DOI:
10.1039/C5RA19037A
(Paper)
RSC Adv., 2015,
5, 88539-88547
Miscible polypeptide blends of polytyrosine and poly(γ-methyl L-glutamate) with rigid-rod conformations†
Received
16th September 2015
, Accepted 12th October 2015
First published on 14th October 2015
Abstract
In this study, a new miscible rod–rod polypeptide blend system comprising polytyrosine (PTyr) and poly(γ-methyl L-glutamate) (PMLG) was developed. Differential scanning calorimetry revealed significant positive deviations in the glass transition temperatures of these PTyr/PMLG blends, arising from intermolecular hydrogen bonding between the phenolic OH groups of PTyr and the side chain C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of PMLG, as well as secondary structural changes of the polypeptides confirmed using solid state nuclear magnetic resonance spectroscopy.
O groups of PMLG, as well as secondary structural changes of the polypeptides confirmed using solid state nuclear magnetic resonance spectroscopy.
Introduction
Miscible polymer blends are attractive materials for academic research and industrial applications.1–5 Most polymer pairs are immiscible because their large molecular weights ensure a small favorable entropy term. In general, enhancing the miscibility of a polymer blend is strongly dependent on the enthalpic term, requiring favorable specific noncovalent interactions (e.g., hydrogen bonding,6–10 π–π,11,12 and dipole–dipole13,14 interactions).
In addition, miscible polymer blends are generally characterized by coil–coil polymer chains, due to the conformation of a coil polymer being more likely to impart miscible behavior through a larger favorable entropy term, compared with that of a rod-like polymer. Polypeptides, including poly(γ-methyl L-glutamate) (PMLG), poly(γ-ethyl L-glutamate) (PELG), poly(γ-benzyl L-glutamate) (PBLG), and polytyrosine (PTyr), often possess hierarchical ordering and secondary structures including α-helices [rigid-rod structures stabilized through intramolecular (intra-chain) hydrogen bonding between peptide bonds] and β-sheets [lamellar structures stabilized through intermolecular (inter-chain) hydrogen bonding between peptide bonds].15,16 In the past decade, several poly(peptide-b-nonpeptide) structures—so-called rod–coil diblock copolymers—have been investigated for their applications in drug delivery and tissue engineering.17–21 In contrast, relatively few polypeptide-based diblock copolymers (rod–rod) have been examined.22,23 The preparation of diblock copolymers is, however, time-consuming and it can be difficult to modify functional polypeptides; polymer blending has, therefore, become a relatively effective and convenient approach toward the development of such functional polypeptides.
In previous studies, we investigated the blending of polyglutamate homopolymers (as hydrogen bond acceptors) with various coil polymers, including poly(vinyl phenol) (PVPh) and phenolic resin (as hydrogen bonding donors).24,25 Painter et al.26 and we25 have reported the miscibility and hydrogen bonding interactions of PMLG, PELG, and PBLG, as rigid-rod polypeptides, with various hydrogen bond donor coil polymers (e.g., PVPh, phenolic resin). These rod–coil blend systems all formed miscible polymer blends because of the intermolecular hydrogen bonding between the side chain carbonyl (CO) groups of the polyglutamate and the phenolic hydroxyl (OH) groups of the PVPh or phenolic resin. In addition, the secondary structures of the polypeptides are correlated with the intermolecular hydrogen bonding strength, the rigidity of the side chain functional groups, the composition of the PVPh or phenolic, and the temperature of these blends.24 We have also found that various functionalized polystyrene derivatives, including polystyrene (PS), poly(acetoxystyrene) (PAS), and PVPh, display different miscibility behavior with PBLG. Only partial miscibility arose from the weak π–π interactions in the PS/PBLG blends, with the α-helical conformation of PBLG not changing upon increasing the PS content.25 In contrast, relatively strong dipole–dipole interactions in PAS/PBLG blends and strong hydrogen bonding in PVPh/PBLG led to complete miscibility, with the fraction of α-helical conformations of PBLG increasing upon increasing the PAS and PVPh contents. We have also investigated the blending of rigid-rod PTyr polypeptides, as hydrogen bond donor polymers, with poly(4-vinyl pyridine) (P4VP), a strongly hydrogen bond accepting coil polymer.27 The miscibility, hydrogen bonding interactions, and secondary structure of PTyr were strongly dependent on the solvent polarity, due to the different chain behavior for separated coils [in N,N-dimethylformamide (DMF)] and aggregated chains (in MeOH).27–29
In those previous studies, they had focused only on miscible rod–coil blend systems formed through intermolecular hydrogen bonding of polypeptides and random-coil polymers. Miscible rod–rod blend systems formed from polypeptides have, the best of our knowledge, never been reported previously. In this paper, we present a new miscible rod–rod blend system formed from PTyr and PMLG, stabilized through intermolecular hydrogen bonding between the phenolic OH groups of PTyr and the side chain CO groups of PMLG. The miscibility, hydrogen bonding interactions, and secondary structures of these PTyr/PMLG blends, as investigated using differential scanning calorimetry (DSC), Fourier transform infrared (FTIR) spectroscopy, wide-angle X-ray diffraction (WAXD), and solid state nuclear magnetic resonance (NMR) spectroscopy.
Experimental section
Materials
γ-Methyl L-glutamate N-carboxyanhydride (MLG-NCA) and L-tyrosine N-carboxyanhydride (Tyr-NCA) were synthesized according to literature procedures.24,27 Triphosgene (TCI), DMF, acetonitrile (MeCN), hexane, and tetrahydrofuran (THF) were purchased from Aldrich.
Poly(γ-methyl L-glutamate) (PMLG) and poly-L-tyrosine (PTyr)
A solution of MLG-NCA (2.00 g) and anhydrous DMF (40 mL) was stirred for 30 min and then butylamine (50 μL) was added using a N2-purged syringe. After stirring the solution for 48 h at room temperature, PMLG was precipitated in ether, isolated, and dried in an oven. A solution of Tyr-NCA (4.00 g) in anhydrous DMF (20 mL) was stirred for 30 min and then propargylamine (0.25 mL) was added using a N2-purged syringe. After stirring the solution for 48 h at 0 °C, PTyr was precipitated in ether, isolated, and dried in an oven. Table 1 summarizes the properties of these PTyr and PMLG samples.
Table 1 Characterization data for the PTyr and PMLG polymers prepared in this study
| Polymer |
Mna |
Mnb |
PDIb |
Mnc |
PDIc |
| Determined from 1H NMR spectrum (Fig. S1 and S2). Determined from GPC analysis (Fig. S3 and S4). Determined from MADLI-TOF mass spectrum (Fig. S5). |
| PTyr8 |
1030 |
3300 |
1.05 |
1350 |
1.05 |
| PMLG10 |
1430 |
6500 |
1.08 |
1500 |
1.08 |
Blend preparations
PTyr/PMGL blends of various compositions were prepared through solution blending. A DMF solution containing 10 wt% of the blend sample was stirred for 24 h, and then the solvent was evaporated slowly at 60 °C over 3 days. The polymer blend sample was then dried at 110 °C for 48 h under high vacuum to ensure removal of residual DMF.
Characterization
DSC (TA-Q20) was employed to determine the thermal properties of polymer blends. The heating and cooling rates during these measurements were both 20 °C min−1; the samples were analyzed under a N2 atmosphere. WAXD data were collected from beamline BL17A1 at the National Synchrotron Radiation Research Center (NSRRC), Taiwan. The wavelength (λ) was 1.33 Å, based on a Si (111) single crystal. FTIR spectra of the samples were recorded using the potassium bromide (KBr) disk method on a Bruker Tensor 27 apparatus, with a resolution of 4 cm−1 and 32 scans. The KBr disks including the blend samples were dried at 80 °C for 24 h prior to measurement. Solid state NMR spectra were recorded using a Bruker AVIII 600 WB spectrometer at 600 MHz and 14.1 Tesla for 13C nuclei.
Results and discussion
Thermal analyses
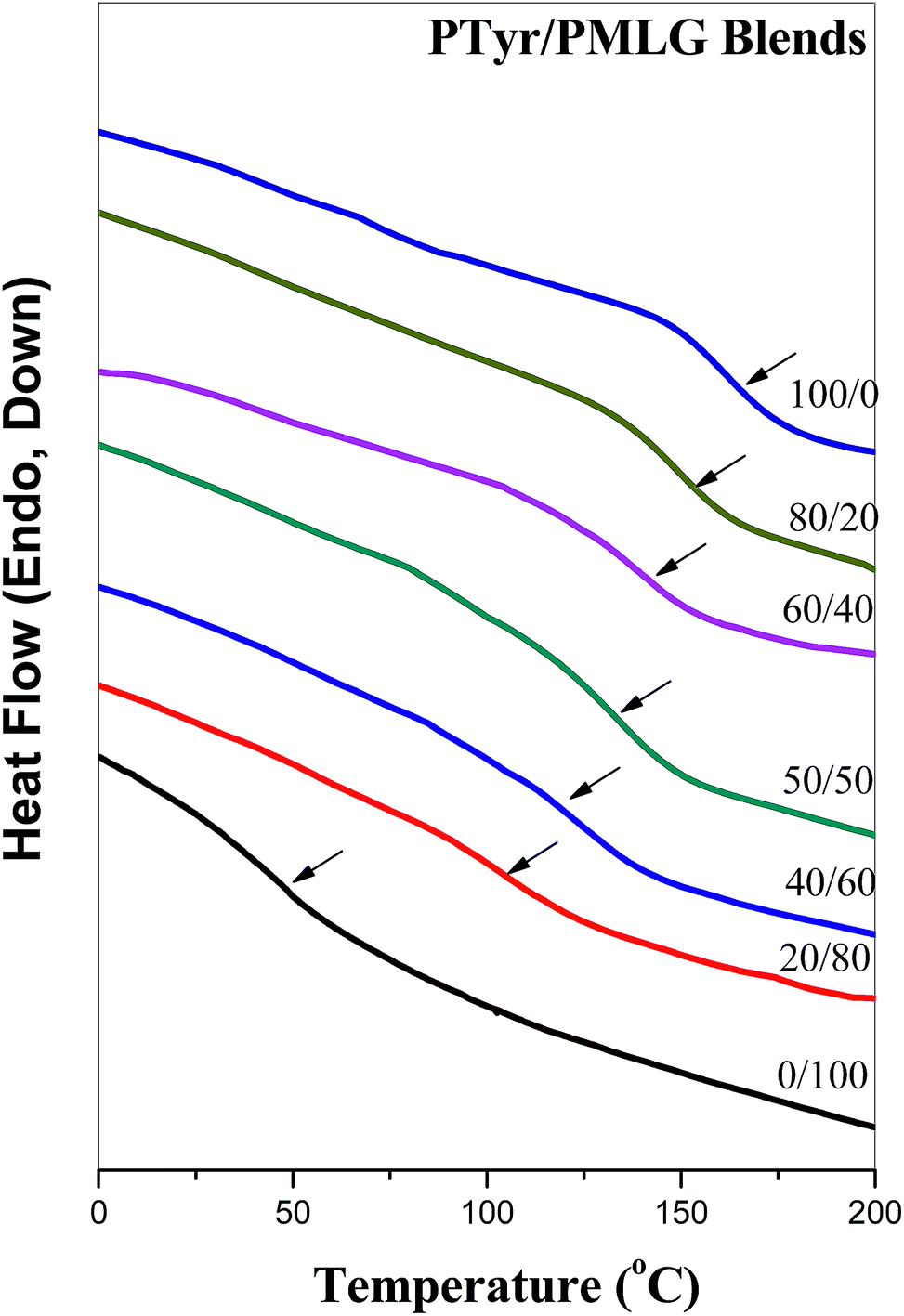
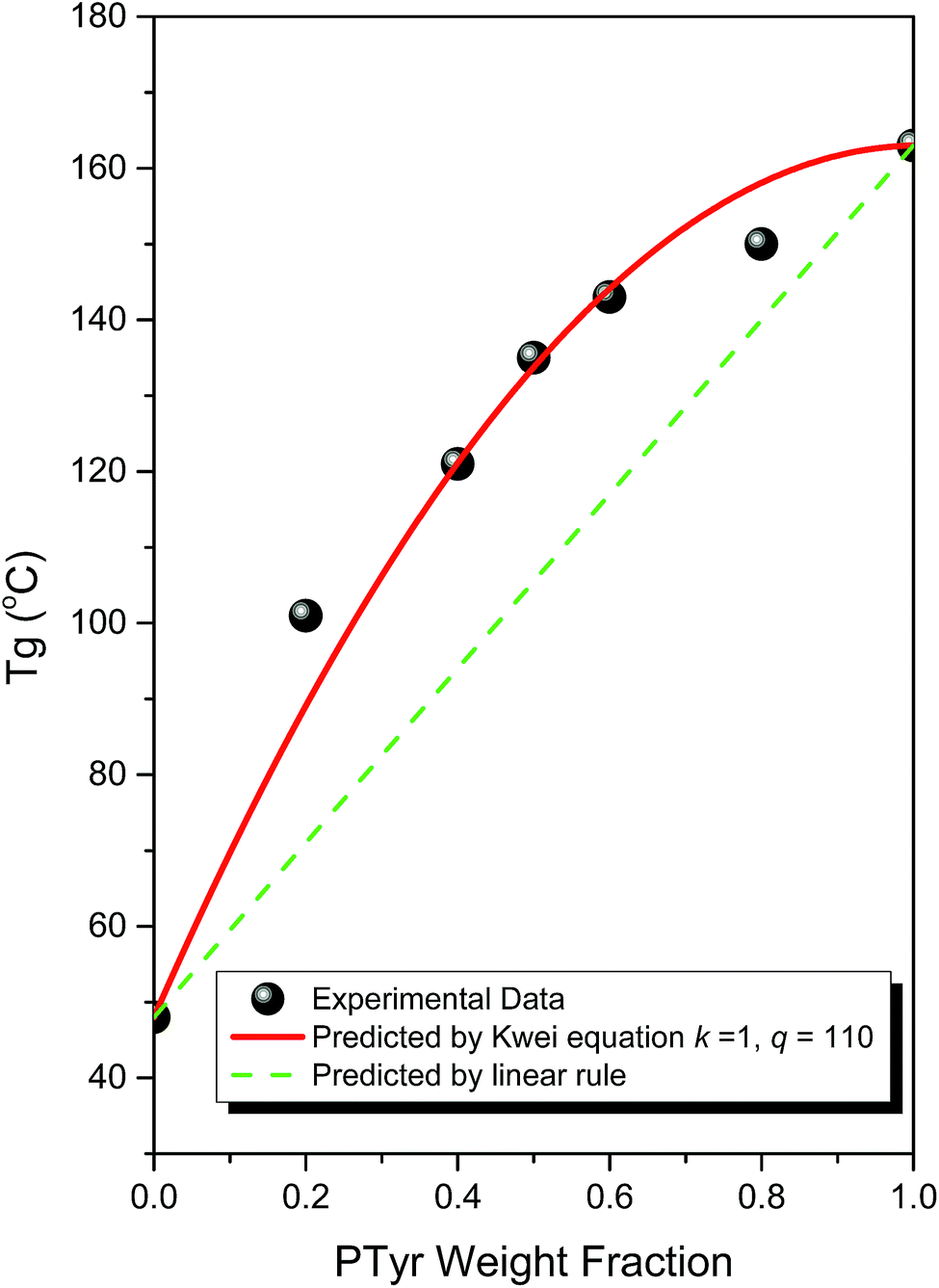
DSC can be used as a general method for determining the miscibility of hydrogen bonded polymer blend systems. Fig. 1 displays the second heating scans of PTyr/PMLG blends of various compositions. Pure PTyr and pure PMGL possessed glass transition temperatures near 163 and 48 °C, respectively. All of the binary PTyr/PMLG blends exhibited only a single glass transition temperature, implying complete miscibility with a homogeneous phase. The glass transition temperature increased significantly upon increasing the PTyr content. Fig. 2 presents the glass transition temperature composition curve of this miscible PTyr/PMGL blend; the values of the glass transition temperatures are significantly higher than those predicted using the linear rule. We used the Kwei equation to predict the glass transition temperature composition relationship for this miscible polymer blend:30| |
 | (1) |
where Wi and Tgi are the weight fractions and glass transition temperatures of each component, respectively, and k and q are fitting constants. The value of the parameter q describes the average intermolecular hydrogen bonding strength of a miscible blend system. From nonlinear fitting, we obtained values for k and q of 1 and 110, respectively. The value of q for this blend system is quite high, close to those of PVPh/P4VP (k = 1; q = 100)31 and PVPh/poly(vinylpyrrolidone) (PVPh/PVP) (k = 1; q = 140)32 blends. The pyridine groups of P4VP and the pyrrolidone groups of PVP make these coil polymers very strong hydrogen bonding acceptors, with the inter-association equilibrium constants (KA) of PVPh/P4VP and PVPh/PVP blends being 1200 and 6000, respectively; these groups are significantly better at accepting hydrogen bonds than are the CO groups of PCL (KA = 90; k = 1, q = −85)33,34 and PMMA (KA = 37, k = 1; q = −10).35 As a result, the strength of the hydrogen bonding interactions between the phenolic OH groups of PTyr and the CO groups of PMLG should be close to those found in PVPh/PCL or PVPh/PMMA blend systems. We have found, however, that the large value of q was observed in this study arose not only from strong intermolecular hydrogen bonding but also from a secondary structural change in the PTyr/PMLG blend system.
 |
| | Fig. 1 DSC thermograms of PTyr/PMLG blends of various compositions. | |
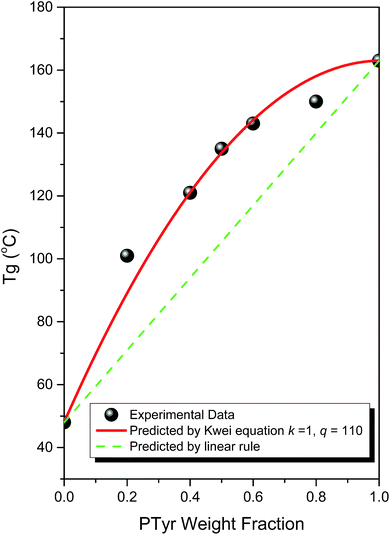 |
| | Fig. 2 Glass transition temperatures of PTyr/PMLG blends, analyzed based on the Kwei equation and the linear rule. | |
FTIR spectroscopic analyses
In general, the hydrogen bonding interactions in polymer blend systems and the secondary structures of polypeptides can be determined both qualitatively and quantitatively using infrared spectroscopy. Fig. 3 presents FTIR spectra, recorded in the range from 1850 to 1580 cm−1, displaying the signals of the side chain CO groups of PMLG and the amide I groups of PMLG and PTyr. In a previous study, the signals of pure PTyr can be resolved into eight major peaks: at 1597 and 1615 cm−1 for ring vibrations, at 1655 cm−1 for the α-helical conformation, at 1630 cm−1 for the β-sheet conformation, at 1670 cm−1 for the β-turn conformation, and at 1643, 1683, and 1700 cm−1 for the random coil conformation.27 Furthermore, four major peaks have been observed for pure PMLG: at 1740 cm−1 for the free CO groups, at 1655 cm−1 for the α-helical conformation, at 1626 cm−1 for the β-sheet conformation, and at 1692 cm−1 for the β-turn conformation.24 Because so many peaks existed in this range, it was difficult to determine the exact area fractions of the CO and amide I groups; thus, we had to ignore the some of these signals. Accordingly, for curve fitting as shown in Fig. S6† by using second derivative curve, we considered the signals of pure PTyr as five major peaks: for the ring vibrations at 1597 and 1615 cm−1, the α-helical conformation at 1655 cm−1, the β-sheet conformation at 1630 cm−1, and a combination of the signals at 1643, 1670, 1683, and 1700 cm−1 into a single signal at 1692 cm−1 for the β-turn and random-coil conformations; the latter is similar to the signal at 1692 cm−1 for the β-turn conformation of pure PMLG. Fig. 3 and Table 2 summarize the results of our curve fitting. Interestingly, a new signal for hydrogen-bonded side-chain CO groups of PMLG appeared near 1720 cm−1 at higher PTyr contents (60 and 80 wt%), implying the presence of hydrogen bonds between the side chain CO groups of PMLG and the phenolic OH groups of PTyr. At PTyr contents of 60 and 80 wt%, the area fractions of the hydrogen-bonded CO groups of PMLG were 32.6 and 42.1%, respectively.
 |
| | Fig. 3 FTIR spectra (CO and amide I stretching bands) of PTyr/PMLG blends of various compositions at room temperature, with corresponding results of curve fitting. | |
Table 2 Curve fitting data for the CO and amide I groups in the PTyr/PMLG blends at 25 °C
| Blend |
CO group of PMLG |
Amide I group in polypeptide |
| Free CO |
H-bonded CO |
β-turn |
α-helix |
β-sheet |
| PTyr/PMLG |
ν (cm−1) |
Af (%) |
ν (cm−1) |
Af (%) |
ν (cm−1) |
Af (%) |
ν (cm−1) |
Af (%) |
ν (cm−1) |
Af (%) |
| 0/100 |
1741 |
100 |
— |
— |
1694 |
19.66 |
1658 |
39.8 |
1625 |
40.6 |
| 20/80 |
— |
— |
— |
— |
1694 |
20.0 |
1658 |
35.8 |
1626 |
44.2 |
| 40/60 |
— |
— |
— |
— |
1692 |
29.1 |
1660 |
32.9 |
1627 |
35.0 |
| 50/50 |
— |
— |
— |
— |
1695 |
27.0 |
1660 |
40.8 |
1627 |
32.2 |
| 60/40 |
1741 |
67.4 |
1716 |
32.6 |
1695 |
14.1 |
1658 |
53.8 |
1628 |
32.1 |
| 80/20 |
1741 |
57.9 |
1720 |
42.1 |
1696 |
11.4 |
1661 |
69.3 |
1628 |
19.3 |
| 100/0 |
— |
— |
— |
— |
1695 |
9.5 |
1659 |
74.1 |
1630 |
16.4 |
Fig. 4 summarizes the area fractions of the secondary structures in the PTyr/PMLG blends at room temperature. Because the degrees of polymerization (DPs) of our PTyr and PMLG polymers were both less than 18, we observed both α-helical and β-sheet secondary structures for PTyr and PMLG.15 Upon blending PTyr into PMLG, the area fraction of the α-helical conformation decreased initially (at 20 wt% PTyr) and then increased thereafter. In contrast, the fraction of β-sheet conformations increased initially and then decreased thereafter. Floudas et al. reported a similar phenomenon, with the fraction of β-sheet conformations of polyalanine (PALa) decreasing in a PBLG-b-PALa diblock copolymer because of the enthalpic interaction of different peptide blocks.36 The rigid-rod conformation of the α-helix and hydrogen bonding between the side chain CO groups of PMLG and the phenolic OH groups of PTyr may have been responsible for our observed (DSC) increase in the glass transition temperature of PTyr when blended into PMLG.
 |
| | Fig. 4 Area fractions of secondary structures of PTyr/PMLG blends, based on amide I absorption signals. | |
WAXD analyses
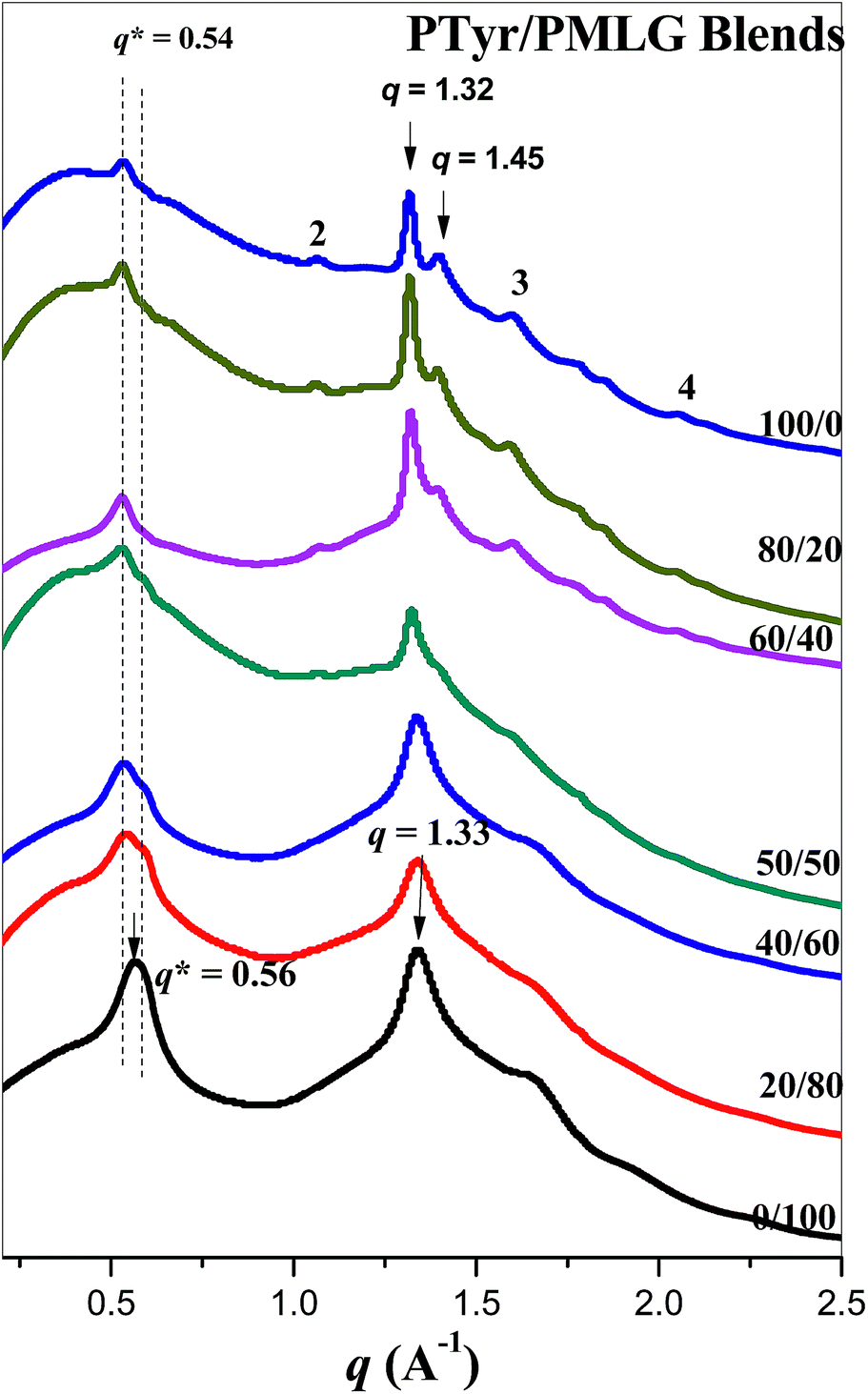
Fig. 5 presents WAXD patterns that we used to identify the secondary structures in the PTyr/PMLG blends. The diffraction pattern of pure PTyr reveals a β-sheet secondary structure, where the first peak at a value of scattering vector (q) of 0.54 (d = 1.16 nm) corresponds to the distance between the backbones in the antiparallel β-pleated sheet structure.27 The long-range order of a lamellar structure is revealed from the peak ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3:4 relative to the first-order diffraction peak. In addition, the values of q of 1.32 and 1.45 represent the intermolecular distance between adjacent peptide chains within one lamella (d = 0.475 nm) and the repeated residues of the peptide chain (d = 0.433 nm), respectively. The diffraction pattern of pure PMLG also reveals a β-sheet secondary structure, where the first peak at a value of q of 0.56 (d = 1.12 nm) also corresponds to the distance between the backbones in the antiparallel β-pleated sheet structure. In addition, the value of q of 1.33 (d = 0.472 nm) also represents the intermolecular distance between adjacent peptide chains within one lamella, similar to that for pure PTyr. The WAXD patterns of pure PTyr remained almost unchanged upon blending with PMLG at weight percentages of less than 40 wt%, implying that the PTyr remained in its β-sheet structure. A further increase in the PMLG content (>50 wt%) resulted in the long-range order of the lamellar structure disappearing, replaced by a combination of β-sheet conformations of PTyr and PMLG. Because the diffraction patterns of these two polypeptides overlapped considerably, we could not observe any clear secondary structural changes of PTyr and PMLG.
:2:3:4 relative to the first-order diffraction peak. In addition, the values of q of 1.32 and 1.45 represent the intermolecular distance between adjacent peptide chains within one lamella (d = 0.475 nm) and the repeated residues of the peptide chain (d = 0.433 nm), respectively. The diffraction pattern of pure PMLG also reveals a β-sheet secondary structure, where the first peak at a value of q of 0.56 (d = 1.12 nm) also corresponds to the distance between the backbones in the antiparallel β-pleated sheet structure. In addition, the value of q of 1.33 (d = 0.472 nm) also represents the intermolecular distance between adjacent peptide chains within one lamella, similar to that for pure PTyr. The WAXD patterns of pure PTyr remained almost unchanged upon blending with PMLG at weight percentages of less than 40 wt%, implying that the PTyr remained in its β-sheet structure. A further increase in the PMLG content (>50 wt%) resulted in the long-range order of the lamellar structure disappearing, replaced by a combination of β-sheet conformations of PTyr and PMLG. Because the diffraction patterns of these two polypeptides overlapped considerably, we could not observe any clear secondary structural changes of PTyr and PMLG.
 |
| | Fig. 5 WAXD patterns of PTyr/PMLG blends of various compositions, measured at room temperature. | |
Solid state NMR spectroscopic analyses
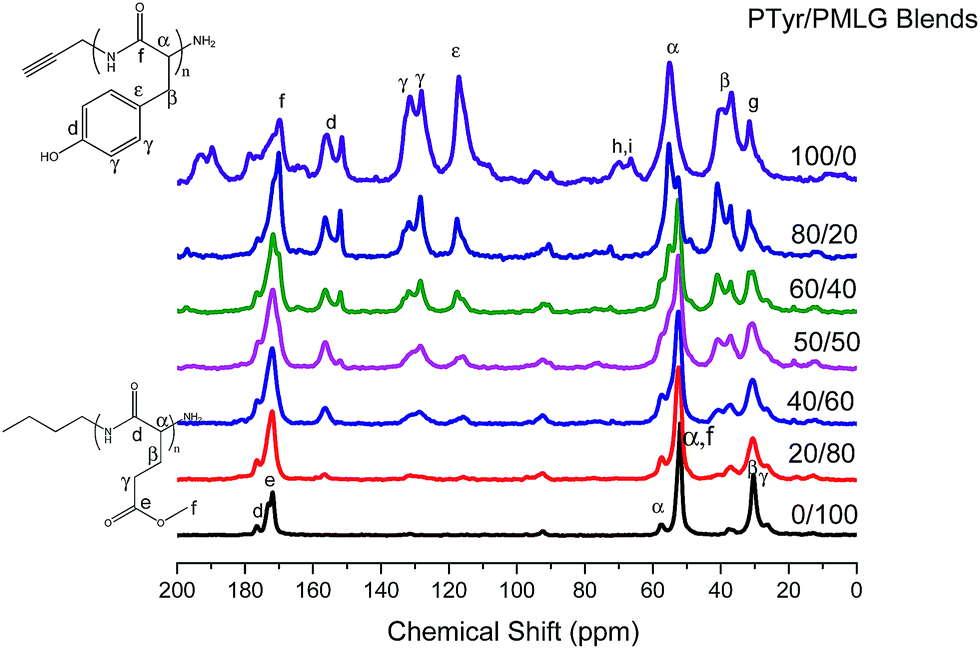
In our FTIR spectroscopic and WAXD analyses, we observed that the fraction of the β-sheet conformations decreased upon increasing the PTyr or PMLG content. Nevertheless, the signals for the secondary structures determined through FTIR spectroscopic and WAXD analyses were strongly overlapped, making it difficult to calculate the exact area fractions of secondary structures for each PTyr or PMLG segment. Thus, solid state NMR spectroscopy (Fig. 6) was used to attempt to calculate the fractions of the secondary structures in the PTyr/PMLG blends. For pure PTyr, the signals of the phenolic carbon atom (Cd) appeared at 155.9 and 151.4 ppm, corresponding to α-helical and β-sheet conformations, respectively; this phenolic OH group could stabilize the α-helical conformation of PTyr. The signals of the methylene carbon atom (Cβ) appeared at 36.1 and 39.3 ppm, corresponding to α-helical and β-sheet conformations, respectively. The signals of the corresponding Cα and amide CO carbon atoms appeared at 58.0 and 177.0 ppm, respectively, for the α-helical conformation; these signals appeared at 52.0 and 169.0 ppm, respectively, shifted upfield by approximately 3–7 ppm, for the β-sheet conformation.15,16 For pure PMLG, the signals of the Cα and the amide CO carbon atoms appeared at 57.5 and 176 ppm for the α-helical conformation; for the β-sheet conformation, they were shifted upfield by approximately 4–5 ppm, appearing at 52.7 and 172 ppm, respectively.15,16 Fig. 6 provides some of the other peak assignments; Table 3 summaries the chemical shifts observed in the solid state NMR spectra of our PTyr/PMGL blends.
 |
| | Fig. 6 High-resolution 13C CP/MAS solid state NMR spectra of PTyr/PMLG blends of various compositions, recorded at room temperature. | |
Table 3 Chemical shifts of signals in the 13C CP/MAS/DD NMR spectra of PTyr/PMGL blends
| PTyr/PMLG |
PTyr |
| Cβ |
Cα |
Cε |
Cγ |
Cd |
Cf |
| 100/0 |
36.8/39.9 |
52.7/55.1/57.7 |
116.7 |
127.9/131.4 |
151.4/155.8 |
169.7/176.5 |
| 80/20 |
37.1/40.9 |
52.5/55.1/57.7 |
117.2 |
128.3/131.7 |
151.9/156.3 |
170.0/173.8/176.3 |
| 60/40 |
37.1/40.9 |
52.4/55.1/57.7 |
117.9 |
128.4/131.8 |
151.9/156.3 |
170.0/173.5/176.2 |
| 50/50 |
37.2/40.9 |
52.4/55.1/57.7 |
117.0 |
128.4/131.6 |
152.0/156.3 |
169.8/173.9/175.9 |
| 40/60 |
37.2/40.9 |
52.4/54.8/57.3 |
116.4 |
128.6/131.6 |
152.7/156.5 |
172.0/173.5/176.5 |
| 20/80 |
37.3/41.0 |
52.4/57.3 |
116.1 |
— |
153.3/156.6 |
171.9/173.5/176.5 |
| PTyr/PMLG |
PMLG |
| Cβ and Cγ |
Cf and Cα |
Cd and Ce |
| 0/100 |
30.3 |
52.0/57.5 |
171.7/173.2/176.5 |
| 20/80 |
30.5 |
52.4/57.3 |
171.9/173.5/176.5 |
| 40/60 |
30.5 |
52.4/54.8/57.3 |
172.0/173.5/176.5 |
| 50/50 |
30.5 |
52.4/55.1/57.7 |
170.0/173.5/175.9 |
| 60/40 |
30.5 |
52.4/55.1/57.7 |
170.0/173.6/176.2 |
| 80/20 |
30.5 |
52.5/55.1/57.7 |
170.0/173.8/176.3 |
For our analysis, first we focus on the secondary structures determined from the signals of the Cα and CO amide atoms (Fig. 7). The signals were observed for the CO groups [Fig. 7(a)] into three major peaks for pure PTyr [at 169.7 (β-sheet conformation) and 172.5 and 176.5 (α-helical conformation) ppm] and three major peaks for pure PMLG (at 171.7, 173.2, and 176.5 ppm for the β-sheet conformation, side chain CO groups, and α-helical conformation, respectively). Interestingly, the signal for the side chain CO groups of PMLG shifted slightly from 173.2 to 173.8 ppm (Table 3) upon blending, suggesting that intermolecular interactions existed between the CO groups of PMLG and the phenolic OH groups of PTyr. Fig. 7(a) also summarizes the results of curve fitting. Fig. 7(b) presents the signals of the Cα atoms in the PTyr/PMLG blends. The spectrum of pure PTyr featured three major peaks at 52.7, 55.1 and 57.7 ppm: the first two corresponding to the β-sheet conformation and the third corresponding to the α-helical conformation. Two major peaks appeared for pure PMLG at 52.0 and 57.5 ppm, representing the β-sheet and α-helical conformations, respectively. Fig. 7(b) also summarizes the results of curve fitting; here, the fraction of the β-sheet conformation at 52.0 ppm was difficult to calculate because the signals of the carbon atoms of the methyl groups (Cf) overlapped strongly at 52.0 ppm.
 |
| | Fig. 7 Results of curve fitting of the signals in the solid state NMR spectra of PTyr/PMLG blends of various compositions, recorded at room temperature: (a) CO and amide I groups; (b) Cα groups. | |
Second, we turned our attention to the secondary structures determined from the signals for the Cd and Cβ atoms of PTyr (Fig. 8), because these two peaks did not overlap with any from PMLG. For the Cd (C–OH) carbon atoms of pure PTyr, we observed [Fig. 8(a)] two clear major peaks at 151.4 and 155.8 ppm, corresponding to the β-sheet and α-helical conformations, respectively. These two signals underwent downfield shifts to 153.3 and 156.5 ppm, respectively, upon increasing the PMLG content, suggesting that these phenolic OH groups could interact with the side chain CO groups of PMLG. The hydrogen bonding interactions in polymer blends can vary the chemical environments of carbon nuclei and cause downfield chemical shifts of their signals. Fig. 8(a) also summarizes the results of curve fitting. Fig. 8(b) displays the signals of the Cβ atoms in the PTyr/PMLG blends. The spectrum of pure PTyr also featured two major peaks at 36.8 and 39.9 ppm, corresponding to α-helical and β-sheet conformations, respectively. When hydrogen bonding existed between PTyr and PMLG, these two peaks also underwent downfield chemical shifts, even though the Cβ carbon atoms were not suitably functionalized to form any hydrogen bonds. Thus, the chemical environment of the neighboring molecules induced these changes in chemical shift through intermolecular interaction. Fig. 8(b) also summarizes the results of the curve fitting.
 |
| | Fig. 8 Results of curve fitting of the signals in the solid state NMR spectra of PTyr/PMLG blends of various compositions, recorded at room temperature: (a) phenolic-OH groups; (b) Cβ groups. | |
Fig. 9 provides a summary of the curve fitting results of the signals for the CO amide, phenolic OH (Cd), and Cβ atoms; we do not present the results for the Cα atoms because their signals overlapped with those of the Cf atoms of PMLG, making it difficult to determine the secondary structures in the blend system. The signals of all three atoms revealed similar trends in that the β-sheet conformation increased initially at 20 wt% of PMLG but then decreased thereafter upon increasing the PMLG content. The β-sheet conformation was almost completely absent at a PMLG content of 80 wt%, consistent with the WAXD analysis; the long-range order of the lamellar structure from the β-sheet conformation disappeared at higher PMLG contents. In other words, the α-helical conformation initially decreased at 20 wt% of PMLG and then increased upon increasing the PMLG content furthers. At 80 wt% PMLG, the fraction of α-helical conformations for PTyr was close to 90%, significantly different from that of pure PTyr. α-Helical and β-sheet secondary structures are stabilized through intra- and inter-chain hydrogen bonding, respectively. When blending PMLG into PTyr, we observed miscible blends through DSC analysis; thus, the inter-chain hydrogen bonding of PTyr was disrupted upon blending with PMLG, through the appearance of intermolecular hydrogen bonding between the phenolic OH groups of PTyr and the side chain CO groups of PMLG, favoring the possibility of intra-chain hydrogen bonding interactions for PTyr. As a result, a partial fraction of the β-sheet conformations transformed to α-helical conformations, as displayed in Scheme 1. This result explains why the value of Tg of PMLG increased significantly from 48 to 101 °C at a PTyr content of only 20 wt%, as revealed in Fig. 1. Accordingly, the larger value of q in Fig. 2 arose not only from intermolecular hydrogen bonding but also from the greater fraction of rigid-rod conformations of the α-helices from the polypeptide main chains in the PTyr/PMLG blends.
 |
| | Fig. 9 Area fractions of the secondary structures of PTyr, based on analyses of solid state NMR spectral data. | |
 |
| | Scheme 1 Cartoon representation of possible secondary structural changes and intermolecular hydrogen bonding in PTyr/PMLG blends. | |
Conclusion
PTyr is totally miscible with PMLG over their entire range of compositions. Using the Kwei equation, DSC analyses revealed a positive deviation in the behavior of the values of Tg as a result of intermolecular hydrogen bonding between the phenolic OH groups of PTyr and the side chain CO groups of PMLG; analyses of FTIR spectra, WAXD patterns, and solid state NMR spectra revealed that the content of rigid-rod conformations of α-helices increased accordingly in the polypeptide blends. To the best of our knowledge, this report is the first to describe how two polypeptides with rigid-rod conformations can be miscible, mediated through intermolecular hydrogen bonding of their polypeptide side chains.
Acknowledgements
This study was financially supported by the Ministry of Science and Technology, Taiwan, under contracts MOST 103-2221-E-110-079-MY3.
References
- J. Colmenero and A. Arba, Soft Matter, 2007, 3, 1474–1485 RSC.
- S. H. Goh, Polymer Blends Handbook, ed. L. A. Ultracki and C. A. Wilkie, Springer, Netherlands, 2014, ch. 21, pp. 875–918 Search PubMed.
- E. M. Masnada, G. Julien and D. R. Long, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 419–443 CrossRef CAS PubMed.
- P. Shi, R. Schach, E. Munch, H. Montes and F. Lequeux, Macromolecules, 2013, 46, 3611–3620 CrossRef CAS.
- P. Xiavier, K. Sharma, K. K. Elayaraja, K. S. Vasu, A. K. Sood and S. Bose, RSC Adv., 2014, 4, 12376–12387 RSC.
- M. M. Coleman and P. C. Painter, Prog. Polym. Sci., 1995, 20, 1–59 CrossRef CAS.
- M. Jiang, L. Mei, M. Xiang and H. Zhou, Adv. Polym. Sci., 1999, 146, 121–196 CrossRef CAS.
- Y. He, B. Zhu and Y. Inoue, Prog. Polym. Sci., 2004, 29, 1021–1051 CrossRef CAS PubMed.
- S. W. Kuo, J. Polym. Res., 2008, 15, 459–486 CrossRef CAS.
- Y. Xu, W. Yu and C. Zhou, RSC Adv., 2014, 4, 55435–55444 RSC.
- S. W. Kuo, C. F. Huang, P. H. Tung, W. J. Hunag, J. M. Hunag and F. C. Chang, Polymer, 2005, 46, 9348–9361 CrossRef CAS PubMed.
- K. Jin and J. M. Torkelson, Polymer, 2015, 65, 233–242 CrossRef CAS PubMed.
- S. W. Kuo, W. J. Huang, C. F. Huang, S. C. Chan and F. C. Chang, Macromolecules, 2004, 37, 4164–4173 CrossRef CAS.
- C. T. Pan, C. K. Yen, H. C. Wu, L. W. Lin, Y. S. Lu, J. C. C. Huang and S. W. Kuo, J. Mater. Chem. A, 2015, 3, 6835–6843 CAS.
- P. Papadopoulous, G. Floudas, H. A. Klok, I. Schnell and T. Pakula, Biomacromolecules, 2004, 5, 81–91 CrossRef PubMed.
- H. A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217–1229 CrossRef CAS.
- L. Zhao, N. Li, K. Wang, C. Shi, L. Zhang and Y. Luan, Biomaterials, 2014, 35, 1284–1301 CrossRef CAS PubMed.
- B. Tian, X. Tao, T. Ren, Y. Weng, X. Lin, Y. Zhang and X. Tang, J. Mater. Chem., 2012, 22, 17404–17414 RSC.
- S. W. Kuo, H. F. Lee, W. J. Huang, K. U. Jeong and F. C. Chang, Macromolecules, 2009, 42, 1619–1626 CrossRef CAS.
- Y. Cheng, C. He, J. Ding, C. Xiao, X. Zhuang and X. Chen, Biomaterials, 2013, 34, 10338–10347 CrossRef CAS PubMed.
- Y. C. Lin and S. W. Kuo, Polym. Chem., 2012, 3, 882–891 RSC.
- M. P. Bhatt, P. Sista, J. Hao, N. Hundt, M. C. Biewer and M. C. Stefan, Langmuir, 2012, 28, 12762–12770 CrossRef CAS PubMed.
- F. Zhou, T. Ye, L. Shi, C. Xie, S. Chang, X. Fan and Z. Shen, Macromolecules, 2013, 46, 8253–8263 CrossRef CAS.
- S. W. Kuo and C. J. Chen, Macromolecules, 2011, 44, 7315–7326 CrossRef CAS.
- S. W. Kuo and C. J. Chen, Macromolecules, 2012, 45, 2442–2452 CrossRef CAS.
- P. C. Painter, W. L. Tang, J. F. Graf, B. Thomson and M. M. Coleman, Macromolecules, 1991, 24, 3929–3936 CrossRef CAS.
- Y. S. Lu, Y. C. Lin and S. W. Kuo, Macromolecules, 2012, 45, 6547–6556 CrossRef CAS.
- K. Y. Shih, Y. C. Lin, T. S. Hsiao, S. L. Deng, S. W. Kuo and J. L. Hong, Polym. Chem., 2014, 5, 5765–5774 RSC.
- M. G. Mohamed, F. H. Lu, J. L. Hong and S. W. Kuo, Polym. Chem., 2015, 6, 6340–6350 RSC.
- T. K. Kwei, J. Polym. Sci., Polym. Lett. Ed., 1984, 22, 307–313 CrossRef CAS PubMed.
- S. W. Kuo, P. H. Tung and F. C. Chang, Macromolecules, 2006, 39, 9388–9395 CrossRef CAS.
- S. W. Kuo and F. C. Chang, Macromolecules, 2001, 34, 5224–5228 CrossRef CAS.
- S. W. Kuo, C. F. Huang and F. C. Chang, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 1348–1359 CrossRef CAS PubMed.
- S. W. Kuo, S. C. Chan and F. C. Chang, Macromolecules, 2003, 36, 6653–6661 CrossRef CAS.
- C. L. Lin, W. C. Chen, C. S. Liao, Y. C. Su, C. F. Huang, S. W. Kuo and F. C. Chang, Macromolecules, 2005, 38, 6435–6444 CrossRef CAS.
- A. Gitsas, G. Floudas, M. Mondeshki, H. W. Spiess, T. Aliferis, H. Iatrou and N. Hadjichristidis, Macromolecules, 2008, 41, 8072–8080 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19037a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.