
Dynamic layer-by-layer films linked with Schiff base bond for sustained drug release
Lin Zhouab,
Mao Chenab,
Ying Guana and
Yongjun Zhang*a
aState Key Laboratory of Medicinal Chemical Biology and Key Laboratory of Functional Polymer Materials, Institute of Polymer Chemistry, College of Chemistry, Nankai University, and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China. E-mail: yongjunzhang@nankai.edu.cn
bInstitute of Chemical Materials, China Academy of Engineering Physics, Mianyang 621999, Sichuan, China
First published on 25th September 2015
Abstract
Dynamic layer-by-layer films were fabricated from a dextran–doxorubicin conjugate, PO-Dex–DOX, and glycol chitosan (GC), via the in situ Schiff base reaction between the aldehyde groups on PO-Dex–DOX and the amino groups on GC. Because of the reversible, dynamic nature of Schiff base bonds in the films, the films disintegrate gradually when soaked in aqueous solutions, and thus release DOX into the media. The release mechanism is different from ordinary drug carriers, in which the drug is usually released via diffusion or polymer degradation. The drug release rate decreases with increasing molecular weight of dextran. More importantly, because of the stimuli-responsivity of Schiff base bonds, the drug release rate can be tuned via external stimuli. Faster hydrolysis of Schiff base bonds at a lower pH and/or a higher temperature results in accelerated film disintegration and thus faster drug release. In vitro cytotoxicity test suggests that the released DOX retains its antitumor activity.
Introduction
Controlled drug release systems offer advantages such as improved efficacy, reduced toxicity, and improved patient compliance and convenience.1 In recent years, a lot of effort has been made to construct drug carriers using layer-by-layer (LbL) assembly,2,3 a simple method for thin film fabrication in which two polymers with complementary functional groups are deposited alternately. In some efforts hollow microcapsules were fabricated via LbL assembly and the therapeutics were encapsulated inside. As the permeability of the capsule wall can be tuned by the bilayer number of LbL coatings, and/or external stimuli, such as pH and temperature, the release rate of the therapeutics can be controlled.4 In a second approach, the drugs were incorporated into macroscopic planar LbL films and released either via diffusion5,6 or the degradation of the LbL films.7–10 Particularly Hammond et al.7–10 fabricated drug-loaded LbL films using hydrolytically degradable polymers, for example, poly(β-amino ester)s, as one of the film components. With the degradation of the degradable polymer, the films disintegrate, and thus release the drugs from the films. Because LbL films can be coated onto substrates with complex morphologies, this method is highly desirable for the localized delivery of bioactive compounds from surfaces of biomedical devices, such as intravascular stents.2Recently we proposed that drug release can also be achieved via the gradual disintegration of dynamic LbL films.11 Dynamic LbL films are films fabricated using dynamic bonds, i.e., interactions that can undergo reversible breaking and reformation, usually under equilibrium conditions,12 as driving forces.13 Like “dynamers” or dynamic polymers containing dynamic bonds,14,15 dynamic LbL films also exhibit a range of dynamic properties.13 Specially dynamic LbL films were found to undergo a gradual disintegration when soaked in aqueous solutions.16–19 When drugs are incorporated in the films, sustained drug release can be achieved as a result of the film disintegration.11,20,21 It is noteworthy that this strategy is different from the one Hammond et al. developed.7,8 Although in both strategies the drug is released as a result of film disintegration, in Hammond et al.'s strategy the films disintegrate via the degradation of the polymer, while the dynamic LbL films disintegrate because of the breakage of the dynamic bonds linking the two polymers.
In our previous works, dynamic LbL films were fabricated using hydrogen bonds16,20 or phenylboronate ester bonds19,21 as driving forces. Schiff base bond is another type of dynamic bond which forms under mild conditions with a high reaction rate.22 Because of its dynamic nature, Schiff base bond has been used to construct various dynamic nanoarchitectures23 and hydrogels.24–27 Here we show that dynamic LbL films based on Schiff base bonds can be used for sustained drug release. The films were fabricated from dextran and chitosan, both of which are of natural origin and widely used in biomedical areas. Doxorubicin (DOX), a representative anthracycline antibiotic and one of the most widely used anticancer drugs, was used as a model drug. We show that sustained DOX release could be achieved via the gradual disintegration of the dynamic film. The released drug can be internalized by cells and remains its anti-cancer activity. More importantly, the drug release rate can be tuned by pH and temperature, thanks to the sensitivity of the Schiff base bonds to these external stimuli. It is noteworthy that Schiff base bonds have been previously used to construct various drug carriers, such as injectable hydrogels.25,27–29
Experimental section
Materials
Dextran (Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 40000 and 70000) was purchased from Aladdin. Glycol chitosan (GC) (deacetylation degree: >60%) was purchased from Sigma-Aldrich. NaIO4 and 3-aminopropyl-triethoxysilane (APTES) were purchased from Alfa Aesar. Doxorubicin hydrochloride (DOX·HCl) was supplied by Huafeng Lianbo Technology Co. (Beijing, China). These reagents were used without further purification unless otherwise noted.
000, 40000 and 70000) was purchased from Aladdin. Glycol chitosan (GC) (deacetylation degree: >60%) was purchased from Sigma-Aldrich. NaIO4 and 3-aminopropyl-triethoxysilane (APTES) were purchased from Alfa Aesar. Doxorubicin hydrochloride (DOX·HCl) was supplied by Huafeng Lianbo Technology Co. (Beijing, China). These reagents were used without further purification unless otherwise noted.
Preparation of partially oxidized dextran (PO-Dex)
PO-Dex with a theoretic oxidation degree of 30% was prepared according to a previously reported method.30,31 Briefly, to a beaker wrapped with aluminum foil dextran solution and NaIO4 solution were charged. The final concentration was 10 mg mL−1 for dextran and the molar ratio of NaIO4 to anhydroglucopyranoside subunit in dextran is 0.6. The reaction mixture was stirred for 24 h in the dark at 30 °C. After dialysis against double-distilled water for three days (cutoff: 8000–15000), the solution was freeze-dried to yield a white product. Oxidation degree of the product was determined to be ∼28.9% from the pH change of the reaction mixture, which is close to the theoretic one.30,31
Synthesis of dextran–doxorubicin conjugate PO-Dex–DOX
DOX was conjugated onto PO-Dex via Schiff base reaction between the amino group on DOX and the aldehyde group on PO-Dex according to a previously reported method.32,33 Briefly, 0.5 g of PO-Dex was dissolved in 50 mL of water, to which 25.2 mg of DOX·HCl was added (the molar ratio of aldehyde to amino group is 40:1). After being stirred overnight in the dark at room temperature, 17.6 mg of NaBH4 (10 equimolar amount of DOX) was added to the reaction mixture to reduce the formed Schiff base. After incubated for 2 hours, the reaction mixture was dialyzed against water for 3 days with frequent water changing. The final product was obtained by lyophilization as red powder. DOX content in the conjugate was determined to be 4.2 wt% by measuring the absorbance at 490 nm of the PO-Dex–Dox solution with known concentration, with the help of a standard curve plotted using DOX·HCl.
The residual aldehyde content in PO-Dex–DOX was determined by hydroxylamine hydrochloride titration.34 Briefly, 0.10 g of PO-Dex–DOX was dissolved in 25 mL of 0.25 M solution of hydroxylamine hydrochloride. The mixture was stirred at room temperature for 3.5 h and then titrated with 0.05 M standardized NaOH solution. The change of pH with volume of added NaOH solution was recorded. The equivalent volume was determined by first derivative of the titration curve.
Fabrication of LbL films
Quartz slides with a size of 44 mm × 10 mm × 1 mm were used as substrates for the fabrication of the LbL films. The substrates were first cleaned in boiling piranha solution (3:7 v/v H2O2–H2SO4 mixture) (caution: this solution is extremely corrosive!), rinsed with deionized (DI) water thoroughly and dried. To introduce amino groups, the substrates were immersed in a 1.0 wt% toluene solution of APTES for 8 h at 25 °C, washed with toluene and dried at about 100 °C in an oven. To fabricate the LbL films, the amino-modified slides were dipped alternately into a 0.2% solution of PO-Dex or PO-Dex–DOX and a 0.1% solution of GC, each for 30 min, washed in DI water in between for 5 min. The temperature of the solutions was remained at 20 °C using a refrigerated circulator. The assembly cycle was repeated until the desired numbers of bilayer were reached.
In vitro DOX release
To measure the release kinetics of DOX, a PO-Dex–DOX/GC film (60 bilayer, unless otherwise specified) was immersed in 6 mL of release media (20 mM pH 7.4 phosphate buffer, unless otherwise specified). The temperature was controlled with a refrigerated circulator. At appropriate intervals, the release media were removed, and the same volume of fresh media with the same temperature was added. Concentration of DOX in the release media was determined using fluorescence spectroscopy. To measure the total amount of doxorubicin in the films, the films were completely disintegrated by immersion in 0.1 M HCl solution. The experiments were carried out in triplicate. The results were reported as mean ± standard deviation.Cytotoxicity and cellular uptake
Cytotoxicity of the polymers was evaluated using MTT assay. Briefly, HepG2 cells were seeded on 96-well plates at a density of 1.0 × 104 cells per well. The cells were cultured in a media containing 90% Dulbecco's modified Eagle medium (DMEM, GIBCO), 10% heat-inactivated fetal calf serum and 100 units per mL of penicillin/streptomycin. The cultures were maintained in an incubator at 37 °C with a humidified atmosphere of 5% CO2. After 24 h culture, the media were changed with media containing various concentrations of the polymers (GC, PO-Dex, or PO-Dex–DOX). After cultured for 48 h, the cell viability was measured using MTT assay (Sigma-Aldrich). For this purpose, 20.0 μL of MTT (5.0 mg mL−1) was added into each well and the cells were incubated for 4 h at 37 °C. After washing with PBS three times, 150.0 μL of DMSO was added to each well to dissolve the resultant formazan crystals. OD values at 492 nm were then measured with a microplate reader (Infinite F50, TECAN).The cytotoxicity of the materials released from the LbL films was evaluated in the same way. For this purpose, a 300 bilayer PO-Dex–DOX/GC or PO-Dex/GC film was first sterilized by soaking in 70% ethanol. It was then soaked in fresh culture media for 10 days to obtain media containing released materials.
Cellular uptake of DOX was examined employing a confocal laser scanning microscopy (CLSM). In this case, the cells were seeded on 24-well plate with coverslips at a density of 1.0 × 105 cells per well. After 24 h culture, the media was changed with media containing released materials. After 48 h incubation, the cells were washed with PBS three times and fixed by 4% paraformaldehyde in PBS at 37 °C for 10 min. They were then permeabilized in 0.1% Triton X-100 for 5 min and washed again with PBS. The samples were then treated with 1% bovine serum albumin at 37 °C for 30 min. Finally, they were incubated in PBS containing 5 μg mL−1 isothiocyanate (FITC)-phalloidin and 0.2 mg mL−1 4′,6-diamidino-2-phenylindole (DAPI) for 10 min. The samples were washed with PBS and imaged using a confocal microscope (Leica TSC SP8).
Other characterizations
UV-vis absorption spectra were measured on a TU 1810 PC UV-vis spectrophotometer (Purkinje General, China). Fluorescence spectra were measured on a Shimadzu RF-5301 PC spectrofluorometer using an excitation wavelength of 495 nm and a bandwidth of 5 nm. Fourier transform infrared (FTIR) spectra were measured on a Bio-Rad FTS-6000 spectrometer operating at 8 cm−1 resolution. 1H NMR spectra were recorded on a Varian UNITY-plus 400 NMR spectrometer using DMSO-d6 as solvent.Results and discussion
Synthesis of PO-Dex–DOX conjugate
The dynamic LbL films were fabricated from dextran bearing aldehyde groups and chitosan bearing amino groups, using in situ formed Schiff base bonds as driving force. Glycol chitosan was chosen because it is soluble in neutral aqueous solutions. To introduce aldehyde groups dextran was first partially oxidized. Then the model drug doxorubicin (DOX) was conjugated onto the polymer.As shown in Scheme 1, partially oxidized dextran (PO-Dex) was synthesized by NaIO4 oxidation.30,31,35,36 According to previous studies, the extent of oxidation of dextran (percentage of anhydroglucopyranoside (Glc) units oxidized) depended on the molar ratio of NaIO4 to Glc units and the reaction time.35 Here a PO-Dex, with theoretic oxidation degree of 30%, was synthesized. The real oxidation degree was determined to be ∼28.9% from the pH change of the reaction mixture, which is close to the theoretic one.31 DOX was conjugated onto PO-Dex according to a previously reported method.32,33 For this purpose, DOX was first linked with PO-Dex via the formation of Schiff base bond between the aldehyde group on PO-Dex and the amino group on DOX. The linkage between PO-Dex and DOX was then fixed by NaBH4 reduction.
 | ||
| Scheme 1 Synthesis of partially oxidized dextran, PO-Dex, and PO-Dex–DOX conjugate. | ||
Fig. 1A compares the absorption spectra of PO-Dex, DOX, and the PO-Dex–DOX conjugate. The appearance of the absorption band centered at 490 nm in the spectra of PO-Dex–DOX confirms the successful introduction of DOX moiety onto dextran chain. With the help of a standard curve of DOX, DOX content in the conjugate was determined to be ∼4.2 wt%, corresponding to 1 mole of DOX per 76 mole of glucosidic residues. Conjugates with higher DOX contents were also synthesized. Unfortunately they exhibit a poor water-solubility possibly because of the hydrophobicity of the DOX moieties. Therefore the conjugate with a relatively low DOX content was chosen for the following studies. The FTIR spectra of PO-Dex, DOX, and the PO-Dex–DOX conjugate were collected in Fig. 1B. Compared to PO-Dex, the PO-Dex–DOX conjugate presents some new bands, which can all be found in the spectra of DOX, indicating again the successful conjugation of DOX onto PO-Dex. Fig. 1C compares the 1H NMR spectra of PO-Dex and PO-Dex–DOX. The new peaks appeared at 7.68 and 7.92 ppm in the spectra of PO-Dex–DOX are assigned to the aromatic protons in DOX,37 confirming again the successful conjugation of DOX onto dextran. The peaks at 13.8 and 14.3 ppm are assigned to the exchangeable phenolic hydroxyl protons in DOX. According to Song et al.,38 the appearance of the two peaks suggests DOX is chemically bonded with the polymer. The peak at 9.64 ppm in the spectra of PO-Dex is assigned to the proton in aldehyde groups.39 This peak shifts to 9.29 ppm after DOX conjugation, probably because of change in its environment.40,41 Meanwhile the intensity of the peak decreases significantly, suggesting a decreased amount of aldehyde groups after DOX conjugation.
 | ||
| Fig. 1 (A and B) UV-vis (A) and FTIR (B) spectra of PO-Dex, DOX and PO-Dex–DOX. (C) 1H NMR spectra of PO-Dex and PO-Dex–DOX. Solvent: DMSO-d6. | ||
Fabrication of PO-Dex–DOX/GC films
From the NMR spectra of PO-Dex–DOX, there is still a small amount of aldehyde groups left after DOX conjugation (Fig. 1C). Because the aldehyde groups usually exist as hemiacetals and/or internal hemiacetals,31,42 the exact content of aldehyde groups cannot be determined from the NMR spectra. Therefore we turned to hydroxylamine hydrochloride titration, an accurate method proposed by Zhao and Heindel.34 Using this method, the degree of aldehyde substitution of the PO-Dex–DOX conjugate was determined to be ∼22%. Note the degree of aldehyde substitution of the parent PO-Dex is ∼60% (each oxidized Glc unit in PO-Dex bears two aldehyde groups). The significantly decreased aldehyde content in PO-Dex–DOX conjugate suggests NaBH4 reduces not only the Schiff base bonds but also some of the free aldehyde groups.The residual aldehyde groups in PO-Dex–DOX conjugate allow the fabrication of LbL films from it and GC via the in situ Schiff base reaction between the aldehyde groups on PO-Dex–DOX and the amino groups on GC (Scheme 2). For this purpose, amino-modified quartz slides were soaked in PO-Dex–DOX and GC solutions alternately. The fabrication process was monitored by UV-vis absorption spectrometry. As shown in Fig. 2, the resulting films present the characteristic absorption band of DOX centered at ∼490 nm, indicating the successful incorporation of the PO-Dex–DOX conjugate in the film. Like many other LbL films, the PO-Dex–DOX/GC film grows linearly as indicated by a linear relationship between the film absorbance and bilayer numbers (inset of Fig. 2).
 | ||
| Scheme 2 Schiff base reaction between PO-Dex–DOX and GC. | ||
 | ||
| Fig. 2 UV-vis absorption spectra of PO-Dex–DOX/GC films with various bilayer numbers (2–20). Inset: plot of absorbance at 495 nm against bilayer number. | ||
In vitro DOX release from the LbL films
Since PO-Dex–DOX and GC are linked with reversible Schiff base bonds, the PO-Dex–DOX/GC film disassembles gradually when soaked in aqueous solutions. As a result, free PO-Dex–DOX chains are released into the solutions (Scheme 3). The accumulation of PO-Dex–DOX chains in the media results in the gradual increase in the fluorescence intensity of the media, as shown in Fig. 3A. | ||
| Scheme 3 Release of DOX via the disintegration of PO-Dex–DOX/GC film. | ||
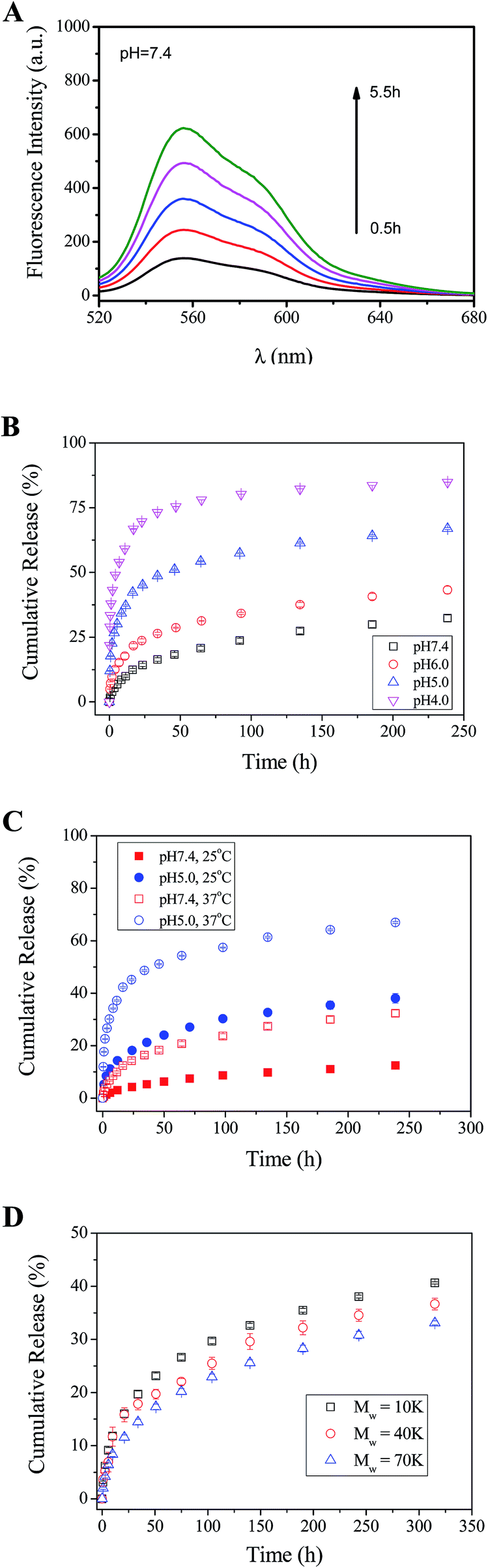 | ||
| Fig. 3 (A) Fluorescence spectra of a phosphate buffer in which a PO-Dex–DOX/GC film was soaked. The excitation wavelength is 495 nm. pH = 7.4, T = 37 °C. (B) Release profile of DOX from PO-Dex–DOX/GC films soaked in 20 mM phosphate buffers of various pHs. T = 37 °C. (C) Release profile of DOX from PO-Dex–DOX/GC films soaked in 20 mM phosphate buffers. pH = 5.0 or 7.4. T = 25 °C or 37 °C. (D) Release of DOX from PO-Dex–DOX/GC films in 20 mM pH 7.4 phosphate buffer. T = 37 °C. The films were fabricated from dextran with various molecular weights as indicated. | ||
To study the effect of pH on DOX release, the PO-Dex–DOX/GC films were soaked in phosphate buffers with various pHs. As shown in Fig. 3B, the release of DOX becomes faster when soaked in a lower pH medium. For example, the film releases about 71% of DOX after a 24 h immersion in pH 4.0 buffer, while only 14% of DOX is released in pH 7.4 buffer. The accelerated DOX release at a lower pH could be attributed to the increased protonation of the free amino groups on GC (pKa of amino groups on chitosan is ∼6.3 (ref. 43)). Enhanced protonation of the amino groups results in more positively charged ammonium groups and therefore enhanced electrostatic repulsion among these charged chains. Accordingly the film disintegrates at a faster rate. On the other hand, the pH-controlled DOX release could also be explained by the faster breakage of Schiff base bond in the film. As mentioned above, the film disintegration (and DOX release) is actually a direct result of this reaction, which can be written as follows:
R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–R′ + H2O ⇌ R–NH2 + R′–CHO CH–R′ + H2O ⇌ R–NH2 + R′–CHO
| (1) |
It is well-known that Schiff base bonds hydrolyze at a faster rate at a lower pH.25,28,29,44–47 They are relatively stable in alkaline solution, but broken in acidic solutions.22 Therefore they are widely used to design pH-responsive materials.44–46 It is very likely that the Schiff base bonds between PO-Dex–DOX and GC also becomes less stable in a medium with a lower pH. Therefore the film disassembly becomes faster and DOX release becomes faster accordingly. The accelerated drug release under acidic conditions is a desirable feature for a drug carrier, because tumor and inflammatory tissues and the endosomes and lysosomes of cells have a more acidic environment than normal tissues.
To study the effect of temperature on the release of DOX, the release profiles of DOX from the PO-Dex–DOX/GC films were measured at two temperatures, i.e., 25 and 37 °C (Fig. 3C). In pH 7.4 media, when temperature increases from 25 °C to 37 °C, the release amount of DOX in the first 24 h increases from ∼4.3% to ∼14.3%. A similar accelerated DOX release was observed in pH 5.0 media. The release amount of DOX in the first 24 h increases from ∼18.2% to ∼45.1% when temperature increases from 25 °C to 37 °C. The faster release of DOX at a higher temperature may be explained by a faster hydrolyzation of the Schiff base bonds between PO-Dex–DOX and GC. Recently Wang et al.29 coupled 6-hydroxy-chromone-3-carbaldehyde as a model drug onto Fe3O4 nanoparticles via Schiff base bond. Similarly they found the drug releases faster at 37 °C than at 25 °C and attributed it to a faster hydrolyzation of Schiff base bond at higher temperature.
In the above studies, PO-Dex–DOX was synthesized using dextran with a Mw of 40 K. PO-Dex–DOX conjugates were also synthesized using dextran with different molecular weights. Similarly LbL films were fabricated and the release profiles of DOX were measured. As shown in Fig. 3D, with the decrease of molecular weight of dextran, DOX release becomes faster. The result suggests that the DOX release rate could be tuned not only via external stimuli, including pH and temperature, but also via changing the molecular weight of the polymer. It was understandable that a polymer chain with a higher molecular weight could provide more binding sites to interact with other polymer chains, and the intermolecular interaction among the polymer chains increases with increasing molecular weight. Therefore the film disintegration rate decreases with increasing molecular weight of the polymer. Previously we showed that the erosion rate of the hydrogen-bonded poly(acrylic acid)/poly(vinyl pyrrolidone) films in water decreases with increasing molecular weight.16 Jang et al.48 also reported that the time for complete disintegration of LPEI/PMAA (linear poly (ethylenimine)/poly(methacrylic acid)) LbL film in pH 2 solution dramatically increases from 2 min to 5 days when Mw of PMAA increases from 15 K to 226 K.
In vitro cytotoxicity
We then studied if the drugs released from the films remain their activities. Before the test, the cytotoxicity of the polymers involved in the film fabrication was first evaluated. For this purpose, they were added to cell culture media at various concentrations. HepG2 liver cancer cells were cultured in the media and the viability of the cells was assessed using the MTT assay. The results were normalized to that of the control group, in which the cells were cultured in the absence of the polymers. As shown in Fig. 4, the cell viability maintains at ∼100% for all the GC concentrations tested, indicating GC is non-toxic to the cells. The result is not surprising because it is well-known that chitosan is highly biocompatible.49,50 In contrast, PO-Dex becomes toxic against the cells when its concentration exceeds 100 μg mL−1. The toxic effect of low molecular weight aldehydes, such as formaldehyde and glutaraldehyde, is well-known. As a high molecular weight aldehyde, PO-Dex was previously proved to be toxic against the RAW 264.7 cell line.51 Similar to low molecular weight aldehydes, aldehyde groups are responsible for the cytotoxicity of PO-Dex. IC50 of PO-Dex against the RAW 264.7 cell line was reported to be 3 μmol mL−1 in terms of aldehyde concentration.51 Here the viability of HepG2 cells reduced to be ∼50% when PO-Dex concentration increases to be 1000 μg mL−1. Aldehyde concentration in the media (3.75 μmol mL−1) is close to the reported IC50, despite that different cell lines were used for the test. As expected, addition of PO-Dex–DOX into the media reduces the cell viability when its concentration exceeds a critical value. At the same concentration, PO-Dex–DOX is more toxic than PO-Dex, especially at a high concentration. The cytotoxicity of the conjugate could be attributed to both aldehyde groups and the DOX moieties coupled on the chains. Previous study shows that DOX retains its antitumor activity when conjugated onto dextran.32 Considering a much lower aldehyde content in the conjugate, the cytotoxicity of PO-Dex–DOX may be mainly attributed to the DOX moieties. | ||
| Fig. 4 Percentage viability of Hep G2 cells after incubation with GC, PO-Dex, or PO-Dex–DOX at different concentrations for 48 h. The superscript letters indicate significant difference (P < 0.05): acompared to GC value; bcompared to PO-Dex value. | ||
Next we test if DOX released from the films remains its antitumor activity. For this purpose, a 300 bilayer PO-Dex–DOX/GC film was soaked in cell culture media for 10 days to allow the film materials to be released into the media. From the change in UV-vis absorption, DOX content in the media was roughly determined to be ∼10 μg mL−1. For comparison, a 300 bilayer PO-Dex/GC film was fabricated and cell culture media containing its release material were prepared in the same way. These media were then used for cell culture, and the viability of the cells was assessed using the MTT assay. As a control, the cells were also cultured with normal media, and the cell viabilities were normalized to the control group. As shown in Fig. 5, the cell viability reduces to be ∼65% in PO-Dex/GC film-treated media. Similar to PO-Dex–DOX/GC film, the PO-Dex/GC film could also disintegrate and release PO-Dex into the media, which is toxic to the cells as shown in Fig. 4. In PO-Dex–DOX/GC film-treated media, the cell viability further reduces to be ∼24%. The result clearly shows that PO-Dex–DOX released from the dynamic films remains to be toxic to the cells.
 | ||
| Fig. 5 Percentage viability of Hep G2 cells after 48 h incubation in culture media containing materials released from PO-Dex–DOX/GC or PO-Dex/GC film. The star indicates significant difference (P < 0.05). | ||
The cell uptake of PO-Dex–DOX released from PO-Dex–DOX/GC films was investigated by confocal laser scanning microscopy (CLSM). For this purpose, HepG2 cells were seeded on coverslips. After 24 h culture, the cells were incubated with media with or without released materials for 48 h. Before imaging, the cell nuclei were stained blue with DAPI, while F-actin in the cellular cytoskeleton was stained green with FITC-phalloidin. As shown in Fig. 6, compared to the control group in which the cells were incubated with normal media, a lower cell density was observed for the PO-Dex/GC and PO-Dex–DOX/GC group, especially for the latter. These results confirm again the cytotoxicity of the polymeric materials released from the films. The cells incubated with normal media or media treated with PO-Dex/GC film do not emit red fluorescence. In contrast, the cells incubated with the PO-Dex–DOX/GC film-treated media emit red fluorescence, suggesting that the media contained PO-Dex–DOX which was released from the film and the conjugate was internalized by the cells. It is noteworthy that DOX fluorescence was detected in both nucleus and cytoplasm. According to previous studies, when incubating the cells with free DOX, the drug is mainly distributed in the nuclear region of cells.52 These results may suggest that the PO-Dex–DOX conjugate is internalized via a mechanism different from free DOX, possibly via endocytosis.52
 | ||
| Fig. 6 Confocal images of immunohistochemically stained Hep G2 cells. Cell nuclei and F-actin were stained blue and green, respectively. The cells were incubated for 48 h with normal media (control), PO-Dex/GC film-treated media, and PO-Dex–DOX/GC film-treated media, respectively. | ||
Conclusions
In conclusion, dynamic LbL films were fabricated from PO-Dex–DOX and GC via the in situ Schiff base reaction between the two polymers. Because the polymers were linked with reversible, dynamic Schiff base bonds, the films disintegrate gradually when soaked in aqueous solutions and thus release DOX into the media. The released DOX retains its ability to inhibit the proliferation of cancer cells. Different from ordinary drug carriers in which drug is released via either diffusion or the degradation of polymers, here drug release was achieved by exploiting the dynamic properties of the PO-Dex–DOX/GC films. The stimuli-responsibility of Schiff base bond further allows for tuning the release rate using external stimuli, including pH and temperature. Considering the biodegradability of the polymers involved, the ability to release drug in pH and temperature-controlled manner, and the ability to be coated onto substrates with complex morphologies, the new drug carrier developed here will be a good candidate for sustained release of bioactive compounds from surfaces of biomedical devices.Acknowledgements
We thank financial support for this work from the National Natural Science Foundation of China (Grants Nos 21174070, 21274068, and 21374048), Tianjin Public Health Bureau (13KG110), ICCAS State Key Laboratory of Polymer Physics and Chemistry (PPCL-2014CX-6), and PCSIRT program (IRT1257).Notes and references
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
- S. Pavlukhina and S. Sukhishvili, Adv. Drug Delivery Rev., 2011, 63, 822–836 CrossRef CAS PubMed.
- B. M. Wohl and J. F. J. Engbersen, J. Controlled Release, 2012, 158, 2–14 CrossRef CAS PubMed.
- B. G. de Geest, N. N. Sanders, G. B. Sukhorukov, J. Demeester and S. C. de Smedt, Chem. Soc. Rev., 2007, 36, 636–649 RSC.
- X. Chen, W. Wu, Z. Guo, J. Xin and J. Li, Biomaterials, 2011, 32, 1759–1766 CrossRef CAS PubMed.
- Z. B. Ding, Y. Guan, Y. J. Zhang and X. X. Zhu, Polymer, 2009, 50, 4205–4211 CrossRef CAS PubMed.
- E. Vazquez, D. M. Dewitt, P. T. Hammond and D. M. Lynn, J. Am. Chem. Soc., 2002, 124, 13992–13993 CrossRef CAS PubMed.
- B. B. Hsu, S. R. Hagerman, K. Jamieson, J. Veselinovic, N. O'Neill, E. Holler, J. Y. Ljubimova and P. T. Hammond, Biomacromolecules, 2014, 15, 2049–2057 CrossRef CAS PubMed.
- R. C. Smith, M. Riollano, A. Leung and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 8974–8977 CrossRef CAS PubMed.
- M. Macdonald, N. M. Rodriguez, R. Smith and P. T. Hammond, J. Controlled Release, 2008, 131, 228–234 CrossRef CAS PubMed.
- X. Zhang, Y. Guan and Y. Zhang, J. Mater. Chem., 2012, 22, 16299–16305 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- Y. Guan and Y. Zhang, J. Appl. Polym. Sci., 2014, 131, 40918 Search PubMed.
- J. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS PubMed.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS PubMed.
- Y. Guan, S. G. Yang, Y. J. Zhang, J. Xu, C. C. Han and N. A. Kotov, J. Phys. Chem. B, 2006, 110, 13484–13490 CrossRef CAS PubMed.
- Y. Guan, Y. J. Zhang, T. Zhou and S. Q. Zhou, Soft Matter, 2009, 5, 842–849 RSC.
- W. Lin, Y. Guan, Y. J. Zhang, J. Xu and X. X. Zhu, Soft Matter, 2009, 5, 860–867 RSC.
- Z. B. Ding, Y. Guan, Y. Zhang and X. X. Zhu, Soft Matter, 2009, 5, 2302–2309 RSC.
- L. Zhou, M. Chen, L. Tian, Y. Guan and Y. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 3541–3548 CAS.
- Y. Zhao, Q. Yuan, C. Li, Y. Guan and Y. Zhang, Biomacromolecules, 2015, 16, 2032–2039 CrossRef CAS PubMed.
- Y. Xin and J. Yuan, Polym. Chem., 2012, 3, 3045–3055 RSC.
- C. Givelet, J. Sun, D. Xu, T. J. Emge, A. Dhokte and R. Warmuth, Chem. Commun., 2011, 47, 4511–4513 RSC.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS.
- C. Ding, L. Zhao, F. Liu, J. Cheng, J. Gu, S. Dan, C. Liu, X. Qu and Z. Yang, Biomacromolecules, 2010, 11, 1043–1051 CrossRef CAS PubMed.
- X. Geng, X. Mo, L. Fan, A. Yin and J. Fang, J. Mater. Chem., 2012, 22, 25130–25139 RSC.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed.
- H. Saito, A. S. Hoffman and H. I. Ogawa, J. Bioact. Compat. Polym., 2007, 22, 589–601 CrossRef CAS PubMed.
- B. Wang, C. Xu, J. Xie, Z. Yang and S. Sun, J. Am. Chem. Soc., 2008, 130, 14436–14437 CrossRef CAS PubMed.
- D. Miksa, E. R. Irish, D. Chen, R. J. Composto and D. M. Eckmann, Biomacromolecules, 2006, 7, 557–564 CrossRef CAS PubMed.
- L. Zhou, M. Chen, Y. Guan and Y. Zhang, Polym. Chem., 2014, 5, 7081–7089 RSC.
- S. Mitra, U. Gaur, P. C. Ghosh and A. N. Maitra, J. Controlled Release, 2001, 74, 317–323 CrossRef CAS.
- A. Bernstein, E. Hurwitz, R. Maron, R. Arnon, M. Sela and M. Wilchek, J. Natl. Cancer Inst., 1978, 60, 379–384 CAS.
- H. Zhao and N. Heindel, Pharm. Res., 1991, 8, 400–402 CrossRef CAS.
- L. Dai, H. A. W. StJohn, J. Bi, P. Zientek, R. C. Chatelier and H. J. Griesser, Surf. Interface Anal., 2000, 29, 46–55 CrossRef CAS.
- S. Martwiset, A. E. Koh and W. Chen, Langmuir, 2006, 22, 8192–8196 CrossRef CAS PubMed.
- L. Wong, M. Kavallaris and V. Bulmus, Polym. Chem., 2011, 2, 385–393 RSC.
- C. Chun, S. M. Lee, C. W. Kim, K. Hong, S. Y. Kim, H. K. Yang and S. Song, Biomaterials, 2009, 30, 4752–4762 CrossRef CAS PubMed.
- J. X. Gu, W. P. Cheng, J. G. Liu, S. Y. Lo, D. Smith, X. Z. Qu and Z. Z. Yang, Biomacromolecules, 2008, 9, 255–262 CrossRef CAS PubMed.
- L. Zhu, L. Zhao, X. Qu and Z. Yang, Langmuir, 2012, 28, 11988–11996 CrossRef CAS PubMed.
- H. Kawai, T. Umehara, K. Fujiwara, T. Tsuji and T. Suzuki, Angew. Chem., 2006, 118, 4387–4392 CrossRef PubMed.
- K. H. Bouhadir, E. Alsberg and D. J. Mooney, Biomaterials, 2001, 22, 2625–2633 CrossRef CAS.
- M. Kumar, R. Muzzarelli, C. Muzzarelli, H. Sashiwa and A. J. Domb, Chem. Rev., 2004, 104, 6017–6084 CrossRef PubMed.
- Y. Zhao, Z. Li, S. Kabehie, Y. Y. Botros, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 13016–13025 CrossRef CAS PubMed.
- Y. Jia, J. B. Fei, Y. Cui, Y. Yang, L. A. Gao and J. B. Li, Chem. Commun., 2011, 47, 1175–1177 RSC.
- N. Giuseppone and J. Lehn, Chem.–Eur. J., 2006, 12, 1715–1722 CrossRef CAS PubMed.
- M. A. Vazquez, J. Donoso, F. Muñoz, F. G. Blanco, M. A. G. Del Vado and G. Echevarria, Helv. Chim. Acta, 1990, 73, 1991–1998 CrossRef CAS PubMed.
- Y. Jang, J. Seo, B. Akgun, S. Satija and K. Char, Macromolecules, 2013, 46, 4580–4588 CrossRef CAS.
- M. Dash, F. Chiellini, R. M. Ottenbrite and E. Chiellini, Prog. Polym. Sci., 2011, 36, 981–1014 CrossRef CAS PubMed.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS PubMed.
- M. Sokolsky-Papkov, A. J. Domb and J. Golenser, Biomacromolecules, 2006, 7, 1529–1535 CrossRef CAS PubMed.
- C. X. Ding, J. X. Gu, X. Z. Qu and Z. Z. Yang, Bioconjugate Chem., 2009, 20, 1163–1170 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |