7 kJ mol−1 according to the Arrhenius equation (Fig. 9B). This activation energy is consistent with calculated parameters of related organocatalyzed reactions.27 | ||
| Fig. 9 (A) Reaction rate versus time at different temperature. (B) Arrhenius diagram. | ||
The empirical rate law
Based on the above kinetic studies, the investigated organocatalytic reaction could be divided into a non-saturated and a saturated regime. If none of the substrates are in large excess, the reaction rate shows approximately linear behaviour, while in the case of elevated concentration of one of the substrates the reaction shows a saturation kinetic profile. Furthermore, the reaction rate shows first order dependence on the electrophilic substrate and the catalyst, near first order dependence of the nucleophilic substrate and the product has inhibitory effect.| Rate = d[4]/dt = k·[1]·[2]m·[3]·[4]−0.5, |
The observed deviations from simple first-order kinetic behaviour of 2 at elevated concentration of substrate 1 indicate non-productive interactions with the bifunctional catalyst. Nevertheless, the data consistent with the formation of a ternary 1:1:1 complex, 1:2:3, in the rate-determining step.
Mechanistic considerations
The above kinetic experiments corroborate the previous theoretical works that suggest the decisive role of the ternary complex in the rate-determining step.11 Furthermore, based on the above studies, the following catalytic cycle is proposed (Scheme 4).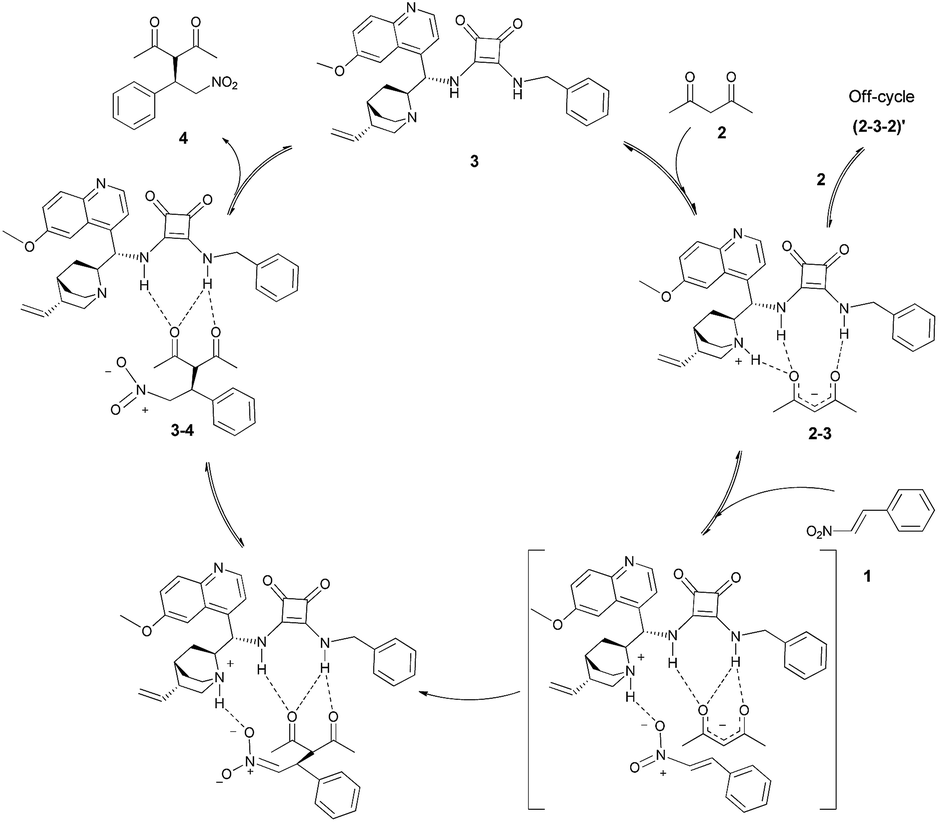 | ||
| Scheme 4 Catalytic cycle of the Michael addition of β-nitrostyrene (1) to acetylacetone (2) using a cinchona based squaramide catalyst 3. | ||
The first step of this multistep reaction is the formation of the adduct 2–3 of acetylacetone (2) and the catalyst 3. The strength of association constant of catalyst–nucleophile complex (Ka = 98.89 ± 1.02 M−1) indicates a substantial complexation between the catalyst and the nucleophile, that seems to be the pre-complex of the rate-determining step. The non-integer rate order dependence on acetylacetone (2) suggests that this pre-complex 2–3 and acetylacetone (2) may also be involved in non-productive association. The formation of this off-cycle product 2–3–2 is also implies the inhibition of the catalytic process when large excess of acetylacetone (2) is at present.
Afterwards, the other reactant, the β-nitrostyrene (1) reacts with the pre-complex 2–3via composing a ternary complex and forming a new C–C bond. In this case the limiting factor is the amount of 1 and the adduct of 2–3. So this step is the rate-determining step of the overall process as first order kinetics was observed for β-nitrostyrene (1). Next, the catalyst reprotonates the anionic Michael adduct and the product dissociates from the catalyst 3. This step may also be equilibrium, as proved by the inhibition and complexation studies. In summary, the access to the active sites of the catalyst is the limiting factor of the reaction rate. At high concentration of any of substrates, or added product, there is competition for the active site of the catalyst and not every complexation is productive.
Conclusions
In this article, we have conducted both kinetic and complexation studies on Michael reaction of acetylacetone and β-nitrostyrene using a cinchona based squaramide catalyst, at a synthetically relevant concentration range ([1]0 = 0.30–1.10 M; [2]0 = 0.15–1.50 M, [3] = 0.003–0.021 M). The main conclusions of our investigations:1. The reaction rate shows nearly first order dependence on both substrates at a concentration range ([1]0 = 0.30–0.80 M; [2]0 = 0.15–0.75 M). Out of this range substrate inhibitory effect was observed, the reaction rate did not increase further because of the competition of the substrates for the same active site.
2. Based on our study, a reaction mechanism was proposed. The pre-complex formation of the catalyst 3 and nucleophile 2 is the first step of the catalytic cycle. In the second step the electrophile 1 binds to the catalyst–nucleophile pre-complex. The simultaneous activation of both substrates by bifunctional catalyst was conceived in accordance with the empirical rate law.
3. We observed that the bifunctional organocatalyst (3) could convert back the Michael adduct (4) to β-nitrostyrene (1) and acetylacetone (2) in a stereoselective manner.
Experimental
Materials
Reagents are commercially available, 9-amino-(9-deoxy)-epihydroquinine9b and catalyst 3 (ref. 14) were prepared as described in literature, CHCl3 was distilled from CaCl2 prior to use.Methods
Nuclear magnetic resonance spectra (NMR) were acquired on a Bruker DRX-500 (500 MHz) instrument using TMS as internal standard. All assignments of the catalyst 3 are confirmed by 2D-COSY, 2D-HSQC, 2D-HMBC and NOESY measurements. Chemical shifts (δ) are reported in ppm relative to residual solvent signals (CHCl3, 7.26 ppm for 1H-NMR). The following abbreviations are used to indicate the multiplicity in 1H NMR spectra: s, singlet; d, doublet; t, triplet; m, multiplet; bs, broad signal. 13C-NMR spectra were acquired on a broad band decoupled mode. IR spectra were recorded on ReactIR 1000 Reaction Analysis System and are reported in wavenumbers (cm−1). The enantiomeric excess (ee) of the products was determined by chiral stationary phase HPLC (Daicel Chiralpak IA columns).Infrared spectra were recorded with a ReactIR 1000 Reaction Analysis System attached to atmospheric pressure silicon (SiComp™) probe head. In situ IR experiments were performed in a three necked 25 mL glass flask. In every 2 minutes 64 scans were recorded between 4000–600 cm−1, with resolution of 4 cm−1 and the mean was converted to a single spectrum. The reaction was stopped after 1–3 h depending on the conversion. In some cases the spectra were recorded in every 1 minute registering 32 scans.
General procedure for the Michael reaction followed by in situ IR
Stock solutions of reactants were used for all reaction. The concentrations of the solutions were: acetylacetone ([2] = 3.24 M, 3338 μL in 10 mL abs. CHCl3); β-nitrostyrene ([1] = 2.47 M, 3696 mg in 10 mL abs. CHCl3), catalyst (the required amount of catalyst was dissolved in variable amount solvent due the end volume of the reaction were 5 mL). First, the catalyst solution was charged into the three necked flask equipped with IR probe head and closed with a septum. The temperature was continuously controlled. The measurement was started, and after 3 spectra the required acetylacetone (2) solution was added via needle into the flask. Further three spectra were recorded and the other reactant, solution of β-nitrostyrene (1) was added and the reaction was started. The enantiomeric excess of Michael product was determined by chiral HPLC analysis. The enantiomeric purity was determined by using IA column (hexanes/EtOH, 90:10, flow rate 1.0 mL min−1, 25 °C, 220 nm); tR = 10 min (major); tR = 14 min (minor).
Acknowledgements
The donation of the ReactIR 1000 instrument by Applied Systems Inc, a Mettler-Toledo Company, is greatly appreciated. T. Soós is grateful for the financial support from Lendület Program. L. T. Mika is grateful for the support of János Bolyai Research Scholarship of the Hungarian Academy of Sciences.Notes and references
- For some recent books and reviews, see:
(a) H. Pellissier, Tetrahedron, 2007, 63, 9267 CrossRef CAS
; (b) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC
- Representative examples of organocatalyzed reactions: with proline catalysts:
(a) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed., 1971, 10, 496 CrossRef CAS
- D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed
-
(a) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS PubMed
-
(a) H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304 CrossRef CAS PubMed
- Selected examples for kinetic studies in organocatalysis
(a) N. Halland, M. Alstrup, L. A. Kjærsgaard, M. Marigo, B. Schiøtt and K. A. Jørgensen, Chem.–Eur. J., 2005, 11, 7083 CrossRef CAS PubMed
-
(a) C. G. Swain and J. F. Brown, J. Am. Chem. Soc., 1952, 74, 2534 CrossRef CAS
-
(a) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS PubMed
-
(a) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem.
Soc., 2003, 125, 12672 CAS
- Recent reviews:
(a) S. J. Connon, Chem. Commun., 2008, 2499 RSC
-
(a) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed
- B. Kótai, G. Kardos, A. Hamza, V. Farkas, I. Pápai and T. Soós, Chem.–Eur. J., 2014, 20, 5631 CrossRef PubMed
-
(a) G. Tárkányi, P. Király, S. Varga, B. Vakulya and T. Soós, Chem.–Eur. J., 2008, 14, 6078 CrossRef PubMed
- G. Kardos and T. Soós, Eur. J. Org. Chem., 2013, 4490 CrossRef CAS
- Selected examples for application of in situ IR spectroscopy in mechanism investigation:
(a) S. Csihony, L. T. Mika, G. Vlád, K. Barta, C. P. Mehnert and I. T. Horváth, Collect. Czech. Chem. Commun., 2007, 72, 1094 CrossRef CAS
- Selected examples for application of in situ spectroscopy in the field of organocatalysis:
(a) C. A. Smith, H. Cramail and T. Tassaing, ChemCatChem, 2014, 6, 1380 CrossRef CAS
- For details see ESI†.
- Comparison of crystal structure of molecule 3 and other quinine-squaramide structures from the literature can be found in ESI†.
- Theoretical and experimental work: see ref. 11b, e, f and h.
- For nonlinear curve fitting method see: E. Joseph Billo, Excel® for Chemists: A Comprehensive Guide, John Wiley & Sons, Inc., 2001, ISBN: 0-471-39462-9; 0-471-22058-2 Search PubMed
- L. Fielding, Tetrahedron, 2000, 56, 6151 CrossRef CAS
- Large excess of substrate can lead to 2:1 substrate/catalyst off-cycle complex with the bifunctional organocatalyst, for similar phenomenon, see ref. 6b.
- Similarly, no complex or very weak complex was observed in bifunctional organocatalysis, see ref. 11e and g.
- The enantiomer (S-4) was synthetized by known process using pseudo-enantiomeric catalyst of 3, see ref. 14.
- Chiral catalysts screening based on microscopic reversibility:
(a) A. Teichert and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 3360 CrossRef CAS PubMed
- Review on catalytic enantioselective stereoablative reactions, see:
(a) J. T. Mohr, D. C. Ebner and B. M. Stoltz, Org. Biomol. Chem., 2007, 5, 3571 RSC
-
(a) E. Vedejs, O. Daugulis, L. A. Harper, J. A. MacKay and D. R. Powell, J. Org. Chem., 2003, 68, 5020 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1412465. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra19593d |
| This journal is © The Royal Society of Chemistry 2015 |