
Highly sensitive determination of perphenazine on a carbon nanocomposite ionic liquid electrode†
Farshid Fasihi,
Fatemeh Farjami* and
Gholam Hossein Shafiee
Department of Chemistry, Marvdasht Branch, Islamic Azad University, Marvdasht, Iran. E-mail: fatemehfarjami@gmail.com
First published on 15th October 2015
Abstract
Carbon nanomaterial–ionic liquid nanocomposites have provided an impressive class of materials expressly for electrochemical applications. In this work the electrocatalytic oxidation of perphenazine (PPZ) was studied at a carbon nanocomposite electrode, fabricated using multi-walled carbon nanotubes (MWCNT) and the ionic liquid 1-octylpyridinium hexaflourophosphate (OPFP). The surface of the proposed electrode was characterized by scanning electron microscopy. Adsorptive stripping voltammetry (AdSV) was applied as a highly sensitive method for the quantification of sub-micromolar amounts of PPZ. Various parameters were optimized for practical applications. It was found that the electrode showed a sensitive voltammetric response to PPZ. Cyclic voltammetry (CV) was also applied to obtain information about the reaction mechanism and for calculating the kinetic parameters. The oxidation was irreversible and exhibited adsorption controlled behavior. The electron transfer coefficient (α) value was found to be 0.57 from the slope of a Tafel plot. The anodic peak current was linear to PPZ concentration in the ranges of 5.0 × 10−8 to 3.0 × 10−5 M and 3 × 10−5 to 1.5 × 10−4 M, with correlation coefficients of 0.9987 and 0.9917, respectively. The detection limit was 2.3 × 10−8 M. Also for the amperometric procedure a linear dynamic range of 1.0 × 10−5 to 1.2 × 10−4 M with a detection limit of 2.1 × 10−6 M was obtained. The method was successfully applied for determination of PPZ content in pharmaceuticals and blood serum samples.
Introduction
More than half a century ago the neuroleptic properties of some drugs were discovered. This discovery caused further investigation to clarify antipsychotic drugs.1 Phenothiazines are traditional antipsychotics used to treat mental disorders. Their derivatives are characterized by tricyclic rings with sulfur and nitrogen atoms at positions 5 and 10. They often work by blocking dopamine D2 receptors.1,2 Perphenazine which is chemically known as 4-[3-(2-chlorophenothiazine-10-yl)propyl]-1-piperazine-ethanol belongs to the phenothiazines family. It is usually prescribed for the treatment of psychotic symptoms such as schizophrenia, Parkinson’s disease and schizoaffective psychoses in order to decrease restlessness, aggressiveness and impulsive behavior in psychotic patients.3,4 PPZ is extensively metabolized in the liver to a number of metabolites by sulfoxidation, hydroxylation, dealkylation and glucuronidation.5,6 Therefore, the determination of PPZ is important in medical screening, clinical diagnostics, and in the pharmaceutical industry to obtain the optimum therapeutic dosage in bodily fluids to minimize the risk of toxicity. Some efforts have been made over the last several decades to develop a simple, reliable and sensitive method for the determination of PPZ in pharmaceuticals and biological samples. Though for the good selectivity of high performance liquid chromatography7–11 and gas chromatography-mass spectroscopy12–14 methods, costly apparatus are needed and the experimental conditions are greatly affected by environmental factors. Spectrophotometric methods have also been applied to determine PPZ content, however the sensitivity of these methods is poor.15,16 In addition, the chemiluminescence method developed for this propose lacks selectivity.17PPZ is an electroactive molecule and electrochemical methods that are simple, sensitive, inexpensive and fast can be applied for the determination of PPZ content. To the best of our knowledge only a few electrochemical methods have been reported. A glassy carbon electrode using methylene blue as the mediator was employed to study and sense the electrocatalytic oxidation of perphenazine.18 Graphene oxide (GO), a low-dimensional carbon material which is extensively used for the biosensing of various compounds,19,20 was applied for the modification of a glassy carbon electrode and PPZ content was determined in the millimolar range.21
A carbon ionic liquid electrode (CILE) was introduced in 2006 for the first time as a new and high performance carbon composite electrode.22 The main idea for the fabrication of this new electrode was the replacement of the conventional nonconductive organic binders in carbon paste electrodes (CPEs) with a pyridinium-based ionic liquid. Some interesting features of CILEs include a wide potential window in aqueous solutions, low background current, renewable surface, resistance toward bio-molecule fouling and a rapid electron transfer.23
Carbon nanotubes (CNTs) have been used for a large number of significant applications in electroanalytical chemistry, including electrochemical sensors that are reported in the literature.24–27 CNT/IL composites have also been explored for the modification of the surface of electrodes by drop casting28 or layer by layer self-assembly.29 CNTs are also incorporated into IL modified CPEs which remarkably improves the electrochemical responses.30,31
In this work we have employed the unique properties of multi-walled carbon nanotube modified CILEs for the fabrication of a PPZ electrochemical sensor. Adsorptive stripping voltammetry was used as a very sensitive analytical method. The results illustrate that the nanocomposite electrode exhibits excellent sensitivity, selectivity, stability and very low background currents for the detection of PPZ.
Experimental
Reagents and chemicals
Graphite powder, ascorbic acid, uric acid, catechol, potassium dihydrogen phosphate, and dipotassium hydrogen phosphate were purchased from Merck and were used as received. Multi-walled carbon nanotubes with a 95% purity, o.d. = 6–9 nm and 5 μm length were obtained from Sigma-Aldrich. Perphenazine was kindly supplied by Darou Pakhsh Pharmaceutical Company (Tehran, Iran) and was used without prior purification. The ionic liquid, 1-octylpyridinium hexafluorophosphate, was synthesized as described elsewhere.32 OPFP was obtained by anion exchange of octylpyridinium iodide with ammonium hexafluorophosphate. A 1.00 × 10−2 M stock solution of the drug was prepared by dissolving an appropriate amount of PPZ in methanol (Merck) and was stored at 4 °C. Standard solutions were prepared using this stock solution. Phosphate buffer (PBS) 0.1 M, pH = 2.0 was used as the supporting electrolyte. All the solutions were freshly prepared with double distilled water. The drug-free serum samples were kindly supplied by the Blood Transfusion Organization (Fars, Iran) and belonged to healthy male volunteers. The serum samples were stored frozen until the assay.Electrode preparation
The carbon ionic liquid electrode (CILE) was fabricated by thoroughly hand-mixing the graphite powder and OPFP with a ratio of 50/50 (w/w) in a mortar and pestle, followed by packing the resulting paste firmly into the electrode cavity (1.8 mm i.d.) of a Teflon holder. In order to have better homogeneity in the composite and to the lower background current, the electrode was heated for 2 min in an oven, to a temperature above the melting point of the IL (mp ∼ 65 °C) prior to use.33 A copper wire was inserted into the carbon paste providing the electrical contact. Before each measurement, pushing an excess of paste out of the tube and then polishing the freshly exposed paste with weighing paper provided a new surface. The carbon nanotube ionic liquid nanocomposite electrode (MWCNT/CLE) was prepared in similar way with the weighted amounts of graphite powder, ionic liquid and multi walled carbon nanotubes (40%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50%:10%, wt%), respectively. The multi-walled carbon nanotube paste electrode (MWCNT/CPE) was prepared by mixing 60% graphite powder, 30% mineral oil and 10% MWCNT. The classical carbon paste electrode (CPE) was made in the same way but without adding MWCNTs to the mixture.
:50%:10%, wt%), respectively. The multi-walled carbon nanotube paste electrode (MWCNT/CPE) was prepared by mixing 60% graphite powder, 30% mineral oil and 10% MWCNT. The classical carbon paste electrode (CPE) was made in the same way but without adding MWCNTs to the mixture.
Apparatus
Voltammetric measurements were performed using an Autolab electrochemical system (Eco-Chemie, Utrecht, The Netherlands) equipped with Autolab PGSTAT-302N, GPES software (Eco-Chemie, Utrecht, The Netherlands). The electrochemical cell was assembled with a conventional three electrode system: an Ag/AgCl/KCl (3 M) reference electrode (Metrohm) and a platinum disk as the counter electrode. The working electrodes used in this study were CILE, MWCNT/CILE, MWCNT/CPE and CPE. The cell was a one compartment cell with an internal volume of 100 mL. All experiments were typically conducted at 25 °C without removing the dissolved oxygen. Scanning electron microscopy (SEM) images were obtained by using a HITACHI S-4160 field emission electron microscope (Japan). The FTIR studies were performed using a Perkin Elmer FT-IR spectrometer spectrum RX-1.Results and discussion
To gain more insight into the structural characterization of the material composite, FTIR spectra of the graphite powder, MWCNTs, IL and the nanocomposite (graphite/MWCNT/IL) were investigated and the results are shown in Fig. S-1.† The peaks appeared at 2930 cm−1 (aliphatic C–H stretching), 1660 cm−1 (stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1490 cm−1 (in-plane bending vibration of –CH3, –CH2–)34,39 and 830 cm−1 (PF6−)38 which is in keeping with the literature data showing the synthesis of the OPFP ionic liquid. Because MWCNTs do not support a static dipole moment, a relatively weak dynamic dipole moment is responsible for the observation of the infrared-active modes of MWCNTs. Thus the characteristic IR absorption peaks of MWCNTs and also graphite are very weak.38 Therefore, the observed spectral features of the composite came from only the IL. The strong band at 833 cm−1 is assigned to the PF6− anion and can be seen in both the IL and the composite spectra.38
N), 1490 cm−1 (in-plane bending vibration of –CH3, –CH2–)34,39 and 830 cm−1 (PF6−)38 which is in keeping with the literature data showing the synthesis of the OPFP ionic liquid. Because MWCNTs do not support a static dipole moment, a relatively weak dynamic dipole moment is responsible for the observation of the infrared-active modes of MWCNTs. Thus the characteristic IR absorption peaks of MWCNTs and also graphite are very weak.38 Therefore, the observed spectral features of the composite came from only the IL. The strong band at 833 cm−1 is assigned to the PF6− anion and can be seen in both the IL and the composite spectra.38
In order to characterize and investigate the nature of MWCNT/CILE, scanning electron microscopy (SEM) images were taken of this composite together with three different pastes CPE, MWCNT/CPE and CILE. As shown in Fig. 1 the electrodes using OPFP (C, D) as the binder have a clearly different morphology compared to those using mineral oil as the binder which has also been reported previously by other researchers.22,33 The electrodes containing OPFP displayed a uniform surface morphology and exclusive structure, indicating that the IL could fit well into the space between graphite particles or CNTs.
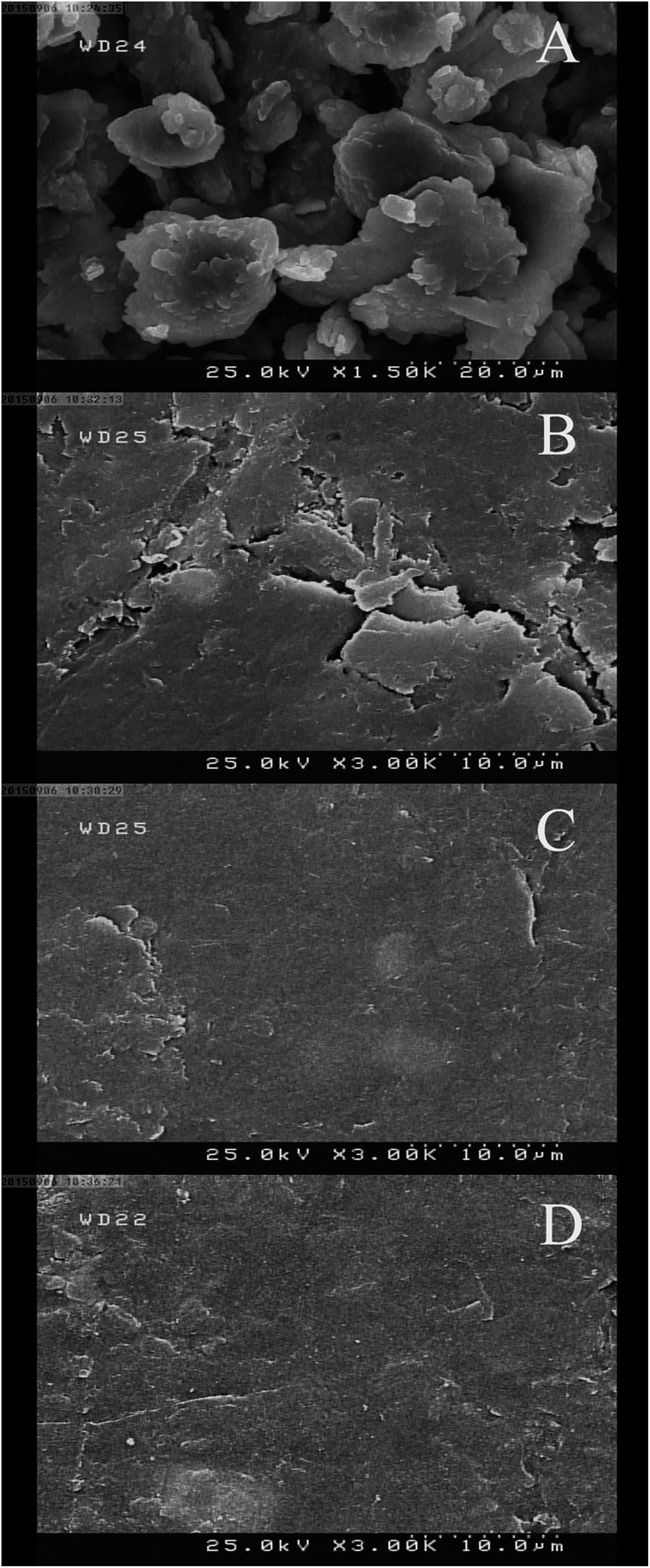 | ||
| Fig. 1 Scanning electron microscopy images of CPE (A), MWCNT/CPE (B), CILE (C) and MWCNT/CILE (D) surfaces. | ||
The interesting behavior of MWCNTs and OPFP toward the oxidation of PPZ is illustrated in Fig. 2. To elucidate the properties of the MWCNT/CILE it was essential to compare it with other electrodes. Fig. 2 shows the typical cyclic voltammograms of CPE (curve a), MWCNT/CPE (b), CILE (c) and MWCNT/CILE (d) in PBS (0.1 M pH = 2.0), containing 100 μM PPZ. In the absence of PPZ, no obvious peak (inset of Fig. 2) was observed during the potential scan. After the addition of PPZ, three oxidation peaks appeared at about +0.73 V (peak I), +0.95 V (peak II) and +1.09 V (peak III) on all electrodes. No cathodic peak was observed on the reverse scan. The oxidation current of PPZ on both CILE and MWCNT/CILE was much higher than CPE. This result clearly shows the significant effect of the IL used as a binder in both CILE and MWCNT/CILE which also increased the sensitivity of these electrodes toward the oxidation of PPZ. It is interesting to note that an increase in faradic currents at CILE and MWCNT/CILE in comparison with classic CPE and MWCNT/CPE is caused by the ionic conductivity of the binder (IL) which leads to a larger electroactive area. Before electron transfer occurs, some fraction of the polar reactant is transferred across the IL/aqueous electrolyte and the electrode reaction takes place at the carbon/IL interface within the CILE body. This is not possible for a classic CPE with a nonpolar binder, where the electrode reaction occurs at the surface of the carbon particles next to the aqueous electrolyte.35
 | ||
| Fig. 2 Cyclic voltammograms of different electrodes in PBS with pH = 2 at a scan rate of 50 mV s−1; using CPE (a) MWCNT/CPE (b) CILE (c) and MWCNT/CILE (d) in the presence of 100 μM PPZ. Inset shows the voltammograms of the corresponding electrodes in the absence of PPZ. | ||
The use of carbon nanotubes has been limited by the difficulty in handling due to their propensity to agglomerate.36 In this regard, OPFP appeared as a suitable material for the dispersion of carbon nanotubes.37 Scheme 1 demonstrates the molecular view of the fabricated nanocomposite. The strong π–π interactions between the carbon nanotubes are shielded by the ionic liquid, which ultimately hinders the rebundling of the nanotubes.37 Possible “cation–π” interactions between the surface of the MWCNTs and the cations of the ionic liquid37 (Scheme 1B) or weak van der Waals interactions38 are known as the reason for the dispersion of nanotubes by ILs such as OPFP. Therefore, the exclusive performance of the MWCNT/CILE toward the oxidation of PPZ is not only because of the MWCNTs but also due to the mixture of MWCNTs and the IL OPFP.
 | ||
| Scheme 1 Schematic representation of the dispersion process for MWCNTs in OPFP (A), the “cation–π” interactions between the surface of the MWCNTs and the cations of the ionic liquid (B) and the oxidation of PPZ on the nanocomposite surface (C). | ||
Consequently, this electrode was selected for further studies.
The influence of the MWCNT amount on the voltammetric response of the MWCNT/CILE was evaluated. Four electrodes containing different amounts of MWCNTs (1, 5, 10 and 25% weight percent ratio) were prepared and examined under identical conditions. Increasing the amount of MWCNTs up to 10% caused an improvement of the PPZ accessibility to the electrode surface active sites, therefore the oxidation peak currents were obviously improved. However composites with higher amounts of MWCNTs were unstable in solution and increased the background current. This could be due to the low mechanical stability of the composite when it contains proportionally more nanotubes compared to the IL which results in an increase in the surface area of the composite and an increase in the double layer capacitance. By decreasing the amount of MWCNTs and having more of the binder, better filling between the nanotubes was achieved which caused higher mechanical stability and lower background currents.33
The accumulation step is mainly a simple and effective way to enhance the sensitivity of the determination. The effect of the accumulation potential on the peak current of PPZ (100 μM) was examined over the range of −0.1 to 0.6 V by keeping the accumulation time of 100 s. The oxidation peak current increased up to 0 V. An increase in the accumulation time improves the sensitivity of the determination. The accumulation time was studied in the range of 0 to 450 s. The peak height increased with increasing accumulation time up to 100 s and then it levelled off. It could be concluded that the adsorption of PPZ on MWCNT/CILE became saturated. Thus all experiments were performed under an accumulation potential of 0 V and accumulation time of 100 s. In addition the optimum conditions for the DPV response were recognized by measuring the current dependence on some instrumental parameters such as modulation amplitude, step potential, interval time and modulation time to obtain the maximum signal to noise ratio and the optimum amounts were 70 mV, 10 mV, 0.1 s and 0.05 s, respectively.
In general, pH is one of the most important variables that can influence the current and shape of voltammograms. Therefore, the electro-oxidation of PPZ was studied over the pH range of 2.0–7.5 using PBS as the electrolyte. At pH values higher than 7.5 the solubility of the drug was partially decreased due to the hydrophobic behaviour of the deprotonated molecule of PPZ. Differential pulse voltammograms of 100 μM PPZ at different pH values in the mentioned range were recorded. The effect of pH on the peak currents is shown in Fig. 3A. With the raising of the pH of the solution, the peak current decreased. The greatest anodic peak current was obtained for pH = 2. Therefore, phosphate buffer with pH = 2.0 was used as the supporting electrolyte in all voltammetric determinations. The second oxidation peak disappeared at pH = 7.5. According to the previous work the cation radicals of phenothiazine derivatives are more stable in acidic solution.40
 | ||
| Fig. 3 (A) Plot of Ipa (peak I, peak II) versus pH. (B) Plot of Epc (peak I, peak II) versus pH. Recorded for a 100 μM concentration of PPZ under optimized conditions. pH values: 2.0, 3.0, 4.0, 5.0, 6.0 and 7.5. | ||
To acquire more insight about the mechanism of the electrocatalytic oxidation of PPZ, the dependence of peak potential versus pH was studied. As shown in Fig. 3B, both catalytic peak potentials shift toward less positive potentials with an increase in the pH of the solution. This behaviour suggests the involvement of protons in the overall electrode reaction. A plot of peak potential versus pH for both anodic peaks I and II is shown in Fig. 3B. For peak I a linear portion is observed in the pH range of 2.0 to 7.5, with a slope of 0.0292 V/pH. The following equation displays correlation between the peak potential and pH: Epa (V) = 0.7729 − 0.0292 pH; and R2 = 0.9898. The slope of 0.0296 V/pH implies that in the electrode reaction the number of electrons transferred was two times that of protons according to the following equation:41
| 0.0592(h/n)V/pH | (1) |
 | ||
| Scheme 2 The proposed mechanism for the electrooxidation of PPZ. | ||
To study the prevailing type of mass transport, the effect of the sweep rate on the electrooxidation of PPZ at the MWCNT/CILE was investigated by cyclic voltammetry. Fig. 4 shows the cyclic voltammograms corresponding to the response of MWCNT/CILE in presence of 100 μM PPZ at different sweep rates. Both anodic peak currents were linearly proportional to the sweep rate over the range 5–100 mV s−1 which indicates that the oxidation is controlled by an adsorptive process (Fig. 4 inset). In addition, with increasing sweep rate, the oxidation peak potentials shift to more positive values. This positive shift in the peak potential also confirms the irreversibility of the electrooxidation process. The plot of log anodic peak current versus log sweep rate was also linear over the range 5–100 mV s−1 for both anodic peaks I and II. A slope of 1.00 is expected for an ideal reaction of surface species and the slopes of 0.91 and 0.89 for the anodic peaks I and II, respectively, are close to the expected value establishing that the PPZ electrooxidation reaction is an adsorption controlled process.
 | ||
| Fig. 4 Cyclic voltammograms of MWCNT/CILE in PBS (pH = 2.0) containing 100 μM PPZ at various sweep rates; sweep rate: 5, 10, 20, 30, 40, 50, 60, 70, 80, 100, 200, 300, 400, 500, 600 and 800 mV s−1. Inset: linear dependence of peak current I and II on sweep rate in the range of 5–100 mV s−1. | ||
There is also a linear relation between the peak potential and the log of the sweep rate with a correlation coefficient of 0.9958 from the following equation: Epa = 0.0354logν + 0.6141. The Tafel slope (b) can be obtained from the slope of anodic peak potential versus log of sweep rate using eqn (2):43
|
Epa = b/2logν + constant
| (2) |
The Tafel slope was found to be 70 mV. Using the number of electrons that were involved during the PPZ oxidation in 0.01 M PBS at MWCNT/CILE, and the Tafel slope, the value of the transfer coefficient was calculated according to following equation:44
| b = (2.303RT)/[(1 − α)nF] | (3) |
The value of α was found to be 0.57.
After optimization of the operating conditions, the calibration curve for PPZ was characterized by differential pulse voltammetry. Fig. 5A illustrates the differential pulse voltammograms obtained for a series of PPZ solutions with different concentrations in PBS (pH = 2) and the respective analytical curve (inset).
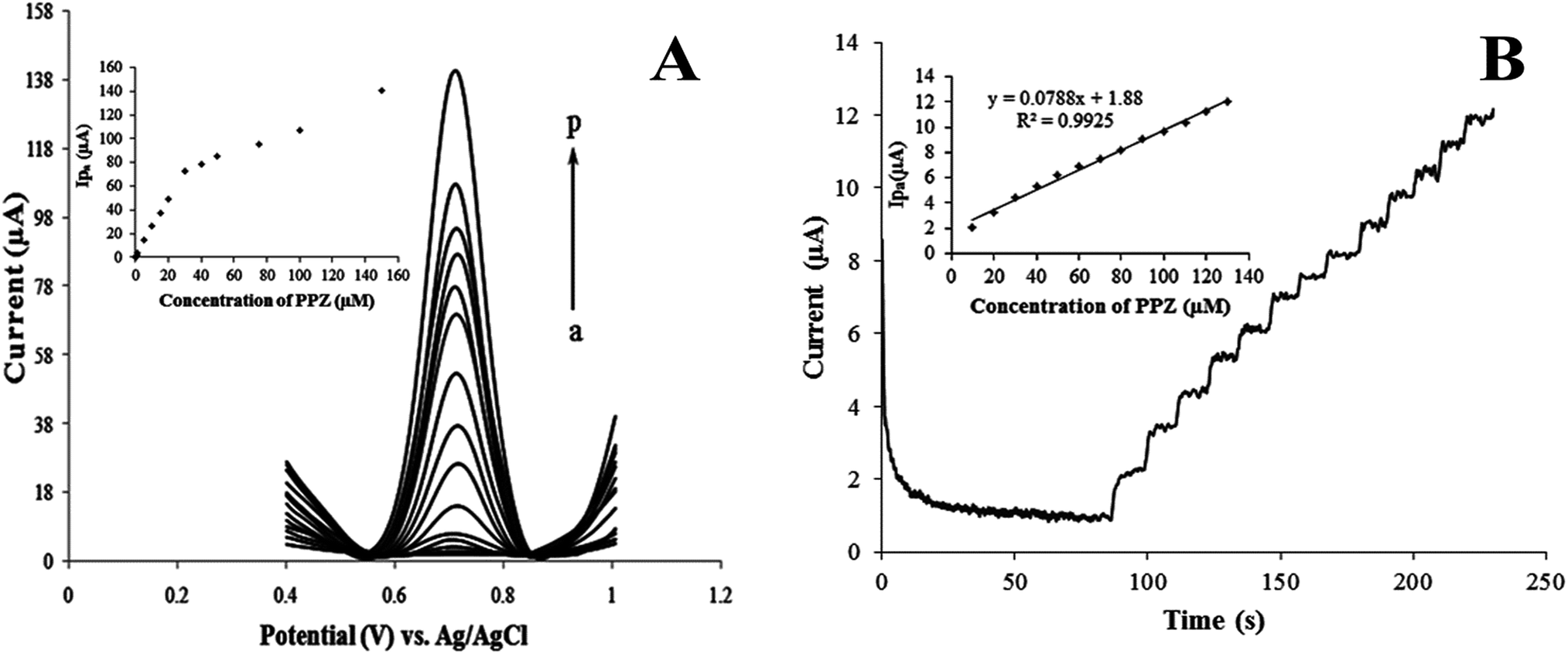 | ||
| Fig. 5 (A) Differential pulse voltammograms for various concentrations of PPZ (a) 0.05, (b) 0.1, (c) 0.2, (d) 0.4, (e) 0.8, (f) 1, (g) 5, (h) 10, (i) 15, (j) 20, (k) 30, (l) 40, (m) 50, (n) 75, (o) 100 and (p) 150 μM, at MWCNT/CILE in 0.1 M PBS (pH = 2), and accumulation time of 100 s. Inset: dependence of peak I currents on the concentration of PPZ. (B) Typical hydrodynamic amperogram of MWCNT/CILE at a constant electrode potential of 0.7 V by the successive addition of 10 μM PPZ. Inset: corresponding calibration graph. | ||
Because the relationship between the peak currents of peak II and the concentration of PPZ was not linear in any range, the data of peak I were selected. The anodic peak current was linear to PPZ concentration in the ranges of 5.0 × 10−8 to 3.0 × 10−5 M and 3 × 10−5 to 1.5 × 10−4 M. The regression equations were: ip (μA) = 2.4186CPPZ (μM) + 0.8527 for the range of 5.0 × 10−8 to 3.0 × 10−5 M (R2 = 0.9987) and ip (μA) = 0.5520CPPZ (μM) + 55.72 for the range of 3 × 10−5 to 1.5 × 10−4 M (R2 = 0.9917). It is clear that good linearity was found at lower concentrations compared with the linearity observed at higher concentrations. The difference between the slopes of the two calibration lines can be attributed to the change of accumulation efficiency.45
The sensitivity of the differential pulse voltammetry was quantified on the basis of the values obtained for the detection and quantification limits. The limit of detection (LOD) and limit of quantification (LOQ) were calculated using the parameters obtained from the analytical curve, using LOD = 3Sb/s and LOQ = 10Sb/s, where Sb is the standard deviation of the blank (n = 7) and s is the slope of the calibration curve. Under the given conditions, the calculated LOD and LOQ of PPZ were found to be 2.32 × 10−8 and 7.73 × 10−8 M, respectively. The relative standard deviation (RSD) of the seven times repeated measurement of 5 × 10−6 M PPZ with the same electrode was 1.9%, whereas the RSD was 2.4% with 5 different electrodes.
The analytical properties of the proposed methods are compared with the previously reported ones for the determination of PPZ in Table 1. As stated before, the electrochemical determination of PPZ was rarely reported. However, the MWCNT/CILE can be applied conveniently for this purpose with its higher calibration sensitivity and wider linear range compared with previous works. The easy preparation and low cost material of the proposed electrode should also be considered.
| Method | Linear range (M) | Calibration sensitivitya (μA μM−1) | Detection limit (M) | Reference |
|---|---|---|---|---|
| a NR not reported. | ||||
| Stripping voltammetry (gold electrode modified with decanethiol SAM) | 6.0 × 10−9 to 5.0 × 10−7 and 5.0 × 10−7 to 3.0 × 10−6 | 0.79 and 0.65 | NR | 45 |
| Cyclic voltammetry (glassy carbon electrode plus methylene blue) | 5.0 × 10−6 to 2.1 × 10−4 | NR | 2.08 × 10−6 | 18 |
| Amperometry (carbon fiber micro-disk bundle electrode) | 1.0 × 10−7 to 1.0 × 10−4 | NR | 5.0 × 10−8 | 3 |
| Differential pulse voltammetry and amperometry (glassy carbon electrode modified with graphene oxide nanosheets) | 8.0 × 10−4 to 8.0 × 10−3 and 7.0 × 10−4 to 7.0 × 10−3 | NR | 4.7 × 10−5 and 3.8 × 10−5 | 21 |
| Differential pulse voltammetry (MWCNT/CILE) | 5.0 × 10−8 to 3.0 × 10−5 and 3.0 × 10−5 to 1.5 × 10−4 | 2.41 and 0.55 | 2.3 × 10−8 | This work |
Hydrodynamic amperometry was also applied for the evaluation of the nanocomposite electrode susceptibility as an amperometric sensor for PPZ. Fig. 5B shows the typical steady-state amperometric response of the MWCNT/CILE with the successive addition of 10 μM PPZ into the continuously stirred PBS solution. The applied potential was kept at 0.7 V during the measurement. The linear regression equation was: ip (μA) = 0.0788CPPZ (μM) + 1.88 (R2 = 0.9925) over the range of 10–120 μM with a detection limit of 2.1 μM. This demonstrates the applicability of the MWCNT/CILE as a chromatographic detector.
To investigate the contaminant effect, various species, especially biological compounds commonly existing in serum, including glucose, ascorbic acid and uric acid were examined. The tolerance limit was defined as the maximum concentration ratio of interferent/PPZ causing an error of less than ±5.0% for the determination of PPZ. The DPV curves of the electrode toward 2 μM PPZ (PBS 0.1 M, pH = 2) in the presence of different concentrations of ascorbic acid (AA) and uric acid (UA) are shown in the ESI (Fig. S-2 and S-3†). As reported previously,46 the oxidation overpotential of ascorbic acid and uric acid significantly decreases at carbon ionic liquid electrodes due to the electrocatalytic effect of an IL used as the paste binder for the construction of these electrodes. Therefore, the anodic peaks for the oxidation of ascorbic acid and uric acid occurred at a less positive potential (0.13 V for ascorbic acid and 0.50 V for uric acid) compared to the corresponding anodic peak for PPZ oxidation (0.73 V). As shown in Fig. S-2 and S-3,† there is a significant difference between their peak potentials, and it is quite possible to detect PPZ oxidation responses with minimal interference from ascorbic acid and uric acid. As illustrated in Fig. S-2† increasing the concentration of AA up to 100 fold of the drug concentration did not affect the electrode response toward the oxidation of PPZ and also increasing the concentration of UA up to 50 fold of PPZ concentration did not interfere with PPZ determination. The effect of catechol (1,2-dihydroxybenzene) on the electrochemical response of PPZ was also studied. The oxidation of catechol was characterized by a well-defined peak at a potential of 450 mV (Fig. S-4†). As seen, increasing the concentration of catechol up to 40 fold of the drug concentration did not affect the electrode response toward the oxidation of PPZ. The results demonstrated good selectivity of the proposed electrode.
To evaluate the applicability of the proposed method for the analysis of real samples, it was applied for the determination of PPZ content in pharmaceutical preparations (nominal contents of 4 mg PPZ/tablet). Five tablets of PPZ (Daruo Pakhsh Pharmaceutical Company, Tehran, Iran) were accurately weighed and triturated to fine powder in a mortar. Then, a definite amount of the powdered sample corresponding to a solution of 1 × 10−4 M PPZ was dissolved in methanol by sonication for 10 min, filtered into a 50 mL volume calibrated flask and diluted with double distilled water. A known volume of this solution was used to spike a 25 mL aliquot of the supporting electrolyte in a volumetric flask, followed by spikes with the standard PPZ solution. A standard addition method was used in these experiments. The amounts of PPZ obtained for the pharmaceutical formulations agree well with the label contents (Table 2). Furthermore, the MWCNT/CILE was applied for the analysis of a human blood serum sample. A 10 mL human blood serum sample was deproteinized by adding 2 mL of 10% (w/w) trichloroacetic acid. Then the solution was centrifuged and diluted 10 times with 0.01 M PBS of pH = 2.0. An appropriate amount of this diluted sample was transferred to the electrochemical cell for the determination of PPZ. The sample was spiked by the drug and the DPV measurements were done as described before. The results are presented in Table 3. The results show good quantitative recoveries which imply the successful applicability of the proposed method for real sample analysis.
Conclusions
Using the unique properties of ionic liquid–MWCNT nanocomposites, we have introduced an effective electrochemical sensor for the determination of PPZ. The electrode exhibited excellent sensitivity toward the oxidation of PPZ over wide concentration ranges of 0.05–30 μM and 30–150 μM with a detection limit of 23 nM and a high calibration sensitivity of 2.41 μA μM−1 in lower concentrations. In addition the MWCNT/CILE exhibited a good response using hydrodynamic amperometry which makes it applicable as a chromatographic detector. Low background current, high selectivity and reproducibility, associated with easy and rapid preparation made this electrode ideal for the determination of PPZ in pharmaceuticals and blood samples.Acknowledgements
The authors acknowledge the support of this work by Islamic Azad University, branch of Marvdasht.Notes and references
- M. J. Gasic, O. Vukovic, M. Pantovic, T. Cvetic and N. M. Bojovic, Psychiatr. Danubina, 2012, 24, 342 Search PubMed.
- A. Kojlo, J. Karpinska, L. Kuzmicka, W. Misiuk, H. Puzanowska-Tarasiewicz and M. Tarasiewicz, J. Trace Microprobe Tech., 2001, 19, 45 CrossRef CAS.
- D. Liu and W. Jin, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 789, 411 CrossRef CAS.
- P. Norouzi, M. R. Ganjali, A. Mirabi-Semnakolaii and B. Larijani, Russ. J. Electrochem., 2008, 44, 1015 CrossRef CAS.
- H. J. Gaertner, U. Breyer and G. Liomin, Biochem. Pharmacol., 1974, 23, 303 CrossRef CAS PubMed.
- B. G. Katzung, S. B. Masters and A. J. Trevor, Basic and clinical pharmacology, McGraw-Hill Medical, New York, 12th edn, 2012 Search PubMed.
- J. P. Foglia, D. Sorisio, M. A. Kirshner, B. H. Mulsant and J. M. Perel, J. Chromatogr. B: Biomed. Sci. Appl., 1995, 668, 291 CrossRef CAS PubMed.
- G. K. Ferguson, J. Chem. Educ., 1998, 75, 1615 CrossRef CAS.
- H. Kirchherr and W. N. Kuhn-Velten, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 843, 100 CrossRef CAS PubMed.
- N. E. Larsen, J. Chromatogr., 1985, 341, 244 CrossRef CAS PubMed.
- A. Pelander, I. Ojanpera, S. Laks, I. Rasanen and E. Vuori, Anal. Chem., 2003, 75, 5710 CrossRef CAS PubMed.
- N. E. Larsen and J. Naestoft, J. Chromatogr., 1975, 109, 259 CrossRef CAS PubMed.
- E. Turunen, M. Lehtonen, T. Jarvinen and P. Jarho, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 872, 51 CrossRef CAS PubMed.
- R. Ventura, M. Casasampere, R. Berges, J. Fernandez-Moran and J. Segura, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2002, 769, 79 CrossRef CAS.
- F. Belal, F. Ibrahim, S. M. Hassan and F. A. Aly, Microchem. J., 1990, 41, 305 CrossRef CAS.
- L. Guo, Y. Zhang and Q. Li, Spectrochim. Acta, Part A, 2009, 74, 307 CrossRef PubMed.
- Y. Li, W. Niu and J. Lua, Talanta, 2007, 7, 1124 CrossRef PubMed.
- A. A. Ensafi and E. Heydari, Anal. Lett., 2008, 41, 2487 CrossRef CAS.
- X.-P. He, Q. Deng, L. Cai, C.-Z. Wang, Y. Zang, J. Li, G.-R. Chen and H. Tian, ACS Appl. Mater. Interfaces, 2014, 6, 5379 Search PubMed.
- X.-P. He, B.-W. Zhu, Y. Zang, J. Li, G.-R. Chen, H. Tian and Y.-T. Long, Chem. Sci., 2015, 6, 1996 RSC.
- H. Heli, N. Sattarahmady and S. N. Zare, RSC Adv., 2015, 5, 21005 RSC.
- N. Maleki, A. Safavi and F. Tajabadi, Anal. Chem., 2006, 78, 3820 CrossRef CAS PubMed.
- A. Safavi, R. Ahmadi and F. A. Mahyari, Amino Acids, 2014, 46, 1079 CrossRef CAS PubMed.
- F. Xiao, C. Ruan, J. Li, L. Liu, F. Zhao and B. Zeng, Electroanalysis, 2008, 20, 361 CrossRef CAS.
- N. Hameed, J. S. Church, N. V. Salim, T. L. Hanley, A. Amini and B. L. Fox, RSC Adv., 2013, 3, 20034 RSC.
- L. Zhou, J. P. Wang, L. Gai, D. J. Li and Y. B. Li, Sens. Actuators, B, 2013, 181, 65 CrossRef CAS.
- F. Zhao, X. Wu, M. Wang, Y. Liu, L. Gao and S. Dong, Anal. Chem., 2004, 76, 4960 CrossRef CAS PubMed.
- Q. Yan, F. Zhao, G. Li and B. Zeng, Electroanalysis, 2006, 18, 1075 CrossRef CAS.
- C. Xiang, Y. Zou, L. X. Sun and F. Xu, Electrochem. Commun., 2008, 10, 38 CrossRef CAS.
- R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Electroanalysis, 2007, 19, 1483 CrossRef CAS.
- N. Moheimanian, J. B. Raoof, A. Safavi and R. Ojania, Electroanalysis, 2012, 24, 1386 CrossRef CAS.
- E. Fisicaro, A. Ghiozzi, E. Pelizzetti, G. Viscardi and P. L. Quagliotto, J. Colloid Interface Sci., 1996, 182, 549 CrossRef CAS.
- R. T. Kachoosangi, M. M. Musameh, I. Abu-Yousef, J. M. Yousef, S. M. Kanan, L. Xiao, S. G. Davies, A. Russell and R. G. Compton, Anal. Chem., 2009, 81, 435 CrossRef CAS PubMed.
- P. Tian, Y. Kang, L. Liu and J. Lu, Appl. Mech. Mater., 2015, 707, 16 CrossRef CAS.
- M. Opallo and A. Lesniewski, J. Electroanal. Chem., 2011, 656, 2 CrossRef CAS.
- M. Tunckol, J. Durand and P. Serp, Carbon, 2012, 50, 4303 CrossRef CAS.
- T. Fukushima and T. Aida, Chem.–Eur. J., 2007, 13, 5048 CrossRef CAS PubMed.
- J. Wang, H. Chu and Y. Li, ACS Nano, 2008, 2, 2540 CrossRef CAS PubMed.
- A. Deyko, S. G. Hessey, P. Licence, E. A. Chernikova, V. G. Krasovskiy, L. M. Kustov and R. G. Jones, Phys. Chem. Chem. Phys., 2012, 14, 1381 RSC.
- H. Y. Cheng, P. H. Sackett and R. L. McCreery, J. Am. Chem. Soc., 1978, 100, 962 CrossRef CAS.
- A. Abbaspour and R. Mirzajani, J. Pharm. Biomed. Anal., 2007, 44, 41 CrossRef CAS PubMed.
- J. Karpinska, B. Starczewska and H. Puzanowska-tarasiewicz, Anal. Sci., 1996, 12, 161 CrossRef CAS.
- J. A. Harrison and Z. A. Khan, J. Electroanal. Chem., 1970, 28, 131 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- B. Zeng, Y. Yang, X. Ding and F. Zhao, Talanta, 2003, 61, 819 CrossRef CAS PubMed.
- A. Safavi, N. Maleki, O. Moradloi and F. Tajabadi, Anal. Biochem., 2006, 359, 224 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14639a |
| This journal is © The Royal Society of Chemistry 2015 |