
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liquid crystalline dihydroazulene photoswitches†
Anne Ugleholdt
Petersen
a,
Martyn
Jevric
a,
Richard J.
Mandle
b,
Edward J.
Davis
b,
Stephen J.
Cowling
b,
John W.
Goodby
b and
Mogens Brøndsted
Nielsen
*a
aDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark. E-mail: mbn@chem.ku.dk
bDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK
First published on 13th October 2015
Abstract
A large selection of photochromic dihydroazulene (DHA) molecules incorporating various substituents at position 2 of the DHA core was prepared and investigated for their ability to form liquid crystalline phases. Incorporation of an octyloxy-substituted biphenyl substituent resulted in nematic phase behavior and it was possible to convert one such compound partly into its vinylheptafulvene (VHF) isomer upon irradiation with light when in the liquid crystalline phase. This conversion resulted in an increase in the molecular alignment of the phase. In time, the meta-stable VHF returns to the DHA where the alignment is maintained. The systematic structural variation has revealed that a biaryl spacer between the DHA and the alkyl chain is needed for liquid crystallinity and that the one aromatic ring in the spacer cannot be substituted by a triazole. This work presents an important step towards employing the dihydroazulene-vinylheptafulvene photo/thermoswitch in photoactive liquid crystalline materials.
1. Introduction
In the field of liquid crystals, it has been a goal to control the phases and order with an external stimulus. Some of the stimuli which have been applied include temperature, magnetic field, electric field and light.1 Light is an interesting stimulus, since this gives the possibility for remote control. Indeed, it has found use in image storing devices.2,3 Specifically, this has been shown with azobenzene polymer matrix, where an image was stored for 8 months without significant decay.2 Other photochromic molecules such as dithienylethenes, spiropyrans and fulgides have also shown suitable properties for this kind of application.4–6 Here we present a study on liquid crystalline systems based upon the dihydroazulene (DHA) photoswitch (Scheme 1). When irradiated with light, DHA 1 undergoes a ring-opening to the vinylheptafulvene (VHF) 2, which can then close back to the initial DHA via a thermal electrocyclization reaction.7 | ||
| Scheme 1 DHA 1/VHF 2 system. | ||
The main advantage of the DHA–VHF photoswitch system is that only the DHA to VHF conversion is light-induced, which means that a broad spectrum of light can be used without triggering the VHF to DHA back-reaction. The DHA ring-opening is associated with significant changes in the physical properties of the corresponding VHF. Thus, ring-opening is associated with an increase in dipole moment and a change in color from yellow (DHA) to intense red (VHF).8,9 We became interested to elucidate if this difference in properties could give rise to reversible, photochemically induced phase transitions.
Molecules with liquid crystalline properties can have differing constructions, but for the purpose of this work, attention was given to rod-like structures. Typically, rod-like, liquid crystalline structures have two aliphatic chains extending from a rigid ‘core’ unit such as biphenyl or one alkyl chain with an additional terminal substituent such as a nitrile.10 It was decided to incorporate either a cyanophenyl-substituted alkyl in the one end of DHA or an octyl chain. In the literature there are many examples of the cyanobiphenyl being used as the terminal substituent,11 but as this unit has absorption properties which conflict with the wavelength needed for the ring-opening event of DHA, the cyanophenyl substituent was used predominantly in this study, as in structures 3a–e and 4a–e (Fig. 1). Structures 3f–i and 4j instead have an octyl end-group. Both cyanophenyl and octyl groups have been used previously to achieve liquid crystallinity.12
 | ||
| Fig. 1 DHAs employed in this study. | ||
2. Results and discussion
2.1 Synthesis
The most convenient method for the construction of 2-substituted DHAs originates from derivatized acetophenones. Several strategies were undertaken in the pursuit of obtaining the starting acetophenones (Scheme 2). From the simple precursors 5a–e, 6, 7a–e, 8, acetophenones 9a–e were prepared in two successive reactions. Thus, dibromoalkanes 5a–e of various chain lengths were treated with phenols 6 and 8. Conversely, acetophenones containing biaryl units, 9f and 9g, were obtained in good yields from a palladium-catalyzed Suzuki cross-coupling reaction from 4-iodoacetophenone 10 with boronic acids 11f and 11g, respectively, using conditions that proved fruitful in the synthesis of the benzaldehyde analogues.13 Finally, 9h could be formed in high yield by a Friedel–Crafts reaction of 12 with acetyl chloride using a literature procedure.14 Compound 9i was purchased. | ||
| Scheme 2 Synthesis of acetophenone starting materials. Conditions: (a) K2CO3, acetone, reflux; (b) 5% Pd(PPh3)4, K3PO4, toluene/H2O, rt to 80 °C; (c) FeCl3, AcCl, CH2Cl2, 0 °C to rt. | ||
Acetophenones 9a–i could be transformed successfully into the corresponding DHAs 3a–i (Scheme 3) by firstly converting the carbonyl moiety into a crotononitrile unit in a Knoevenagel condensation furnishing intermediates 13a–i, generally in good yields. Intermediates 13a–i were then treated with tropylium tetrafluoroborate in the presence of triethylamine to affix the cycloheptatriene unit to the malonitriles which could be further converted into 3a–i by subsequent oxidation using tritylium tetrafluoroborate followed by treatment with triethylamine. Alternatively, biaryl DHAs 3f and 3g could be obtained directly from DHA 15 in a Suzuki reaction with boronic acids 11f and 11g, using the catalytic system of palladium acetate and RuPhos (Scheme 4). A separate strategy involving the introduction of the extended chains could also be effected using the copper-catalyzed azide–alkyne cycloaddition (CuAAC)15 to form triazole compounds. Triazoles have previously been incorporated successfully into liquid crystalline compounds16 and, synthetically, DHA has been found to withstand the conditions.17 Alkyl bromides 7a–e were converted into their corresponding azides 14a–e (Scheme 5) and when subjected to treatment with 10 mol% cuprous iodide in the presence of 16 and Hünig's base, triazoles 4a–e were achieved. DHA 4j could be made from a CuAAC of octyl azide with 16 in the same manner.
 | ||
| Scheme 3 Synthesis of DHAs. Conditions: (a) CH2(CN)2, NH4OAc, toluene, AcOH, Δ. (b) [C7H7]BF4, NEt3, CH2Cl2, −78 °C. (c) [Ph3C]BF4, CH2ClCH2Cl, Δ, then toluene, NEt3, 0 °C. | ||
 | ||
| Scheme 4 Functionalization of DHA via Suzuki cross-coupling. Conditions: (a) Pd(OAc)2, RuPhos, K3PO4, toluene/H2O. RuPhos = 2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl. | ||
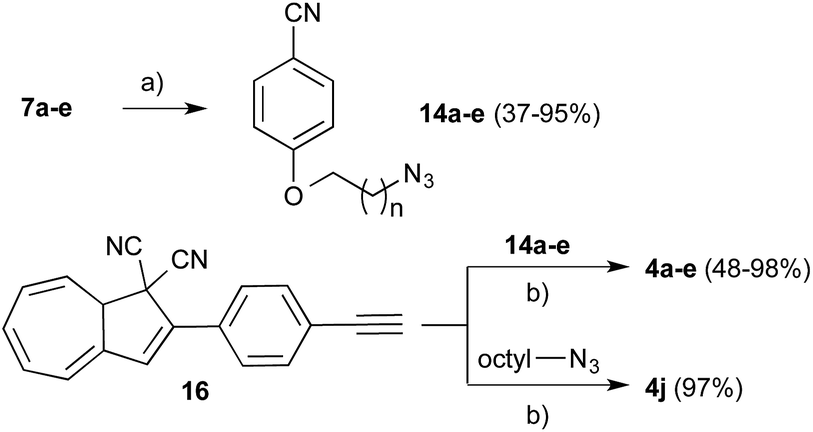 | ||
| Scheme 5 Functionalization of DHA via CuAAC. Conditions: (a) NaN3, DMSO, 50 °C; (b) CuI, Hünig's base, toluene. | ||
We also decided to prepare the corresponding VHFs of two of the DHAs (to study their liquid crystalline properties of relevance for the switching studies, vide infra). It has been shown that DHA can be ring-opened by treatment with AlCl3 to afford the corresponding VHF18 and this method could be used to convert 3f into 17 in good yield (Scheme 6). When the method was used on 3g, not only did the DHA undergo ring-opening to the VHF, but these reaction conditions also resulted in the cleavage of the octyl chain to form phenol 18.
 | ||
| Scheme 6 Synthesis of VHFs. Conditions (a) AlCl3, CH2Cl2, 0 °C/rt, then H2O, 0 °C. | ||
2.2 Photochemical properties
All DHAs made in this study show photoactive properties akin to that of unfunctionalized DHA 1. All DHAs exhibited an absorption maximum at around 360 nm and when irradiated with light of this wavelength, they undergo an electrocyclic ring-opening reaction. Thereby a peak at 475 nm emerges, corresponding to the formation of the VHF (Table 1). The rate of VHF to DHA thermal ring-closure has been found to be influenced greatly by the introduction of electron-donating/accepting functional groups.9,19 The VHFs of DHAs 3a–e all exhibit a rate constant corresponding to that of the p-methoxy substituted derivative of DHA 1. The rate of ring-closure for the VHFs of the triazole derivatives 4a–e and 4j show a small deviation from a formerly reported triazole functionalized DHA,17 which had a rate constant for the VHF ring-closure of 7.45 × 10−5 s−1. The VHFs of 3h and 3i have comparable ring-closure rate constants as those of the VHFs of 3a–e and of formerly reported DHA with a p-methyl substituent. Inclusion of fluorine seems to have no effect for the VHF of 3h, or its effect is cancelled by the electron-donating octyloxy substituent. For the biphenyls 3f and 3g the rate of VHF to DHA conversion is higher and more similar to that of 4a–e. Here the effect of the fluorine is clear; the rate increases to 7.30 × 10−5 s−1 compared to the non-fluorinated compound with a rate of 6.78 × 10−5 s−1.| DHA, λmax (nm) | VHF, λmax (nm) | VHF → DHA, k (10−5 s−1) | VHF → DHA, t1/2 (min) | |
|---|---|---|---|---|
| 3a | 365 | 469 | 5.04 | 229 |
| 3b | 366 | 470 | 5.00 | 231 |
| 3c | 367 | 471 | 4.97 | 232 |
| 3d | 366 | 470 | 4.97 | 232 |
| 3e | 365 | 470 | 4.97 | 232 |
| 3f | 374 | 474 | 6.78 | 170 |
| 3g | 366 | 475 | 7.30 | 158 |
| 3h | 359 | 480 | 5.00 | 231 |
| 3i | 358 | 471 | 4.98 | 232 |
| 4a | 370 | 475 | 6.65 | 174 |
| 4b | 370 | 476 | 6.72 | 172 |
| 4c | 370 | 475 | 6.72 | 172 |
| 4d | 368 | 475 | 6.68 | 173 |
| 4e | 370 | 475 | 6.66 | 174 |
| 4j | 369 | 475 | 6.63 | 174 |
2.3 Physical properties
The synthesized molecules were examined by polarized optical microscopy (POM), but all starting materials en route to and including DHAs 3a–e from the first two approaches did not show any liquid-crystalline properties. The same could also be said for DHAs 4a–e and 4j. Instead, biaryl containing acetophenones 9f and 9g and the corresponding DHAs 3f and 3g exhibited a liquid-crystalline phase and were examined by both POM and differential scanning calorimetry (DSC). Compound 9f was found to exhibit a mesophase. Indeed, 9f (Fig. 2) has been previously reported with two different transitions, where in one, it exhibited a smectic E phase,20 and in the other, a melt was reported at 115–118 °C.21 We found that under the microscope 9f did exhibit a smectic phase (see Fig. 2), in agreement with the former report although definitive phase assignment was not possible due to the short range of supercooling. Hence, we denote it smectic X. Fig. 3 shows the DSC thermogram. | ||
| Fig. 2 Photomicrographs of the smectic X phase of 9f at 118 °C. | ||
 | ||
| Fig. 3 (Top) DSC thermograms at a heating–cooling rate of 10 °C min−1 for compound 9f. (Bottom) DSC thermograms at a heating–cooling rate of 5 °C min−1 for compound 9g, where the dashed line is the first heating. | ||
DSC showed a transition for a mesophase, where the size of the associated enthalpy to the phase transition is indicative of the formation of a smectic mesophase. The partially fluorinated analogue 9g gave a lower melt and did not exhibit a liquid-crystalline phase during the heating process. Upon cooling, a peak corresponding to a liquid-crystalline phase could only be observed in the DSC at 53.8 °C. It was only possible to observe this phase when the sample had been heated to 100 °C prior to cooling. When compound 9g was heated to a lesser extent, such as 67 or 80 °C, the sample underwent crystallization during the cooling phase prior to the temperature where this initial phase transition was observed. When examined by POM, the crystallization occurred prior to the liquid crystal phase transition temperature. Table 2 lists transition temperatures for 9f and 9g and Table 3 the enthalpies and entropies associated with the transitions.
|
|
||
|---|---|---|
| Transition temperatures (°C) | ||
| 9f | Cr 91.0 SmX 128 Iso | Iso 130.2 SmX 79.5 Cr |
| 9g | Cr 63.2 Iso | Iso 53.8 SmX 48.8 Cr |

| Cr-Iso | Cr-SmX | SmX-Iso | Iso-SmX | SmX-Cr | ||
|---|---|---|---|---|---|---|
| 9f | ΔH | — | 23.48 | 13.377 | 12.54 | 24.10 |
| ΔS/R | — | 7.755 | 4.0106 | 3.739 | 8.819 | |
| 9g | ΔH | 25.91 | — | — | 2.713 | 22.95 |
| ΔS/R | 9.265 | — | — | 0.9980 | 8.574 |
The two DHAs 3f and 3g showed liquid crystalline properties, whilst 3h and 3i did not exhibit any mesophases. This suggests that the additional aryl ring is important for the modified DHAs to demonstrate liquid crystalline properties. Neither 3f nor 3g exhibited a mesophase in the first heating cycle, but upon cooling, a nematic phase appeared (Fig. 4). Further cooling did not result in crystallization, instead a glass transition occurred and when reheating the glass, the nematic phase reappeared and the transition from nematic to isotropic liquid could also be observed. DHA 3f gave a melt at 90.6 °C and the nematic phase appeared upon cooling at 75.7 °C, see Fig. 4. On heating, the clearing point was found to be at 75.6 °C. The phase transitions showed small enthalpy changes, which agrees with the changes from nematic to liquid phase and vice versa. For the fluorinated 3g analogue, the melt occurred at a temperature about 10 °C lower than for 3f. The clearing point for the nematic phase of 3g is 52.6 °C, while it is 75.7 °C for 3f. Table 4 lists transition temperatures for 3f and 3g and Table 5 the enthalpies and entropies associated with the transitions.
 | ||
| Fig. 4 DSC thermograms at a heating-cooling rate of 5 °C min−1 for compound 3f (Top) and compound 3g (Bottom). The dashed line corresponds to the first heating. | ||
|
|
|
|---|---|
| Transition temperatures (°C) | |
| 3f | Cr 90.6 (Tg 29.8 N 75.7) Iso |
| 3g | Cr 79.9 (Tg 30.8 N 52.6) Iso |

| N-Iso | Cr-Iso | Iso-N | ||
|---|---|---|---|---|
| 3f | ΔH | 0.4612 | 30.53 | 0.4044 |
| ΔS/R | 0.1590 | 10.09 | 0.1394 | |
| 3g | ΔH | 0.4526 | 31.70 | 0.4057 |
| ΔS/R | 0.1671 | 10.80 | 0.1498 |
When 3f is irradiated (at 365 nm) in the nematic phase, a color change from yellow-orange to red can be observed by the naked eye, but this did not correspond to a change in the phases. When solid VHF 17 was heated to 40 °C for 24 h, neither a change in color nor a phase change was observed, while heating the same material to 80 °C resulted in a simultaneous melt and color change to yellow ascribed to conversion to the DHA state. When 3g was irradiated with light in the nematic phase at 40 °C for 60 min, conversion to the corresponding VHF occurred in a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 (VHF/DHA) as determined by NMR spectroscopy. After one hour of irradiation, using POM one could see an increase in the alignment of the nematic phase, such that the defects disappear (Fig. 5c). Irradiation for a prolonged period (16 hours) afforded no further changes in neither the POM picture nor in the isomer ratio determined by NMR spectroscopy. When left in the dark to reclose to form the DHA, the alignment is maintained. If the top glass slide is sheared horizontally to disrupt the alignment, the nematic texture appears for the DHA, (Fig. 5d). If the same disruption is done with the VHF present, the alignment reforms over several minutes. In addition to the VHF structure being planar, it also has a larger dipole moment, which could in essence lead to this larger degree of alignment of the total system relative to the corresponding neat nematic DHA. There are several examples in the literature22 with photoalignment; the difference is that our experiment was performed in between untreated glass slides, which gives a texture. This texture then disappears upon irradiation. On the other hand, when heating pure VHF 17, no mesophases were observed for VHF 17, instead a rapid ring-closure to 3f occurred.
:7 (VHF/DHA) as determined by NMR spectroscopy. After one hour of irradiation, using POM one could see an increase in the alignment of the nematic phase, such that the defects disappear (Fig. 5c). Irradiation for a prolonged period (16 hours) afforded no further changes in neither the POM picture nor in the isomer ratio determined by NMR spectroscopy. When left in the dark to reclose to form the DHA, the alignment is maintained. If the top glass slide is sheared horizontally to disrupt the alignment, the nematic texture appears for the DHA, (Fig. 5d). If the same disruption is done with the VHF present, the alignment reforms over several minutes. In addition to the VHF structure being planar, it also has a larger dipole moment, which could in essence lead to this larger degree of alignment of the total system relative to the corresponding neat nematic DHA. There are several examples in the literature22 with photoalignment; the difference is that our experiment was performed in between untreated glass slides, which gives a texture. This texture then disappears upon irradiation. On the other hand, when heating pure VHF 17, no mesophases were observed for VHF 17, instead a rapid ring-closure to 3f occurred.
 | ||
| Fig. 5 (a) Photomicrographs of the nematic phase of 3f at 60 °C. (b–d) Photomicrographs of the nematic phase of 3g at 40 °C, (b) before light, (c) after 60 min of light, (d) after dark for 16 h. | ||
3. Conclusions
In order to ascertain the requirements of substitution needed to make the DHA photoswitch liquid crystalline, we have prepared a large selection of compounds with various substituents at position 2 of the DHA core. Of these compounds, two exhibited a nematic liquid crystal phase, both of which had an octyloxybiaryl attached to C2 of DHA. Irradiation of the one compound, with fluorine substitution of the biaryl, in its nematic phase partly formed the VHF. This DHA/VHF mixture exhibited a higher degree of alignment. In time, the DHA was regenerated and the order maintained. Nevertheless, full conversion to the VHF from the DHA in the nematic phase was not possible. This work presents an important step in using the DHA–VHF system in photoactive liquid crystalline materials. Future work will be aiming at exploring the influence of having two substituent groups on the DHA core.4. Experimental
4.1 General methods
Chemicals were used as purchased from commercial sources. Purification of products was carried out by flash chromatography on silica gel (40−63 μm, 60 Å). Thin-layer chromatography (TLC) was carried out using aluminum sheets precoated with silica gel. 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were recorded on an instrument with a noninverse cryoprobe using the residual solvent as the internal standard (CDCl3, 1H 7.26 ppm and 13C 77.16 ppm). All chemical shifts are quoted on the δ scale (ppm), and all coupling constants (J) are expressed in Hz. In APT spectra, CH and CH3 correspond to negative signals and C and CH2 correspond to positive signals. High resolution mass spectra (HRMS) were acquired either using an electrospray method of ionization (ESP) or using MALDI. Melting points are uncorrected. Compounds 15 (ref. 23) and 16 (ref. 24) were made by their respective literature methods.4-((5-Bromopentyl)oxy)benzonitrile (7a). Mp = 54.0–55.9 °C. 1H NMR (500 MHz, CDCl3): δ = 7.57 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 4.01 (t, J = 6.3 Hz, 2H), 3.44 (t, J = 6.7 Hz, 2H), 1.97–1.91 (m, 2H), 1.87–1.81 (m, 2H), 1.67–1.60 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.4, 134.1, 119.4, 115.3, 104.0, 68.1, 33.6, 32.5, 28.3, 24.8 ppm. MS (ESP +ve): m/z = 290 [(M + Na)+]. Analysis calcd (%) for C12H14BrNO (268.15): C 53.75, H 5.26, N 5.22; found: C 53.75, H 4.90, N 5.13.
4-((6-Bromohexyl)oxy)benzonitrile (7b). Mp = 44.0–46.5 °C. 1H NMR (500 MHz, CDCl3): δ = 7.57 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 4.00 (t, J = 6.4 Hz, 2H), 3.43 (t, J = 6.6 Hz, 2H), 1.90 (p, J = 6.6 Hz, 2H), 1.82 (p, J = 6.6 Hz, 2H), 1.53–1.50 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.5, 134.1, 119.4, 115.3, 103.9, 68.3, 33.8, 32.7, 29.0, 28.0, 25.3 ppm. MS (ESP +ve): m/z = 282 [(M + H)+]. Analysis calcd (%) for C13H16BrNO (282.18): C 55.33, H 5.72, N 4.96; found: C 55.66, H 5.72, N 4.98.
4-((8-Bromooctyl)oxy)benzonitrile (7c). R f = 0.50 (toluene). Mp = 68.5–69.8 °C. 1H NMR (500 MHz, CDCl3): δ = 7.57 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 3.99 (t, J = 6.5 Hz, 2H), 3.41 (t, J = 6.8 Hz, 2H), 1.89–1.84 (m, 2H), 1.82–1.77 (m, 2H), 1.49–1.42 (m, 4H), 1.39–1.33 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.5, 134.1, 119.4, 115.3, 103.8, 68.5, 34.1, 32.9, 29.2, 29.1, 28.8, 28.2, 26.0 ppm. MS (ESP +ve): m/z = 310 [(M + H)+]. Analysis calcd (%) for C15H20BrNO (310.24): C 58.07, H 6.50, N 4.51; found: C 58.26, H 6.53, N 4.51.
4-((9-Bromononyl)oxy)benzonitrile (7d). R f = 0.50 (toluene). Mp = 64.1–65.7 °C. 1H NMR (500 MHz, CDCl3): δ = 7.55 (d, J = 8.9 Hz, 2H), 6.91 (d, J = 8.9 Hz, 2H), 3.98 (t, J = 6.5 Hz, 2H), 3.39 (t, J = 6.5 Hz, 2H), 1.86–1.75 (m, 4H), 1.46–1.39 (m, 4H), 1.36–1.29 (m, 6H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.5, 134.0, 119.4, 115.2, 103.7, 68.4, 34.1, 32.8, 29.3, 29.2, 29.0, 28.7, 28.2, 25.9 ppm. MS (ESP +ve): m/z = 324 [(M + H)+]. Analysis calcd (%) for C16H22BrNO (324.26): C 59.27, H 6.84, N 4.32; found: C 59.52, H 6.93, N 4.32.
4-((10-Bromodecyl)oxy)benzonitrile (7e). R f = 0.50 (toluene). Mp = 73.2–74.7 °C. 1H NMR (500 MHz, CDCl3): δ = 7.57 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 3.99 (t, J = 6.5 Hz, 2H), 3.41 (t, J = 6.8 Hz, 2H), 1.88–1.77 (m, 4H), 1.48–1.40 (m, 4H), 1.36–1.28 (m, 8H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.6, 134.1, 119.5, 115.3, 103.8, 68.5, 34.2, 32.9, 29.5, 29.5, 29.4, 29.1, 28.9, 28.3, 26.1 ppm. MS (ESP +ve): m/z = 360 [(M + Na)+]. Analysis calcd (%) for C17H24BrNO (338.29): C 60.36, H 7.15, N 4.14; found: C 60.53, H 7.19, N 4.15.
4-((5-(4-Acetylphenoxy)pentyl)oxy)benzonitrile (9a). R f = 0.39. Mp = 100.8–102.8 °C. 1H NMR (500 MHz, CDCl3): δ = 7.96 (d, J = 8.9 Hz, 2H), 7.60 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 6.92 (d, J = 8.9 Hz, 2H), 4.06 (t, J = 6.2 Hz, 2H), 4.04 (t, J = 6.3 Hz, 2H), 2.55 (s, 3H), 1.92–1.87 (m, 4H), 1.70–1.64 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ = 196.9, 163.0, 162.4, 134.1, 130.7, 130.4, 119.4, 115.3, 114.2, 104.0, 68.2, 68.0, 29.0, 28.9, 26.5, 22.8 ppm. HRMS (MALDI +ve) calcd for C20H22NO3 ([M + H]+): m/z = 324.1594; exp 324.1595. Analysis calcd (%) for C20H21NO3 (323.39): C 74.28, H 6.55, N 4.33; found: C 74.09, H 6.28, N 4.27.
4-((6-(4-Acetylphenoxy)hexyl)oxy)benzonitrile (9b). R f = 0.33. Mp = 99.5–101.3 °C. 1H NMR (500 MHz, CDCl3): δ = 7.92 (d, J = 8.9 Hz, 2H), 7.57 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 6.91 (d, J = 8.9 Hz, 2H), 4.04 (t, J = 6.4 Hz, 2H), 4.02 (t, J = 6.4 Hz, 2H), 2.55 (s, 3H), 1.87–1.82 (m, 4H), 1.57–1.55 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ = 196.9, 163.1, 162.5, 134.1, 130.7, 130.4, 119.4, 115.3, 114.2, 103.9, 68.3, 68.1, 29.2, 29.1, 26.5, 25.9, 25.9 ppm. HRMS (MALDI +ve) calcd for C21H24NO3 ([M + H]+): m/z = 338.1751; exp 338.1751. Analysis calcd (%) for C21H23NO3 (337.42): C 74.75, H 6.87, N 4.15; found: C 74.60, H 6.73, N 3.99.
4-((8-(4-Acetylphenoxy)octyl)oxy)benzonitrile (9c). R f = 0.35. Mp = 91.1–92.3 °C. 1H NMR (500 MHz, CDCl3): δ = 7.92 (d, J = 8.9 Hz, 2H), 7.56 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.9 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 4.02 (t, J = 6.5 Hz, 2H), 3.99 (d, J = 6.5 Hz, 2H), 2.55 (s, 3H), 1.84–1.78 (m, 4H), 1.50–1.45 (m, 4H), 1.41–1.38 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ = 196.9, 163.2, 162.5, 134.1, 130.7, 130.3, 119.4, 115.3, 114.2, 103.8, 68.5, 68.3, 29.4, 29.4, 29.2, 29.1, 26.5, 26.1, 26.0 ppm. HRMS (MALDI +ve) calcd for C23H28NO3Na ([M + Na]+): m/z = 388.1883; exp 388.1884. Analysis calcd (%) for C23H27NO3 (365.47): C 75.59, H 7.45, N 3.83; found: C 75.51, H 7.28, N 3.78.
4-((9-(4-Acetylphenoxy)nonyl)oxy)benzonitrile (9d). R f = 0.35. Mp = 86.4–87.2 °C. 1H NMR (500 MHz, CDCl3): δ = 7.92 (d, J = 8.6 Hz, 2H), 7.57 (d, J = 8.6 Hz, 2H), 6.93 (d, J = 8.6 Hz, 2H), 6.91 (d, J = 8.6 Hz, 2H), 4.02 (t, J = 6.5 Hz, 2H), 3.99 (t, J = 6.5 Hz, 2H), 2.55 (s, 3H), 1.83–1.77 (m, 4H), 1.49–1.43 (m, 4H), 1.39–1.35 (m, 6H) ppm. 13C NMR (125 MHz, CDCl3): δ = 196.9, 163.2, 162.6, 134.1, 130.7, 130.3, 119.5, 115.3, 114.3, 103.8, 68.5, 68.3, 29.6, 29.4, 29.4, 29.2, 29.1, 26.5, 26.1, 26.1 ppm. HRMS (MALDI +ve) calcd for C24H30N3O2 ([M + H]+): m/z = 380.2220; exp 380.2221. Analysis calcd (%) for C24H29NO3 (379.50): C 75.96, H 7.70, N 3.69; found: C 75.82, H 7.68, N 3.55.
4-((10-(4-Acetylphenoxy)decyl)oxy)benzonitrile (9e). R f = 0.40. Mp = 71.5–73.5 °C. 1H NMR (500 MHz, CDCl3): δ = 7.92 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 4.01 (t, J = 6.5 Hz, 2H), 3.99 (d, J = 6.5 Hz, 2H), 2.55 (s, 3H), 1.83–1.76 (m, 4H), 1.48–1.42 (m, 4H), 1.37–1.32 (m, 8H) ppm. 13C NMR (125 MHz, CDCl3): δ = 196.9, 163.2, 162.6, 134.1, 130.7, 130.3, 119.4, 115.3, 114.2, 103.8, 68.5, 68.4, 29.6, 29.4, 29.4, 29.2, 29.1, 26.5, 26.1, 26.1 ppm, 1C masked. HRMS (MALDI +ve) calcd for C25H32NO3 ([M + H]+): m/z = 394.2377; exp 394.2377. Analysis calcd (%) for C25H31NO3 (393.53): C 76.30, H 7.94, N 3.56; found: C 76.29, H 7.90, N 3.52.
1-(4′-(Octyloxy)-[1,1′-biphenyl]-4-yl)ethan-1-one (9f)20,21. To a degassed biphasic mixture of 4-iodoacetophenone 10 (5.03 g, 20.4 mmol), K3PO4 (11.94 g, 56.71 mmol) and 11f (6.43 g, 25.7 mmol) in toluene (120 mL) and water (20 mL) was added Pd(PPh3)4 (1.00 g, 0.86 mmol) and the biphasic mixture was stirred 20 h at rt and heated to 80 °C for 10 h. The reaction mixture was cooled to rt and was diluted with water (80 mL) and the phases were separated. The aqueous phase was extracted with CH2Cl2 (2 × 50 mL) and the combined organic extracts dried over MgSO4, filtered and the solvent removed in vacuo. The solid was triturated with CH2Cl2 and heptane to give 9f (5.73 g, 86%) as an off white solid. Rf = 0.41 (toluene). Mp = Cr 91.0 SmX 128 Iso; Iso 130.2 SmX 79.5 Cr °C. 1H NMR (500 MHz, CDCl3): δ = 8.00 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.6 Hz, 2H), 6.99 (d, J = 8.6 Hz, 2H), 4.01 (t, J = 6.5 Hz, 2H), 2.63 (s, 3H), 1.84–1.78 (m, 2H), 1.50–1.45 (m, 2H), 1.42–1.20 (m, 8H), 0.89 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 197.9, 159.7, 145.6, 135.4, 132.1, 129.1, 128.5, 126.7, 115.1, 68.3, 32.0, 29.5, 29.4, 26.8, 26.2, 22.8, 14.3 ppm, 1C masked. HRMS (MALDI +ve) calcd for C22H28O2Na [(M + Na)+]: m/z = 347.1982; exp 347.1991. Analysis calcd (%) for C22H28O2 (324.46): C 81.44, H 8.70; found: C 80.99, H 8.42.
1-(2′,3′-Difluoro-4′-(octyloxy)-[1,1′-biphenyl]-4-yl)ethan-1-one (9g). To a degassed biphase of 4-iodoacetophenone 10 (5.20 g, 21.1 mmol) in toluene (120 mL) and water (20 mL) were added Pd(PPh3)4 (1.00 g, 0.86 mmol), K3PO4 (14.90 g, 70.2 mmol) and 11g (9.08 g, 31.7 mmol) and the biphasic mixture was stirred 20 h at rt, followed by heating at 80 °C for 7 h. The reaction was allowed to cool to rt and the vessel was diluted with water (80 mL) and the phases were separated. The aqueous phase was extracted with CH2Cl2 (2 × 50 mL) and the combined organic phases dried over MgSO4, filtered and the solvent removed in vacuo. The residue was subjected to flash column chromatography (gradient elution of 50–80% CH2Cl2/heptane) and triturated from CH2Cl2 and heptane to give 9g (5.01 g, 66%) as a white solid. Rf = 0.51 (toluene). Mp = Cr 63.2 Iso; Iso 53.8 SmX 48.8 Cr °C. 1H NMR (500 MHz, CDCl3): δ = 8.02 (d, J = 7.9 Hz, 2H), 7.61 (d, J = 7.9 Hz, 2H), 7.13 (td, J = 8.4, 2.2 Hz, 1H), 6.82 (td, J = 8.4, 1.6 Hz, 1H), 4.09 (t, J = 6.5 Hz, 2H), 2.64 (s, 3H), 1.87–1.82 (m, 2H), 1.51–1.45 (m, 2H), 1.43–1.22 (m, 8H), 0.89 (t, J = 6.9 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 197.8, 149.1 (dd, J = 249.8, 11.22 Hz), 148.8 (dd, J = 8.3, 3.1 Hz), 141.9 (dd, J = 248.2, 14.9 Hz), 139.8 (dd, J = 2.8, 1.9 Hz), 136.2, 129.0 (d, J = 3.1 Hz), 128.7, 123.8 (t, J = 4.1 Hz), 121.8 (d, J = 10.7 Hz), 109.8 (dd, J = 3.2, 0.9 Hz), 70.1, 32.0, 29.4, 29.4, 29.3, 26.8, 26.0, 22.8, 14.2 ppm. HRMS (MALDI +ve) calcd for C22H26F2O2Na [(M + Na)+]: m/z = 383.1793; exp 383.1805. Analysis calcd (%) for C22H26F2O2 (360.44): C 73.31, H 7.27; found: C 73.25, H 7.34.
2,3-Difluoro-4-(octyloxy)acetophenone (9h)14. To a degassed solution of 12 (6.22 g, 25.7 mmol) and FeCl3 (5.32 g, 32.8 mmol) in CH2Cl2 (150 mL), under argon and in an ice bath, was added a solution of acetyl chloride (2.1 mL in 50 mL CH2Cl2, 29.4 mmol) dropwise over a period of 80 min. The resulting mixture was stirred for 22 h during which time the vessel was allowed to reach rt. The reaction was quenched with water (500 mL) and the phases separated. The aqueous phase was extracted with CH2Cl2 (100 mL) and the combined organic extracts were washed with saturated aqueous saturated NaHCO3 (200 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (gradient elution of heptane to toluene) to give 9h (6.14 g, 84%) as a pale yellow oil. Rf = 0.55 (toluene). 1H NMR (500 MHz, CDCl3): δ = 7.65 (ddd, J = 9.0, 6.7, 2.3 Hz, 1H), 6.78 (ddd, J = 9.0, 7.0, 1.8 Hz, 1H), 4.10 (t, J = 6.6 Hz, 2H), 2.61 (d, J = 5.0 Hz, 3H), 1.96–1.78 (m, 2H), 1.52–1.41 (m, 2H), 1.37–1.25 (m, 8H), 0.88 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 193.9 (dd, J = 3.8, 2.4 Hz), 152.8 (dd, J = 8.1, 3.8 Hz), 152.0 (dd, J = 255.7, 11.3 Hz), 141.1 (dd, J = 248.4, 15.6 Hz), 125.0 (dd, J = 4.1, 3.4 Hz), 119.5 (dd, J = 10.5, 2.1), 108.9 (dd, J = 2.7, 1.0 Hz), 70.1, 31.9, 31.3 (d, J = 7.2 Hz), 29.4, 29.3, 29.1, 25.9, 22.8, 14.2 ppm. HRMS (ESP +ve) calcd for C16H22F2O2Na [(M + Na)+]: m/z = 307.1480; exp 307.1483. Analysis calcd (%) for C16H22F2O2 (284.35): C 67.59, H 7.80; found: C 67.69, H 7.71.
2-(1-(4-((5-(4-Cyanophenoxy)pentyl)oxy)phenyl)ethylidene)malononitrile (13a). Yellowish solid. Rf = 0.65 (CH2Cl2). Mp = 83.2–84.9, 95.6–100.3 °C. 1H NMR (500 MHz, CDCl3): δ = 7.60 (d, J = 8.9 Hz, 2H), 7.57 (d, J = 8.9 Hz, 2H) 6.97 (d, J = 8.9 Hz, 2H), 6.94 (d, J = 8.9 Hz, 2H), 4.06 (t, J = 6.2 Hz, 2H), 4.04 (t, J = 6.2 Hz, 2H), 2.61 (s, 3H), 1.92–1.87 (m, 4H), 1.70–1.64 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ = 174.1, 162.7, 162.4, 134.1, 130.0, 127.9, 119.4, 115.3, 115.0, 113.8, 113.5, 104.0, 82.0, 68.2, 28.9, 28.8, 23.9, 22.8 ppm, 1C masked. HRMS (MALDI +ve) calcd for C23H22N3O2 ([M + H]+): m/z = 372.1707; exp 372.1707. Analysis calcd (%) for C23H21N3O2 (371.44): C 74.37, H 5.70, N 11.31; found: C 73.41, H 5.76, N 10.80.
2-(1-(4-((6-(4-Cyanophenoxy)hexyl)oxy)phenyl)ethylidene)malononitrile (13b). White solid. Rf = 0.61 (CH2Cl2). Mp = 62.5–64.5 °C. 1H NMR (500 MHz, CDCl3): δ = 7.60 (d, J = 8.9 Hz, 2H), 7.57 (d, J = 8.9 Hz, 2H) 6.97 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 4.04 (t, J = 6.4 Hz, 2H), 4.02 (t, J = 6.3 Hz, 2H), 2.61 (s, 3H), 1.86–1.83 (m, 4H), 1.57–1.54 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ = 174.1, 162.8, 162.5, 134.1, 130.0, 127.9, 119.4, 115.3, 115.0, 113.8, 113.5, 103.9, 82.0, 68.3, 29.1, 29.1, 25.9, 25.9, 23.9 ppm, 1C masked. HRMS (MALDI +ve) calcd for C24H24N3O2 ([M + H]+): m/z = 386.1858; exp 386.1864. Analysis calcd (%) for C24H23N3O2 (385.47): C 74.78, H 6.01, N 10.90; found: C 74.75, H 5.73, N 10.81.
2-(1-(4-((8-(4-Cyanophenoxy)octyl)oxy)phenyl)ethylidene)malononitrile (13c). White solid. Rf = 0.62 (CH2Cl2). Mp = 67.0–67.7 °C. 1H NMR (500 MHz, CDCl3): δ = 7.64 (d, J = 8.9 Hz, 2H), 7.60 (d, J = 8.9 Hz, 2H) 7.00 (d, J = 8.9 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 4.05 (t, J = 6.5 Hz, 2H), 4.02 (t, J = 6.4 Hz, 2H), 2.64 (s, 3H), 1.87–1.81 (m, 4H), 1.54–1.48 (m, 4H), 1.46–1.41 ppm. 13C NMR (125 MHz, CDCl3): δ = 174.2, 162.8, 162.5, 134.1, 130.0, 127.7, 119.5, 115.2, 114.9, 113.9, 113.6, 103.7, 81.8, 68.4, 29.4, 29.1, 29.1, 26.0, 26.0, 24.0 ppm. HRMS (MALDI +ve) calcd for C26H28N3O2 ([M + H]+): m/z = 414.2176; exp 414.2177. Analysis calcd (%) for C26H27N3O2 (413.52): C 75.52, H 6.58, N 10.16; found: C 75.13, H 6.29, N 10.19.
2-(1-(4-((9-(4-Cyanophenoxy)nonyl)oxy)phenyl)ethylidene)malononitrile (13d). White solid. Rf = 0.68 (CH2Cl2). Mp = 87.1–88.1 °C. 1H NMR (500 MHz, CDCl3): δ = 7.60 (d, J = 8.9 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 4.01 (t, J = 6.5 Hz, 2H), 3.99 (t, J = 6.5 Hz, 2H), 2.61 (s, 3H), 1.83–1.77 (m, 4H), 1.49–1.43 (m, 4H), 1.39–1.35 (m, 6H) ppm. 13C NMR (125 MHz, CDCl3): δ = 174.2, 162.8, 162.5, 134.1, 130.0, 127.7, 119.5, 115.3, 115.0, 113.9, 113.6, 103.7, 81.8, 68.5, 68.5, 29.6, 29.4, 29.1, 29.1, 26.1, 26.0, 23.9 ppm, 1C masked. HRMS (MALDI +ve) calcd for C27H29N3O2Na ([M + Na]+): m/z = 450.2157; exp 450.2153. Analysis calcd (%) for C27H29N3O2 (427.55): C 75.85, H 6.84, N 9.83; found: C 75.50, H 6.81, N 9.80.
2-(1-(4-((10-(4-Cyanophenoxy)decyl)oxy)phenyl)ethylidene)malononitrile (13e). White solid. Rf = 0.65 (CH2Cl2). Mp = 62.6–63.8 °C. 1H NMR (500 MHz, CDCl3): δ = 7.60 (d, J = 8.9 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 4.01 (t, J = 6.5 Hz, 2H), 3.99 (t, J = 6.5 Hz, 2H), 2.61 (s, 3H), 1.83–1.77 (m, 4H), 1.47–1.43 (m, 4H), 1.37–1.31 (m, 8H) ppm. 13C NMR (125 MHz, CDCl3): δ = 174.2, 162.8, 162.5, 134.1, 130.0, 127.7, 199.5, 115.3, 115.0, 113.9, 113.6, 103.7, 81.8, 68.5, 68.5, 29.6, 29.4, 29.1, 29.1, 26.1, 26.0, 23.9 ppm, 2Cs masked. HRMS (MALDI +ve) calcd for C28H32N3O2 ([M + H]+): m/z = 442.2489; exp 442.2491. Analysis calcd (%) for C28H31N3O2 (441.58): C, 76.16; H, 7.08; N, 9.52; found: C 76.30, H 7.07, N 9.50.
2-(1-(4′-(Octyloxy)-[1,1′-biphenyl]-4-yl)ethylidene)malononitrile (13f). White solid. Rf = 0.56 (toluene). Mp = 58.6–59.4 °C. 1H NMR (500 MHz, CDCl3): δ = 7.68 (d, J = 8.5 Hz, 2H), 7.64 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 8.7 Hz, 2H), 6.99 (d, J = 8.7 Hz, 2H), 4.01 (t, J = 6.6 Hz, 2H), 2.67 (s, 3H), 1.84–1.78 (m, 2H), 1.51–1.45 (m, 2H), 1.40–1.26 (m, 8H), 0.90 (t, J = 6.9 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ = 174.7, 159.9, 145.2, 133.8, 131.5, 128.4, 128.3, 127.1, 115.2, 113.2, 83.8, 68.3, 32.0, 29.5, 29.4, 29.4, 26.2, 24.1, 22.8, 14.3 ppm. HRMS (MALDI +ve) calcd for C25H28N2ONa [(M + Na)+]: m/z = 395.2094; exp 395.2102. Analysis calcd (%) for C25H28N2O (372.51): C 80.61, H 7.58, N 7.52; found: C 80.67, H 7.67, N 7.38.
2-(1-(2′,3′-Difluoro-4′-(octyloxy)-[1,1′-biphenyl]-4-yl)ethylidene)malononitrile (13g). White solid. Rf = 0.61 (toluene). Mp = 59.5–60.4 °C. 1H NMR (500 MHz, CDCl3): δ = 7.65 (apparent s, 4H), 7.16–7.10 (ddd, J = 8.7, 8.2, 2.4, 1H), 6.83 (ddd, J = 9.1, 7.5, 1.8 Hz, 1H), 4.09 (t, J = 6.6 Hz, 2H), 2.68 (s, 3H), 1.92–1.81 (m, 2H), 1.54–1.44 (m, 2H), 1.42–1.22 (m, 8H), 0.89 (t, J = 6.8, 1H) ppm. 13C NMR (125 MHz, CDCl3): δ = 174.7, 149.1 (dd, J = 250, 11.4 Hz), 149.0 (dd, J = 8.1, 3.1 Hz), 141.9 (dd, J = 248.2, 14.8 Hz), 139.3, 134.9, 129.4, 129.4, 127.9, 123.8, 123.7, 123.7, 121.2, 121.1, 113.1, 112.9, 109.9, 84.7, 70.1, 31.9, 29.4, 29.4, 29.3, 26.0, 24.3, 22.8, 14.2 ppm. HRMS (MALDI +ve) calcd for C25H27F2N2O [(M + H)+]: m/z = 409.2086; exp 409.2095. Analysis calcd (%) for C25H26F2N2O (408.49): C 73.51, H 6.42, N 6.86; found: C 73.33, H 6.30, N 6.78.
2-(1-(2,3-Difluoro-4-(octyloxy)phenyl)ethylidene)malononitrile (13h). Off-white solid. Rf = 0.62 (toluene); Mp = 59.1–60.5 °C; 1H NMR (500 MHz, CDCl3): δ = 7.13 (ddd, J = 9.9, 7.6, 2.2 Hz, 1H), 6.82 (ddd, J = 9.9, 7.2, 1.8 Hz, 1H), 4.09 (t, J = 6.5 Hz, 2H), 2.61 (d, J = 1.6 Hz, 3H), 1.92–1.74 (m, 2H), 1.50–1.39 (m, 2H), 1.41–1.19 (m, 8H), 0.94–0.76 (m, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 170.3 (dd, J = 2.5, 1.5 Hz), 151.9 (dd, J = 8.1, 3.7 Hz), 148.5 (dd, J = 254.3, 12.1 Hz), 141.6, (dd, J = 251.7, 13.8 Hz), 123.2 (dd, J = 4.5, 3.2 Hz), 117.4 (d, J = 10.6 Hz), 112.2, 112.1, 109.3 (dd, J = 3.1, 1.3 Hz), 87.3, 70.1, 31.8, 29.2, 29.2, 28.9, 25.8, 24.1 (d, J = 4.6 Hz), 22.7, 14.1 ppm; HRMS (MALDI +ve) calcd for C19H22F2N2ONa [(M + Na)+]: m/z: 355.1592, found m/z = 355.1600.
2-(1-(4-Octylphenyl)ethylidene)malononitrile (13i). White solid. Rf = 0.65 (toluene). Mp = 51.5–52.0 °C. 1H NMR (500 MHz, CDCl3): δ = 7.50 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.3 Hz, 2H), 2.66 (t, J = 7.7 Hz, 2H), 2.63 (s, 3H), 1.66–1.1.60 (m, 2H), 1.35–1.27 (m, 10H), 0.88 (t, J = 7.1 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 175.3, 148.5, 133.3, 129.3, 127.7, 113.3, 113.2, 83.7, 36.1, 32.0, 31.2, 29.5, 29.4, 29.3, 24.2, 22.8, 14.2 ppm. HRMS (MALDI +ve) calcd for C19H24N2Na ([M + Na]+): m/z = 303.1831; exp 303.1838. Analysis calcd (%) for C19H24N2 (280.42): C 81.38, H 8.63, N 9.99; found: C 81.60, H 8.62, N 10.05.
2-(4-((5-(4-Cyanophenoxy)pentyl)oxy)phenyl)azulene-1,1(8aH)-dicarbonitrile (3a). To a solution of 13a (882 mg, 2.37 mmol) and [C7H7]BF4 (483 mg, 2.71 mmol) in CH2Cl2 (100 mL) at −78 °C, under an argon atmosphere, was slowly added NEt3 (0.70 mL, 5.0 mmol) and the resulting solution stirred for 30 min. The reaction was quenched by addition of aqueous 1 M HCl (100 mL) and the vessel allowed to reach ambient temperature. The phases were separated and the organic phase dried over MgSO4, filtered and the solvent removed under reduced pressure. The residue was dissolved in CH2ClCH2Cl (50 mL) and to the vessel [Ph3C]BF4 (882 mg, 2.49 mmol) was added whereafter the mixture was heated to reflux point for 2 h. The cooled vessel was placed in an ice bath and NEt3 (0.50 mL, 3.6 mmol) was added slowly to the flask. Toluene (50 mL) was added to the flask and the contents allowed to sit overnight at rt, after which time the solvent was removed in vacuo and the crude residue purified by flash column chromatography (2% EtOAc/toluene) to give 3a (345 mg, 32%), as an orange solid. Rf = 0.30 (2% EtOAc/toluene). Mp = 141.0–145.2 °C. 1H NMR (500 MHz, CDCl3): δ = 7.70 (d, J = 8.9 Hz, 2H), 7.61 (d, J = 8.9 Hz, 2H), 6.99 (d, J = 8.9 Hz, 2H); 6.97 (d, J = 8.9 Hz, 2H), 6.79 (s, 1H), 6.59 (dd, J = 11.3, 6.4 Hz, 1H), 6.47 (dd, J = 11.3, 6.1 Hz, 1H), 6.34–6.31 (m, 2H), 5.84 (dd, J = 10.2, 3.9 Hz, 1H), 4.08 (t, J = 6.3 Hz, 2H), 4.07 (t, J = 6.3 Hz, 2H), 3.80 (ddd, J = 3.9, 1.9, 1.9 Hz, 1H), 1.96–1.89 (m, 4H), 1.74–1.69 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.3, 160.4, 139.9, 139.0, 134.0, 131.0, 130.4, 130.1, 127.8, 127.6, 123.0, 120.0, 119.4, 119.3, 115.3, 115.2, 115.1, 112.9, 103.7, 68.1, 67.8, 51.1, 45.2, 28.8, 28.8, 22.7 ppm. HRMS (MALDI +ve) calcd for C30H26N3O2 ([M + H]+): m/z = 460.2020; exp 460.2021. Analysis calcd (%) for C30H25N3O2 (459.55): C 78.41, H 5.48, N 9.14; found: C 78.11, H 5.40, N 9.05.
2-(4-((6-(4-Cyanophenoxy)hexyl)oxy)phenyl)azulene-1,1(8aH)-dicarbonitrile (3b). To a solution of 13b (1.05 g, 2.72 mmol) and [C7H7]BF4 (527 mg, 2.96 mmol) in CH2Cl2 (100 mL) at −78 °C, under an argon atmosphere, was slowly added NEt3 (1.0 mL, 7.2 mmol) and the resulting solution stirred for 30 min. The reaction was quenched by addition of aqueous 1 M HCl (100 mL) and the vessel allowed to reach ambient temperature. The phases were separated and the organic phase dried over MgSO4, filtered and the solvent removed under reduced pressure. The residue was dissolved in CH2ClCH2Cl (50 mL) and to the vessel [Ph3C]BF4 (1.00 g, 3.03 mmol) was added whereafter the mixture was heated to reflux point for 2 h. The cooled vessel was placed in an ice bath and NEt3 (0.70 mL, 5.0 mmol) was added slowly to the flask. Toluene (50 mL) was added to the flask and the contents allowed to sit overnight at rt, after which time the solvent was removed in vacuo and the crude residue purified by flash column chromatography (2% EtOAc/toluene) to give 3b (603 mg, 47%), as a fluffy yellow solid. Rf = 0.33 (2% EtOAc/toluene). Mp = 122.1–124.0 °C. 1H NMR (500 MHz, CDCl3): δ = 7.67 (d, J = 8.9 Hz, 2H), 7.57 (d, J = 8.9 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 6.75 (s, 1H), 6.55 (dd, J = 11.3, 6.4 Hz, 1H), 6.44 (dd, J = 11.3, 6.1 Hz, 1H), 6.31–6.28 (m, 2H), 5.81 (dd, J = 10.2, 3.9 Hz, 1H), 4.03 (t, J = 6.4 Hz, 2H), 4.02 (t, J = 6.4 Hz, 2H), 3.77 (ddd, J = 3.9, 2.0, 2.0 Hz, 1H), 1.88–1.82 (m, 3H), 1.57–1.54 (m, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.5, 160.6, 140.1, 139.2, 134.1, 131.1, 130.5, 130.2, 128.0, 127.8, 123.1, 120.1, 119.4, 119.4, 115.3, 115.3, 113.0, 68.3, 68.1, 51.3, 45.4, 29.2, 29.1, 25.9, 25.9 ppm, 1C masked. HRMS (MALDI +ve) calcd for C31H28N3O2 ([M + H]+): m/z = 474.2176; exp 474.2176. Analysis calcd (%) for C31H27N3O2 (473.58): C 78.62, H 5.75, N 8.87; found: C 78.31, H 5.42, N 8.69.
2-(4-((8-(4-Cyanophenoxy)octyl)oxy)phenyl)azulene-1,1(8aH)-dicarbonitrile (3c). To a solution of 13c (1.04 g, 2.52 mmol) and [C7H7]BF4 (522 mg, 2.93 mmol) in CH2Cl2 (100 mL) at −78 °C, under an argon atmosphere, was slowly added NEt3 (1.0 mL, 7.2 mmol) and the resulting solution stirred for 30 min. The reaction was quenched by addition of aqueous 1 M HCl (100 mL) and the vessel allowed to reach ambient temperature. The phases were separated and the organic phase dried over MgSO4, filtered and the solvent removed under reduced pressure. The residue was dissolved in CH2ClCH2Cl (50 mL) and to the vessel [Ph3C]BF4 (929 mg, 2.81 mmol) was added whereafter the mixture was heated to reflux point for 2 h. The cooled vessel was placed in an ice bath and NEt3 (0.70 mL, 5.0 mmol) was added slowly to the flask. Toluene (50 mL) was added to the flask and the contents allowed to sit overnight at rt, after which time the solvent was removed in vacuo and the crude residue purified by flash column chromatography (2% EtOAc/toluene) to give 3c (200 mg, 16%) as an orange solid. Rf = 0.35 (2% EtOAc/toluene). Mp = 98.5–101.2 °C. 1H NMR (500 MHz, CDCl3): δ = 7.67 (d, J = 8.9 Hz, 2H), 7.57 (d, J = 8.9 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 6.75 (s, 1H), 6.55 (dd, J = 11.2, 6.4 Hz, 1H), 6.44 (dd, J = 11.3, 6.1 Hz, 1H), 6.31–6.28 (m, 2H), 5.81 (dd, J = 10.2, 3.8 Hz, 1H), 4.01 (t, J = 6.5 Hz, 1H), 4.00 (d, J = 6.5 Hz, 1H), 3.77 (ddd, J = 3.9, 2.0, 2.0 Hz, 1H), 1.84–1.78 (m, 4H), 1.51–1.45 (m, 4H), 1.42–1.39 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.6, 160.7, 140.2, 139.2, 134.1, 131.1, 130.4, 130.2, 127.9, 127.8, 123.0, 120.0, 119.5, 119.4, 115.5, 115.3, 113.0, 103.8, 68.5, 68.3, 51.3, 45.4, 29.4, 29.2, 29.1, 26.1, 26.0 ppm, 2C masked. HRMS (MALDI +ve) calcd for C33H32N3O2 ([M + H]+): m/z = 502.2489; exp 502.2490.
2-(4-((9-(4-Cyanophenoxy)nonyl)oxy)phenyl)azulene-1,1(8aH)-dicarbonitrile (3d). To a solution of 13d (1.02 g, 2.39 mmol) and [C7H7]BF4 (500 mg, 2.81 mmol) in CH2Cl2 (100 mL) at −78 °C, under an argon atmosphere, was slowly added NEt3 (1.0 mL, 7.2 mmol) and the resulting solution stirred for 30 min. The reaction was quenched by addition of aqueous 1 M HCl (100 mL) and the vessel allowed to reach ambient temperature. The phases were separated and the organic phase dried over MgSO4, filtered and the solvent removed under reduced pressure. The residue was dissolved in CH2ClCH2Cl (50 mL) and to the vessel [Ph3C]BF4 (935 mg, 2.83 mmol) was added whereafter the mixture was heated to reflux point for 2 h. The cooled vessel was placed in an ice bath and NEt3 (0.70 mL, 5.0 mmol) was added slowly to the flask. Toluene (50 mL) was added to the flask and the contents allowed to sit overnight at rt, after which time the solvent was removed in vacuo and the crude residue purified by flash column chromatography (2% EtOAc/toluene) to give 3d (258 mg, 21%) as an orange solid. Rf = 0.36 (2% EtOAc/toluene). Mp = 98.9–103.8, 125.6–130.6 °C. 1H NMR (500 MHz, CDCl3): δ 7.67 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 6.75 (s, 1H), 6.55 (dd, J = 11.2, 6.4 Hz, 1H), 6.44 (dd, J = 11.2, 6.1 Hz, 1H), 6.31–6.28 (m, 2H), 5.81 (dd, J = 10.2, 3.8 Hz, 1H), 4.01 (t, J = 6.8 Hz, 2H), 4.00 (t, J = 6.8 Hz, 2H), 3.77 (ddd, J = 3.8, 2.0, 2.0 Hz, 1H), 1.83–1.78 (m, 4H), 1.49–1.44 (m, 4H), 1.40–1.37 (m, 6H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.6, 160.7, 140.2, 139.2, 134.1, 131.1, 130.4, 130.2, 127.9, 127.8, 123.0, 120.0, 119.5, 119.4, 115.5, 115.3, 113.0, 103.8, 68.5, 68.3, 51.3, 45.4, 29.6, 29.4, 29.2, 29.1, 26.1, 26.1 ppm, 2Cs masked. HRMS (MALDI +ve) calcd for C34H34N3O2 ([M + H]+): m/z = 516.2646; exp 516.2649. Analysis calcd (%) for C34H33N3O2 (515.66): C 79.19, H 6.45, N 8.15; found: C 78.68, H 6.18, N 8.11.
2-(4-((10-(4-Cyanophenoxy)decyl)oxy)phenyl)azulene-1,1(8aH)-dicarbonitrile (3e). To a solution of 13e (1.12 g, 2.54 mmol) and [C7H7]BF4 (503 mg, 2.83 mmol) in CH2Cl2 (100 mL) at −78 °C, under an argon atmosphere, was slowly added NEt3 (1.0 mL, 7.2 mmol) and the resulting solution stirred for 30 min. The reaction was quenched by addition of aqueous 1 M HCl (100 mL) and the vessel allowed to reach ambient temperature. The phases were separated and the organic phase dried over MgSO4, filtered and the solvent removed under reduced pressure. The residue was dissolved in CH2ClCH2Cl (50 mL) and to the vessel [Ph3C]BF4 (851 mg, 2.58 mmol) was added whereafter the mixture was heated to reflux point for 2 h. The cooled vessel was placed in an ice bath and NEt3 (0.70 mL, 5.0 mmol) was added slowly to the flask. Toluene (50 mL) was added to the flask and the contents allowed to sit overnight at rt, after which time the solvent was removed in vacuo and the crude residue purified by flash column chromatography (2% EtOAc/toluene) to give 3e (552 mg, 41%), as an orange solid. Rf = 0.38 (2% EtOAc/toluene). Mp = 102.2–121.8 °C. 1H NMR (500 MHz, CDCl3): δ = 7.67 (d, J = 8.9 Hz, 2H), 7.57 (d, J = 8.9 Hz, 2H), 6.97 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 6.75 (s, 1H), 6.55 (dd, J = 11.3, 6.3 Hz, 1H), 6.44 (dd, J = 11.3, 6.1 Hz, 1H), 6.31–6.28 (m, 2H), 5.81 (dd, J = 10.2, 3.8 Hz, 1H), 4.01 (d, J = 6.6 Hz, 2H), 3.99 (d, J = 6.6 Hz, 2H), 3.77 (ddd, J = 3.8, 1.9, 1.9 Hz, 1H), 1.83–1.77 (m, 4H), 1.49–1.43 (m, 4H), 1.39–1.32 (m, 6H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.6, 160.7, 140.2, 139.2, 134.1, 131.1, 130.4, 130.1, 127.9, 127.8, 123.0, 120.0, 119.5, 119.4, 115.5, 115.3, 115.3, 113.0, 103.8, 68.5, 68.3, 51.3, 45.4, 29.6, 29.6, 29.5, 29.5, 29.2, 29.1, 26.1, 26.1 ppm. HRMS (MALDI +ve) calcd for C35H36N3O2 ([M + H]+): m/z = 530.2802; exp 530.2806.
2-(4′-(Octyloxy)-[1,1′-biphenyl]-4-yl)azulene-1,1(8aH)-dicarbonitrile (3f). Method 1: to a solution/suspension of the crude mixture from 13f (3.54 g, 9.50 mmol) and [C7H7]BF4 (1.71 g, 9.61 mmol) in CH2Cl2 (250 mL), at −78 °C under an argon atmosphere, was added NEt3 (1.0 g, 1.4 mL, 10 mmol) over a 30 min period and the reaction mixture stirred at −78 °C for 2 h. Additional NEt3 was added (0.5 mL, 3.58 mmol) and the reaction mixture was left to stir for 1 h and allowed to reach rt and subjected to ultrasonication. HCl (150 mL, 2 M) was added to the vessel and the phases separated. The aqueous phase was extracted with CH2Cl2 (100 mL) and the combined organic phases dried over MgSO4, filtered, and the solvent was removed in vacuo. The residue was taken up in CH2ClCH2Cl (125 mL) and treated with [Ph3C]BF4 (3.85 g, 11.6 mmol) and subjected to reflux for 2 h under inert atmosphere. The reaction mixture was diluted with toluene (125 mL) and cooled to 0 °C and NEt3 (1.2 g, 1.7 mL, 12 mmol) was added, where the mixture was stirred overnight and allowed to reach room temperature. The solvent was removed in vacuum, and the residue was subjected to flash chromatography (40–50% CH2Cl2/heptane) and crystallized from CH2Cl2/ethanol to furnish 3f (502 mg, 11%) as a yellow solid. Method 2: To a degassed solution of 15 (1.54 g, 4.03 mmol) in toluene (60 mL) and water (15 mL) was added Pd(OAc)2 (55 mg, 0.245 mmol), RuPhos (187 mg, 0.400 mmol), K3PO4 (1.31 g, 6.17 mmol) and 11f (1.56 g, 6.24 mmol), and the biphasic mixture stirred for 18 h at 80 °C. TLC indicated an incomplete reaction and more Pd(OAc)2 (60 mg, 0.267 mmol), RuPhos (195 mg, 0.419 mmol) and K3PO4 (2.32 g, 10.9 mmol) and 11f (1.21 g, mmol) were added and the mixture heated to 80 °C for another 18 h. The contents of the vessel were diluted with water (100 mL) and the phases were separated. The aqueous phase was extracted with CH2Cl2 (100 mL) and the combined organic extracts were dried over MgSO4, filtered and the solvent removed in vacuo. The crude residue was subjected to flash column chromatography (gradient elution of 55–70% toluene/heptane) and subsequently recrystallized from CH2Cl2/heptane to give pure 3f (562 mg, 30%). Rf = 0.42 (50% CH2Cl2/heptane). Mp = Cr 90.6 (N 75.7) Iso; Iso 75.6 N 29.8 Tg °C. 1H NMR (500 MHz, CDCl3): δ = 7.78 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 8.7 Hz, 2H), 6.99 (d, J = 8.7 Hz, 2H), 6.91 (s, 1H), 6.57 (dd, J = 11.2, 6.4 Hz, 1H), 6.47 (dd, J = 11.2, 6.1 Hz, 1H), 6.35 (d, J = 6.4 Hz, 1H), 6.31 (ddd, J = 10.1, 6.1, 2.1 Hz, 1H), 5.84 (dd, J = 10.1, 3.8 Hz, 1H), 4.01 (t, J = 6.6 Hz, 1H), 3.81 (ddd, J = 3.7, 2.0, 2.0 Hz, 1H), 1.84–1.78 (m, 2H), 1.51–1.46 (m, 2H), 1.43–1.14 (m, 10H), 0.89 (t, J = 6.9 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ = 159.3, 142.4, 140.0, 138.9, 132.0, 131.7, 131.0, 130.8, 128.5, 128.1, 127.7, 127.2, 126.7, 120.7, 119.5, 115.2, 115.0, 112.8, 68.2, 51.2, 45.1, 31.8, 29.4, 29.3, 29.3, 26.1, 22.7, 14.1 ppm. HRMS (MALDI +ve) calcd for C32H32N2ONa [(M + Na)+]: m/z = 483.2407; exp 483.2406. Analysis calcd (%) for C32H32N2O (460.62): C 83.44, H 7.00, N 6.08; found: C 83.28, H 6.90, N 6.12.
2-(2′,3′-Difluoro-4′-(octyloxy)-[1,1′-biphenyl]-4-yl)azulene-1,1(8aH)-dicarbonitrile (3g). Method 1: to a mixture of 13g (3.35 g, 8.21 mmol) and [C7H7]BF4 (1.53 g, 8.60 mmol) in CH2Cl2 (250 mL) at −78 °C under argon atmosphere was added NEt3 (1.2 mL, 8.6 mmol) portion-wise over 30 min and the reaction mixture stirred at −78 °C for 2 h. Since there were still starting material and tropylium left unreacted, more NEt3 was added (0.5 mL, 3.58 mmol) and the reaction mixture was left to stir for 1 h more and allowed to reach rt while being sonicated. Then it was quenched with HCl (100 mL, 2 M) and diluted with water (50 mL). Subsequently, the phases were separated and the water phase was extracted with CH2Cl2 (100 mL) and the combined organic phase was dried over MgSO4 and the solvent was removed by rotary evaporation. The residue was treated with [Ph3C]BF4 (3.27 g, 9.89 mmol) in DCE (125 mL) under reflux for 2 h under an argon atmosphere, after which time the reaction mixture was cooled to 0 °C and diluted with toluene (125 mL). When the reaction mixture reached 0 °C, NEt3 (1.5 mL, 10 mmol) was added, and the mixture was stirred overnight, while allowing to reach rt, and left to stir for 15 h. The solvent was removed in vacuo and the residue was subjected to flash column chromatography (40–50% CH2Cl2/heptane) and crystallized from CH2Cl2 and ethanol to give 3g (1.104 g, 25%) as a yellow solid. Method 2: To a degassed solution of 15 (2.05 g, 5.36 mmol) in toluene (60 mL) and water (15 mL) were added Pd(OAc)2 (76 mg, 0.339 mmol), RuPhos (262 mg, 0.561 mmol) and K3PO4 (2.51 g, 11.8 mmol) and 11g (1.56 g, mmol). Degassed water (15 mL) was added and the biphasic mixture stirred for 18 h at 90 °C. Additional Pd(OAc)2 (62 mg, 0.276 mmol), RuPhos (206 mg, 0.441 mmol) and K3PO4 (1.80 g, 8.48 mmol) and 11g (1.04 g, mmol) were added and the mixture heated to 80 °C for a further 18 h. The contents of the vessel were diluted with water (100 mL) and the phases were separated. The aqueous phase was extracted with CH2Cl2 (100 mL) and the combined organic components were dried over MgSO4, filtered and the solvent removed in vacuo. The crude residue was subjected to flash column chromatography (20% EtOAc/heptane) and a second flash column (gradient eluent 40–60% CH2Cl2/heptane) and then recrystallized from CH2Cl2/heptane to give 3g (522 mg, 20%). Rf = 0.44 (50% CH2Cl2/heptane). Mp = Cr 79.9 (N 52.6) Iso; Iso 52.6 N 30.8 Tg °C. 1H NMR (500 MHz, CDCl3): δ = 7.80 (d, J = 8.4 Hz, 2H), 7.62 (dd, J = 8.4, 1.3 Hz, 2H), 7.13 (td, J = 8.5, 2.3 Hz, 1H), 6.93 (s, 1H), 6.83 (td, J = 8.5, 1.6 Hz, 1H), 6.58 (dd, J = 11.2, 6.3 Hz, 1H), 6.49 (dd, J = 11.2, 6.1 Hz, 1H), 6.37 (d, J = 6.3 Hz, 0H), 6.32 (ddd, J = 10.2, 6.1, 2.1 Hz, 1H), 5.84 (dd, J = 10.2, 3.8 Hz, 1H), 4.09 (t, J = 6.6 Hz, 2H), 3.81 (ddd, J = 3.8, 2.1, 2.1 Hz, 1H), 1.85 (p, J = 6.6 Hz, 2H), 1.52–1.49 (m, 2H), 1.41–1.20 (m, 10H), 0.89 (t, J = 6.6 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 149.2 (dd, J = 250.6, 11.2 Hz), 148.6 (dd, J = 8.2, 3.0 Hz), 142.0 (dd, J = 247.9, 14.9 Hz), 139.8, 138.8, 136.8 (dd, J = 2.4, 1.5 Hz), 132.6, 131.1 (d, J = 8.5 Hz), 129.7, 129.6 (d, J = 3.3 Hz), 127.8, 126.5, 123.6 (apparent t, J = 4.1 Hz), 121.7 (d, J = 10.6 Hz), 121.3, 119.6, 115.4, 112.9, 109.8 (dd, J = 3.3, 1.2 Hz), 70.1, 51.3, 45.3, 32.0, 29.5, 29.4, 29.3, 26.0, 22.8, 14.3 ppm. HRMS (MALDI +ve) calcd for C32H30F2N2ONa [(M + Na)+]: m/z = 519.2218, found m/z = 519.2217. Analysis calcd (%) for C32H30F2N2O (496.60): C 77.40, H 6.09, N 5.64; found: C 77.59, H 5.88, N 5.70.
2-(2,3-Difluoro-4-(octyloxy)phenyl)azulene-1,1(8aH)-dicarbonitrile (3h). To a mixture of 13h (6.58 g, 19.8 mmol) and [C7H7]BF4 (3.62 g, 20.4 mmol) in CH2Cl2 (250 mL) at −78 °C under an argon atmosphere, was added NEt3 (2.8 mL, 20.4 mmol) over 30 min, and the reaction mixture stirred at −78 °C for 2 h. Additional NEt3 (0.5 mL, 3.58 mmol) was added and the reaction mixture was allowed to stir for a further 60 min. The reaction was quenched cold with HCl (50 mL, 2 M) and allowed to reach rt and diluted with water (50 mL). The phases were separated and the organic phase was washed with water (250 mL), dried over MgSO4, filtered and the solvent removed under reduced pressure. The residue was taken up in CH2ClCH2Cl (125 mL) and treated with [Ph3C]BF4 (7.96 g, 24.1 mmol) and the vessel subjected to reflux for 2 h under argon. The reaction mixture was cooled to 0 °C and diluted with toluene (125 mL) and NEt3 (3.4 mL, 24.4 mmol) was added. The mixture was stirred overnight, allowing the vessel to reach rt. The solvent was removed in vacuo and the residue was subjected to flash chromatography (30–50% CH2Cl2/heptane) to give 3h (2.528 g, 30%) as a yellow solid. Rf = 0.60 (50% CH2Cl2/heptane). Mp = 95.0–97.9 °C. 1H NMR (500 MHz, CDCl3): δ = 7.52 (td, J = 8.5, 2.0 Hz, 1H), 7.04 (s, 1H), 6.92–6.81 (m, 1H), 6.57 (dd, J = 11.3, 6.4 Hz, 1H), 6.48 (dd, J = 11.3, 6.1 Hz, 1H), 6.37 (d, J = 6.4 Hz, 1H), 6.31 (ddd, J = 10.3, 6.1, 1.9 Hz, 1H), 5.80 (dd, J = 10.3, 3.8 Hz, 1H), 4.10 (t, J = 6.6 Hz, 2H), 3.74 (ddd, J = 3.8, 1.9, 1.9 Hz, 1H), 1.88–1.82 (m, 2H), 1.51–1.45 (m, 2H), 1.40–1.25 (m, 8H), 0.90 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 150.51 (dd, J = 254.2, 11.8 Hz), 149.89 (dd, J = 8.2, 3.6 Hz), 142.05 (dd, J = 249.0, 14.8 Hz), 139.27, 135.90 (d, J = 14.9 Hz), 132.86 (dd. J = 4.3, 3.3 Hz), 131.08 (d, J = 1.6 Hz), 127.91, 121.71 (dd, J = 4.3, 3.6 Hz), 121.67, 119.45, 115.22, 112.95 (d, J = 9.1 Hz), 112.83, 109.34 (dd, J = 3.0, 1.2 Hz), 70.03, 50.36, 46.32, 31.91, 29.38, 29.31, 29.09, 25.95, 22.78, 14.22 ppm, 1C masked. HRMS (MALDI +ve) calcd for C26H26F2N2O [(M)+]: m/z = 420.2008; exp 420.2008; analysis calcd (%) for C26H26F2N2O (420.20): C 74.26, H 6.23, N 6.66; found: C 74.14, H 6.35, N 6.54.
2-(4-Octylphenyl)azulene-1,1-(8aH)-dicarbonitrile (3i). To a mixture of 13i (10.01 g, 35.70 mmol) and [C7H7]BF4 (6.35 g, 35.7 mmol) in CH2Cl2 (250 mL), at −78 °C under an argon atmosphere, was added NEt3 (5 mL, 35.9 mmol) over 30 min, and the resulting mixture allowed to stir at −78 °C for 2 h. Saturated aqueous NH4Cl (100 mL) and water (100 mL) were added to the vessel and the contents allowed to reach ambient temperature. The phases were separated and the organic phase was washed with water (200 mL) and brine (200 mL). The organic phase was dried over MgSO4, filtered, and the removal of the solvent gave the crude mixture (13.08 g). Of this crude material (12.84 g, 34.65 mmol) was dissolved in ClCH2CH2Cl (200 mL) and treated with [Ph3C]BF4 (13.73 g, 41.58 mmol), whilst being subjected to reflux for 2 h under argon. After cooling the vessel, toluene (200 mL) was added and the vessel placed in an ice bath, where NEt3 (6.3 mL, 45 mmol) was added and the mixture was stirred overnight, whilst being allowed to reach rt. Aqueous NH4Cl (100 mL) was added to the vessel and the phases separated. The organic phase was washed with water (100 mL) and brine (100 mL), dried with MgSO4, filtered and absorbed onto Celite. The material was purified by dry column vacuum chromatography (gradient elution from heptane to toluene in 10% steps) followed by recrystallization from boiling ethanol to give 3i (1.73 g, 14%). The material was stored in the freezer, as it decomposes at ambient temperature. Rf = 0.65 (50% toluene/heptane). Mp = 54.4–58.6 °C. 1H NMR (500 MHz, CDCl3): δ = 7.58 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 6.77 (s, 1H), 6.49 (dd, J = 11.2, 6.4 Hz, 1H), 6.39 (dd, J = 11.2, 6.1 Hz, 1H), 6.31–6.05 (m, 2H), 5.75 (dd, J = 10.3, 3.8 Hz, 1H), 3.71 (ddd, J = 3.8, 1.9, 1.9 Hz, 1H), 2.57 (t, J = 7.7 Hz, 2H), 1.62–1.52 (m, 2H), 1.34–1.01 (m, 10H), 0.81 (t, J = 7.0 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 145.7, 140.5, 139.1, 131.4, 131.1, 130.7, 129.5, 128.0, 127.8, 126.3, 120.6, 119.6, 115.4, 113.0, 51.3, 45.3, 36.0, 32.0, 31.3, 29.6, 29.5, 29.4, 22.8, 14.3 ppm. HRMS (MALDI +ve) calcd for C26H27N2 [(M − H)+]: m/z = 367.2169; exp 367.2169; analysis calcd (%) for C26H28N2 (368.52): C 84.74, H 7.66, N 7.60; found: C 84.69, H 7.58, N 7.60.
4-((5-Azidopentyl)oxy)benzonitrile (14a). R f = 0.41. Mp = 29.1–30.5 °C. 1H NMR (500 MHz, CDCl3): δ = 7.57 (d, J = 7.6 Hz, 2H), 6.93 (d, J = 7.6 Hz, 2H), 4.01 (t, J = 6.3 Hz, 2H), 3.32 (t, J = 6.7 Hz, 2H), 1.84 (p, J = 6.3 Hz, 2H), 1.68 (p, J = 6.7 Hz, 2H), 1.65–1.51 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.4, 134.1, 119.4, 115.3, 104.0, 68.1, 51.4, 28.7, 28.7, 23.5 ppm. HRMS (ESP +ve) calcd for C12H14N4ONa ([M + Na]+): m/z = 231.1060; exp 253.1060.
4-((6-Azidohexyl)oxy)benzonitrile (14b). R f = 0.42. Mp = 37.4–40.5 °C. 1H NMR (500 MHz, CDCl3): δ = 7.57 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 4.00 (t, J = 6.4 Hz, 2H), 3.29 (t, J = 6.8 Hz, 2H), 1.82 (p, J = 6.4 Hz, 2H), 1.64 (p, J = 6.8 Hz, 2H), 1.57–1.42 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.5, 134.1, 119.4, 115.3, 103.9, 68.3, 51.5, 29.0, 28.9, 26.6, 25.7 ppm. HRMS (ESP +ve) calcd for C13H16N4ONa ([M + Na]+): m/z = 267.1217; exp 267.1216.
4-((8-Azidooctyl)oxy)benzonitrile (14c). R f = 0.44. Mp = 33.9–36.5 °C. 1H NMR (500 MHz, CDCl3): δ = 7.57 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 3.99 (t, J = 6.5 Hz, 2H), 3.26 (t, J = 6.6 Hz, 2H), 1.80 (p, J = 6.5 Hz, 2H), 1.63–1.56 (m, 2H), 1.53–1.16 (m, 8H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.5, 134.1, 119.4, 115.3, 103.8, 68.5, 51.6, 29.3, 29.2, 29.1, 28.9, 26.8, 26.0 ppm. HRMS (ESP +ve) calcd for C15H20N4ONa ([M + Na]+): m/z = 295.1529; exp 295.1530.
4-((9-Azidononyl)oxy)benzonitrile (14d). R f = 0.49. Mp = 42.5–43.2 °C. 1H NMR (500 MHz, CDCl3): δ = 7.57 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 3.99 (t, J = 6.56 Hz, 2H), 3.26 (t, J = 6.8 Hz, 2H), 1.79 (t, J = 6.56 Hz, 2H), 1.60 (t, J = 6.8 Hz, 2H), 1.51–1.25 (m, 10H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.6, 134.1, 119.5, 115.3, 103.8, 68.5, 51.6, 29.5, 29.3, 29.2, 29.1, 29.0, 26.8, 26.0 ppm. HRMS (ESP +ve) calcd for C16H22N4ONa ([M + Na]+): m/z = 309.1687; exp 309.1686.
4-((10-Azidodecyl)oxy)benzonitrile (14e). R f = 0.49. Mp = 46.7–47.7 °C. 1H NMR (500 MHz, CDCl3): δ = 7.57 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 3.99 (t, J = 6.6 Hz, 2H), 3.26 (t, J = 6.9 Hz, 2H), 1.80 (p, J = 6.6 Hz, 2H), 1.60 (p, J = 6.9 Hz, 2H), 1.48–1.31 (m, 12H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.4, 134.0, 119.3, 115.2, 103.7, 68.4, 51.5, 29.4, 29.4, 29.3, 29.1, 29.0, 28.8, 26.7, 25.9 ppm. HRMS (ESP +ve) calcd for C17H24N4ONa ([M + Na]+): m/z = 323.1842; exp 323.1842.
1,1-Dicyano-2-(4-(1-(5-(4-cyanophenoxy)pentyl)-1H-1,2,3-triazol-4-yl)phenyl)-1,8a-dihydroazulene (4a). To a degassed solution of 15 (51 mg, 0.182 mmol), 14a (96 mg, 0.417 mmol) and Hünig's base (10 drops) in toluene (4 mL) was added CuI (8 mg, 0.021 mmol) and the contents stirred for 2 days at rt. The solvent was removed under reduced pressure and the residue purified by flash column chromatography (10% EtOAc/CH2Cl2) to furnish 4a as a yellow solid (91 mg, 98%). Rf = 0.60 (5% EtOAc/CH2Cl2). Mp = 183.6–185.5; 186.9–188.7 °C. 1H NMR (500 MHz, CDCl3): δ = 7.92 (d, J = 8.4 Hz, 2H), 7.82 (s, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.8 Hz, 2H), 6.93 (s, 1H), 6.90 (d, J = 8.8 Hz, 2H), 6.57 (dd, J = 11.3, 6.4 Hz, 1H), 6.48 (dd, J = 11.3, 6.1 Hz, 1H), 6.36 (d, J = 6.4 Hz, 1H), 6.31 (ddd, J = 10.2, 6.1, 1.9 Hz, 1H), 5.82 (dd, J = 10.2, 3.7 Hz, 1H), 4.46 (t, J = 7.0 Hz, 2H), 3.99 (t, J = 6.2 Hz, 2H), 3.80 (ddd, J = 3.7, 1.9, 1.9 Hz, 1H), 2.06 (p, J = 7.0 Hz, 2H), 1.87 (p, J = 6.2 Hz, 2H), 1.59–1.53 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.3, 146.9, 139.7, 138.8, 134.1, 132.5, 132.2, 131.1, 131.1, 130.2, 127.8, 126.9, 126.4, 121.3, 120.2, 119.5, 119.4, 115.3, 112.9, 104.1, 51.2, 50.4, 45.2, 30.1, 28.5, 23.2 ppm. HRMS (MALDI +ve) calcd for C32H27N6O ([M + H]+): m/z = 511.2244; exp 511.2244. Analysis calcd (%) for C32H26N6O (510.60): C 75.27, H 5.13, N 16.46; found: C 75.27, H 4.78, N 16.26.
1,1-Dicyano-2-(4-(1-(6-(4-cyanophenoxy)hexyl)-1H-1,2,3-triazol-4-yl)phenyl)-1,8a-dihydroazulene (4b). To a degassed solution of 15 (104 mg, 0.371 mmol), 14b (117 mg, 0.478 mmol) and Hünig's base (10 drops) in toluene (4 mL) was added CuI (10 mg, 0.053 mmol) and the contents stirred for 2 days at rt. The solvent was removed under reduced pressure and the residue was subjected to flash column chromatography (10% EtOAc/CH2Cl2) and subsequently crystallized (CH2Cl2/heptane) to furnish 4b (141 mg, 72%) as a yellow solid. Rf = 0.66 (5% EtOAc/CH2Cl2). Mp = 54.0–55.7 °C. 1H NMR (500 MHz, CDCl3): δ = 7.92 (d, J = 8.6 Hz, 2H), 7.82 (s, 1H), 7.80 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.9 Hz, 2H), 6.94 (s, 1H), 6.91 (d, J = 8.9 Hz, 2H), 6.58 (dd, J = 11.2, 6.5 Hz, 1H), 6.48 (dd, J = 11.2, 6.1 Hz, 1H), 6.36 (d, J = 6.5 Hz, 1H), 6.31 (ddd, J = 10.2, 6.1, 2.1 Hz, 1H), 5.83 (dd, J = 10.2, 3.8 Hz, 1H), 4.44 (t, J = 7.1 Hz, 2H), 3.98 (t, J = 6.3 Hz, 2H), 3.80 (ddd, J = 3.8, 2.1, 2.1 Hz, 1H), 2.01 (p, J = 7.1 Hz, 2H), 1.85–1.77 (m, 2H), 1.58–1.50 (m, 2H), 1.48–1.40 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.4, 146.9, 139.7, 138.8, 134.1, 132.5, 132.3, 131.1, 131.1, 130.2, 127.8, 126.9, 126.4, 121.3, 120.1, 119.6, 119.4, 115.3, 112.9, 103.9, 68.1, 51.3, 50.5, 45.3, 30.3, 28.9, 26.3, 25.6 ppm. MS (ESP +ve): m/z = 525 ([M + H]+). Analysis calcd (%) for C33H28N6O (524.63): C 75.55, H 5.38, N 16.02; found: C 75.57, H 5.39, N 16.00.
1,1-Dicyano-2-(4-(1-(8-(4-cyanophenoxy)octyl)-1H-1,2,3-triazol-4-yl)phenyl)-1,8a-dihydroazulene (4c). To a degassed solution of 15 (66 mg, 0.235 mmol), 14c (87 mg, 0.318 mmol) and Hünig's base (10 drops) in toluene (4 mL) was added CuI (7 mg, 0.037 mmol) and the contents stirred for 2 days at rt. The solvent was removed under reduced pressure and the residue purified by flash column chromatography (10% EtOAc/CH2Cl2) to furnish 4c (65 mg, 50%) as a yellow solid. Rf = 0.75 (5% EtOAc/CH2Cl2). Mp = 148.6–152.4 °C. 1H NMR (500 MHz, CDCl3): δ = 7.93 (d, J = 8.5 Hz, 2H), 7.81 (s, 1H), 7.79 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 8.8 Hz, 2H), 6.93 (s, 1H), 6.91 (d, J = 8.8 Hz, 2H), 6.57 (dd, J = 11.3, 6.4 Hz, 1H), 6.48 (dd, J = 11.3, 6.1 Hz, 1H), 6.36 (d, J = 6.4 Hz, 1H), 6.31 (ddd, J = 10.2, 6.1, 2.0 Hz, 1H), 5.83 (dd, J = 10.2, 3.8 Hz, 1H), 4.42 (t, J = 7.1 Hz, 2H), 3.97 (t, J = 6.5 Hz, 2H), 3.80 (ddd, J = 3.8, 2.0, 2.0 Hz, 1H), 2.00–1.96 (m, 2H), 1.81–1.75 (m, 2H), 1.53–1.32 (m, 8H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.5, 146.8, 139.7, 138.78, 134.1, 132.5, 132.3, 131.1, 130.1, 127.8, 126.9, 126.4, 121.3, 120.1, 119.6, 119.4, 115.3, 115.3, 112.9, 103.8, 68.4, 51.2, 50.6, 45.2, 30.4, 29.2, 29.0, 29.0, 26.5, 25.9 ppm, 1C masked. HRMS (MALDI +ve) calcd for C35H33N6O ([M + H]+): m/z = 553.2710; exp 553.2716. Analysis calcd (%) for C35H32N6O (552.68): C 76.06, H 5.84, N 15.21; found: C 75.93, H 5.88, N 15.11.
1,1-Dicyano-2-(4-(1-(9-(4-cyanophenoxy)nonyl)-1H-1,2,3-triazol-4-yl)phenyl)-1,8a-dihydroazulene (4d). To a degassed solution of 15 (49 mg, 0.175 mmol), 14d (49 mg, 0.179 mmol) and Hünig's base (10 drops) in toluene (4 mL) was added CuI (7 mg, 0.037 mmol) and the contents stirred for 2 days at rt. The solvent was removed under reduced pressure and the residue was subjected to flash column chromatography (10% EtOAc/CH2Cl2) followed by crystallization to give pure 4d (49 mg, 49%) as a yellow solid. Rf = 0.77 (5% EtOAc/CH2Cl2). Mp = 131.9–148.4 °C. 1H NMR (500 MHz, CDCl3): δ = 7.94 (d, J = 8.4 Hz, 2H), 7.81–7.79 (m, 3H), 7.57 (d, J = 8.77 Hz, 2H), 6.93–6.91 (m, 3H), 6.58 (dd, J = 11.3, 6.3 Hz, 1H), 6.49 (dd, J = 11.3, 6.1 Hz, 1H), 6.36 (d, J = 6.3 Hz, 1H), 6.32 (ddd, J = 10.2, 6.1, 2.0 Hz, 1H), 5.83 (dd, J = 10.2, 3.8 Hz, 1H), 4.42 (t, J = 7.1 Hz, 2H), 3.98 (t, J = 6.5 Hz, 2H), 3.81 (ddd, J = 3.8, 2.0, 2.0 Hz, 1H), 1.99–1.96 (m, 2H), 1.78 (p, J = 6.5 Hz, 2H), 1.44–1.41 (m, 2H), 1.37–1.34 (m, 8H) ppm. 13C NMR (125 MHz, CDCl3): δ = 162.4, 146.7, 139.6, 138.7, 134.0, 132.3, 132.2, 131.0, 130.9, 130.0, 127.7, 126.8, 126.3, 121.1, 119.9, 119.5, 119.3, 115.2, 112.7, 103.7, 68.3, 51.1, 50.5, 45.1, 30.3, 29.3, 29.2, 29.0, 28.9, 26.5, 25.9 ppm. HRMS (MALDI +ve) calcd for C36H35N6O (M+): m/z = 566.2789; exp 566.2794. Analysis calcd (%) for C36H34N6O (566.71): C 76.30, H 6.05, N 14.83; found: C 75.92, H 5.70, N 14.71.
1,1-Dicyano-2-(4-(1-(10-(4-cyanophenoxy)deconyl)-1H-1,2,3-triazol-4-yl)phenyl)-1,8a-dihydroazulene (4e). To a degassed solution of 15 (73 mg, 0.260 mmol), 14e (112 mg, 0.373 mmol) and Hünig's base (10 drops) in toluene (4 mL) was added CuI (15 mg, 0.079 mmol) and the contents stirred for 2 days at rt. The solvent was removed under reduced pressure and the residue was subjected to flash column chromatography (10% EtOAc/CH2Cl2) followed by crystallization to give pure 4e (73 mg, 48%) as a yellow solid. Rf = 0.80 (5% EtOAc/CH2Cl2). Mp = 111.1–115.1 °C. 1H NMR (500 MHz, CDCl3): δ = 7.94 (d, J = 8.4 Hz, 2H), 7.81–7.79 (m, 3H), 7.56 (d, J = 8.9 Hz, 1H), 6.93–6.91 (m, 3H), 6.58 (dd, J = 11.2, 6.4 Hz, 1H), 6.49 (dd, J = 11.2, 6.0 Hz, 1H), 6.36 (d, J = 6.4 Hz, 1H), 6.32 (ddd, J = 10.4, 6.1, 1.8 Hz, 1H), 5.83 (dd, J = 10.4, 3.7 Hz, 1H), 4.42 (t, J = 7.0 Hz, 2H), 3.98 (t, J = 6.4 Hz, 2H), 3.81 (ddd, J = 3.7, 1.8, 1.8 Hz, 1H), 2.16–1.89 (m, 2H), 1.81–1.75 (m, 2H), 1.49–1.16 (m, 12H) ppm. 13C NMR (125 MHz, CDCl3): δ = 146.8, 139.8, 138.8, 134.1, 132.5, 132.4, 131.1, 131.1, 130.1, 127.9, 126.9, 126.4, 121.3, 120.1, 119.6, 119.5, 115.3, 115.3, 112.9, 103.8, 68.5, 51.3, 50.7, 45.3, 30.5, 29.5, 29.4, 29.4, 29.1, 29.1, 26.6, 26.1 ppm. HRMS (MALDI +ve) calcd for C37H36N6O (M+): m/z = 580.2945; exp 580.2951.
1,1-Dicyano-2-(4-octyl-(1H-1,2,3-triazol-4-yl)phenyl)-1,8a-dihydroazulene (4j). To a degassed solution of 16 (98 mg, 0.350 mmol), octylazide (106 mg, 0.683 mmol) and Hünig's base (10 drops) in toluene (4 mL) was added CuI (11 mg, 0.058 mmol) and the contents stirred for 2 days at rt. The solvent was removed under reduced pressure and the residue was subjected to flash column chromatography (10% EtOAc/CH2Cl2) followed by crystallization to give pure 4j (147 mg, 97%) as a yellow solid. Rf = 0.84 (5% EtOAc/CH2Cl2). Mp = 126.1–130.6 °C. 1H NMR (500 MHz, CDCl3): δ = 7.94 (d, J = 8.4 Hz, 2H), 7.81 (s, 1H), 7.79 (d, J = 8.4 Hz, 2H), 6.93 (s, 1H), 6.57 (dd, J = 11.2, 6.3 Hz, 1H), 6.48 (dd, J = 11.2, 6.1 Hz, 1H), 6.35 (d, J = 6.3 Hz, 1H), 6.34–6.28 (m, 1H), 5.83 (dd, J = 10.2, 3.6 Hz, 1H), 4.41 (t, J = 7.2 Hz, 2H), 3.80 (ddd, J = 3.7, 1.9, 1.9 Hz, 1H), 1.99–1.91 (m, 2H), 1.97–1.95 (m, 2H), 1.42–1.18 (m, 8H), 0.87 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 146.6, 139.7, 138.7, 132.3, 130.9, 130.9, 129.9, 127.7, 126.7, 126.3, 121.1, 120.0, 119.5, 115.2, 112.7, 51.1, 50.6, 45.1, 31.7, 30.4, 29.1, 29.0, 26.5, 22.6, 14.1 ppm, 1C masked. HRMS (MALDI +ve) calcd for C28H30N5 ([M + H]+): m/z = 436.2496; exp 436.2495. Analysis calcd (%) for C28H29N5 (435.58): C 77.21, H 6.71, N 16.08; found: C 77.15, H 6.80, N 15.94.
2-(2-(Cyclohepta-2,4,6-trien-1-ylidene)-1-(4′-(octyloxy)-[1,1′-biphenyl]-4-yl)ethylidene)malononitrile (17). To a stirred solution of 3f (418 mg, 0.908 mmol) in CH2Cl2 (50 mL) at rt under an argon atmosphere, was added AlCl3 (341 mg, 2.56 mmol). After continued stirring for approximately 20 min, TLC analysis indicated the presence of unreacted 3f and more AlCl3 (183 mg, 1.37 mmol) was added to the vessel and the reaction allowed to stir a further 20 min. The mixture was cooled on an ice bath and quenched by addition of ice-water (100 mL) and the phases separated. The aqueous phase was extracted with CH2Cl2 (2 × 50 mL) and the combined organic phases dried over MgSO4 and filtered. The solution was diluted with heptane (100 mL) and the CH2Cl2 was removed by rotary evaporation while being kept at 0 °C. The solution was placed in a freezer resulting in the crystallization of 17, which was isolated as a dark red solid (368 mg, 88%). This material was stored in the freezer to prevent ring-closure back to 3f. Mp = 105–109 °C. 1H NMR (500 MHz, CDCl3): δ = 7.35 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 8.7 Hz, 2H), 7.10 (d, J = 8.2 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 6.12 (s, 1H), 5.80 (dd, J = 11.4, 1.5, 1H), 5.65 (dd, J = 12.2, 2.1 Hz, 1H), 5.59–5.49 (m, 2H), 5.46–5.42 (m, 1H), 5.10 (dd, J = 12.2, 7.8 Hz, 1H), 3.69 (t, J = 6.5 Hz, 2H), 1.68–1.63 (m, 2H), 1.39–1.34 (m, 2H), 1.33–1.18 (m, 8H), 0.92 (t, J = 7.0 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 167.8, 160.1, 152.9, 143.7, 142.2, 135.2, 134.7, 134.2, 133.3, 133.2, 132.7, 132.1, 129.3, 128.6, 128.4, 127.6, 119.8, 115.3, 78.4, 68.1, 32.2, 29.8, 29.7, 29.7, 26.4, 23.1, 14.4 ppm, 1C masked. HRMS (MALDI +ve) calcd for C32H32N2ONa [(M + Na)+]: m/z = 483.2407, found m/z = 483.2412. Analysis calcd (%) for C32H32N2O (496.62): C 83.49, H 7.00, N 6.08; found: C 83.38, H 7.11, N 6.14.
2-(2-(Cyclohepta-2,4,6-trien-1-ylidene)-1-(2′,3′-difluoro-4′-hydroxy-[1,1′-biphenyl]-4-yl)ethylidene)malononitrile (18). To a stirred solution of 3g (210 mg, 0.423 mmol) in CH2Cl2 (50 mL) at rt, under an argon atmosphere, was added AlCl3 (1.010 g, 7.58 mmol) and the mixture was stirred for 20 min. TLC analysis indicated the presence of unreacted 3g and more AlCl3 (540 mg, 4.05 mmol) was added, whereby the reaction was allowed to stir a further 20 min. The mixture was cooled on an ice bath and quenched by addition of ice-water (100 mL) after which the phases were separated. The aqueous phase was extracted with CH2Cl2 (2 × 50 mL) and the combined organic phases was dried with MgSO4 and filtered. Heptane was added (100 mL), and the solution concentrated under reduced pressure at 0 °C, whereby the solution was placed at −18 °C for 16 h, resulting in the crystallization of 18 (85 mg, 52%) as a dark red solid. This material was stored in the freezer to prevent ring-closure back to 3g. Mp = 129.7–131.0 (color change to yellow), 168 (decomposes) °C. 1H NMR (500 MHz, CDCl3): δ = 7.61 (d, J = 7.9 Hz, 2H), 7.47 (d, J = 7.9 Hz, 2H), 7.13 (td, J = 8.3, 2.2 Hz, 1H), 6.90–6.84 (m, 1H), 6.75–6.69 (m, 1H), 6.45–6.37 (m, 2H), 6.35 (s, 1H), 6.33–6.27 (m, 1H), 5.94 (d, J = 4.6 Hz, 1H), 5.60 (br s, 1H, exchanges D2O) ppm. 13C NMR (125 MHz, CDCl3): δ = ppm 13C NMR (125 MHz, CDCl3): δ = 168.4, 154.1, 148.4 (dd, J = 250.4, 11.4 Hz), 145.0 (dd, J = 11.3, 2.0 Hz), 142.3, 137.7 (dd, J = 2.5, 1.6 Hz), 135.6, 135.3, 135.2, 134.5, 134.1, 133.7, 129.9 (d, J = 3.3 Hz), 128.6, 124.3 (dd, J = 3.8, 3.8 Hz), 121.4 (d, J = 9.8 Hz), 118.9, 115.1, 114.6, 112.6 (dd, J = 3.6, 1.0 Hz) ppm, 2Cs masked. HRMS (MALDI +ve) calcd for C32H31F2N2O [(M + H)+]: m/z = 358.1147, found m/z = 385.1142.
Acknowledgements
The Danish Council for Independent Research | Technology and Production Sciences (#12-126668) and University of Copenhagen are acknowledged for financial support. Dr Theis Brock-Nannestad, University of Copenhagen, is thanked for helpful discussions.Notes and references
- Y. Li, A. Urbas and Q. Li, J. Am. Chem. Soc., 2012, 134, 9573 CrossRef CAS PubMed.
- T. Ikeda and O. Tsutsumi, Science, 1995, 268, 1873 CAS.
- R. H. Berg, S. Hvilsted and P. S. Ramanujam, Nature, 1996, 383, 505 CrossRef CAS.
- A. Y. Bobrovsky, N. I. Boiko, V. P. Shibaev, M. A. Kalik and M. Krayushkin, J. Mater. Chem., 2001, 11, 2004 RSC.
- T. J. Bunning, R. L. Crane and W. W. Adams, Adv. Mater. Opt. Electron., 1992, 1, 293 CrossRef.
- S. Z. Janicki and G. B. Schuster, J. Am. Chem. Soc., 1995, 117, 8524 CrossRef CAS.
- J. Daub, T. Knöchel and A. Mannschreck, Angew. Chem., Int. Ed. Engl., 1984, 23, 960 CrossRef.
- T. Li, M. Jevric, J. R. Hauptmann, R. Hviid, Z. Wei, R. Wang, N. E. Reeler, E. Thyrhaug, S. Petersen, J. A. S. Meyer, N. Bovet, T. Vosch, J. Nygård, X. Qiu, W. Hu, Y. Liu, G. C. Solomon, H. G. Kjaergaard, T. Bjørnholm, M. B. Nielsen, B. W. Laursen and K. Nørgaard, Adv. Mater., 2013, 25, 4164 CrossRef CAS PubMed.
- S. L. Broman and M. B. Nielsen, Phys. Chem. Chem. Phys., 2014, 16, 21172 RSC.
- D. Demus, J. Goodby, G. W. Gray, H.-W. Spies and V. Vill, Handbook of liquid crystals, Fundamentals, Wiley-VCH, New York, 1998, ch 6 Search PubMed.
- See for example: I. M. Saez and J. W. Goodby, J. Mater. Chem., 2003, 13, 2727 RSC; A. R. E. Brás, S. Henriques, T. Casimoro, A. Aguiar-Ricardo, J. Sotomayor, J. Caldeira, C. Santos and M. Dionísio, Liq. Cryst., 2007, 34, 591 CrossRef CAS; I. M. Saez and J. W. Goodby, Liq. Cryst., 2010, 26, 1101 CrossRef; R. J. Mandle, E. J. Davis, C.-C. A. Voll, D. J. Lewis, S. J. Cowling and J. W. Goodby, J. Mater. Chem. C, 2015, 3, 2380 RSC.
- See for example: H. Sorkin, Mol. Cryst. Liq. Cryst., 1980, 56, 279 CrossRef CAS; M. Hara, Y. Iwakabe, K. Tochigi, H. Sasabe, A. F. Garito and A. Yamada, Nature, 1990, 344, 228 CrossRef; H. Sugisawa, H. Toriumi and H. Watanabe, Mol. Cryst. Liq. Cryst., 1992, 214, 11 CrossRef.
- A. S. Achalkumar, D. S. Shankar Rao and C. V. Yelamaggad, New J. Chem., 2014, 38, 4235 RSC.
- S. M. Kelly, Helv. Chim. Acta, 1989, 72, 594 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS; H. C. Kolb, M. G. Finn and B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef.
- R. Cristiano, D. M. P. de Oliveira Santos, G. Conte and H. Gallardo, Liq. Cryst., 2006, 33, 997 CrossRef CAS.
- H. S. Lissau, S. L. Broman, M. Jevric, A. Ø. Madsen and M. B. Nielsen, Aust. J. Chem., 2014, 67, 531 CrossRef CAS.
- C. R. Parker, C. G. Tortzen, S. L. Broman, M. Schau-Magnussen, K. Kilså and M. B. Nielsen, Chem. Commun., 2011, 47, 6102 RSC.
- S. L. Broman, M. Jevric and M. B. Nielsen, Chem.–Eur. J., 2013, 19, 9542 CrossRef CAS PubMed.
- S. Diele, S. Tosch, S. Mahnke and D. Demus, Cryst. Res. Technol., 1991, 26, 809 CrossRef CAS.
- Z. Puterová, J. Romiszewski, J. Mieczkowski and E. Gorecka, Tetrahedron, 2012, 68, 8172 CrossRef.
- For a review on photoalignment techniques, see: T. Seki, S. Nagano and M. Hara, Polymer, 2013, 54, 6053 CrossRef CAS.
- L. Gobbi, P. Seiler, F. Diederich, V. Gramlich, C. Boudon, J.-P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 2001, 84, 743 CrossRef CAS.
- M. Santella, V. Mazzanti, M. Jevric, C. R. Parker, S. L. Broman, A. D. Bond and M. B. Nielsen, J. Org. Chem., 2012, 77, 8922 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18649h |
| This journal is © The Royal Society of Chemistry 2015 |