
Novel design, facile synthesis and low infrared emissivity properties of single-handed helical polysilanes†
Abstract
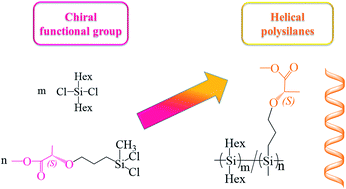
The novel single-handed helical polysilanes (HPS), poly[di-n-hexylsilane-co-(methoxycarbonyl ethyl propyl ether)methylsilane] were synthesized by the Wurtz-type coupling of the chiral functional monomer, dichloro(methoxycarbonyl ethyl propyl ether)methylsilane (DCMMS). The monomer was prepared by the hydrosilylation reaction of methyl (S)-2-(allyloxy)propanoate. The chemical structure of HPS was confirmed by 1H, 13C and 29Si nuclear magnetic resonance (NMR), Fourier transform infrared spectroscopy (FT-IR), ultraviolet-visible spectroscopy (UV-Vis) and circular dichroism (CD). The weight distributions and crystallinity of the polysilanes were demonstrated by gel permeation chromatography (GPC), differential scanning calorimetry (DSC) and polarized light microscopy (POM), respectively. The single-handed helical characterization and polymerization degree of the new-typed polysilanes could be controlled by the co-monomers of DCMMS and di-n-hexyldichlorosilane (DCDHS), respectively. Furthermore, the infrared emissivity values (0.598 at 8–14 μm) of HPS were investigated by an Infrared Emissometer.
 Please wait while we load your content...
Please wait while we load your content...

