DOI:
10.1039/C5RA17679D
(Paper)
RSC Adv., 2015,
5, 83922-83930
Hierarchical microspheres: in situ assembly of CdS quantum dots on Cd2SnO4 nanosheets with enhanced visible light photocatalytic properties†
Received
1st September 2015
, Accepted 28th September 2015
First published on 28th September 2015
Abstract
A CdS/Cd2SnO4 composite was successfully synthesized via a one step solvothermal route synergistically assisted by L-cysteine and diethanolamine. The composite is composed of CdS quantum dots stuffed in hierarchical Cd2SnO4 microspheres with a diameter of around 2 μm, which are assembled from nanosheets with a thickness of 20–50 nm. The band gap of the composite is 2.46 eV, which is higher than pure phase CdS and Cd2SnO4, due to the quantum size effect from CdS QDs. One possible formation mechanism of the composite is presented; the initial precipitation of Sn(OH)2 serves as a template, and the complexes of L-cysteine coordinated with metal ion provide structural direction for the hierarchical microspheres. The CdS/Cd2SnO4 composite showed superior adsorption ability and enhanced visible light photocatalytic activity in the degradation of RhB to CdS, Cd2SnO4, and Degussa P-25, due to its microstructure and the efficient charge separation at the interface of CdS and Cd2SnO4.
1 Introduction
Photocatalysis is a highly appealing process with multiple applications, especially in the fields of pollution removal and fuel production.1–3 As a II–VI semiconductor, cadmium sulfide (CdS) is regarded as one of the most attractive visible light-driven photocatalysts because its band gap is 2.42 eV, which corresponds well with the solar spectrum. Recently, extensive efforts have been made to achieve controlled synthesis of nanostructures, such as nanoribbons,4 nanowires,5 nanorods,6 nanospheres,7 and many kinds of hierarchitectures8–10 for specific properties and applications. CdS quantum dots (QDs) can allow for over 100% quantum efficiency in producing electron–hole pairs through the generation of multiple excitons from the absorption of a single photon.11,12 Nevertheless, the low separation efficiency of electron hole pairs is still an obstacle for the development of CdS in environmental remediation and solar conversion. To improve photocatalytic efficiency, recent attempts have focused on combining CdS photocatalysts with other semiconductor materials. Chang et al. synthesized CdS QDs sensitized TiO2, which exhibited extremely enhanced photocatalytic activity in the reduction of O-chloronitrobenzene under visible-light irradiation.13 Su et al. prepared CdS QDs sensitized branched TiO2 nanoarrays, which demonstrated remarkable photocurrent density and high solar to hydrogen efficiency as anodes for photoelectrochemical water splitting.14 Liu et al. fabricated CdMoO4/CdS hybrid materials, which indicated high photocatalytic activity in decomposition of rhodamine B (RhB).15 In summary, these enhanced photocatalytic performances can be ascribed to the presence of another semiconductor as an electron acceptor that improves the effective charge separation in CdS. Therefore, coupling with suitable materials that have a relatively low conduction band potential in order to transfer photogenerated electrons appears to be an efficient way to improve photocatalytic efficiency.
Cadmium tin oxide (Cd2SnO4) is one of the most widely studied binary oxide systems due to its distinctive optical and electronic properties.16,17 So far, most of the work on Cd2SnO4 has focused on thin film deposition and characterization for applications as transparent conducting oxide (TCO) substrates for optoelectronic devices.18,19 The direct band gap of Cd2SnO4 is 2.5 eV, which appears quite promising for photovoltaic applications. Based on its superior electrical conductivity (1–10 Ω−1 cm−1) and mobility (10–100 cm2 V−1 s−1), favorable band alignment with respect to the redox potential of water, and its chemical stability, Cd2SnO4 is also a potential photoanode material for solar water splitting.20 However, it is difficult to obtain pure phase Cd2SnO4 because the formation of Cd2SnO4 is usually accompanied by the impurities such as SnO2, CdO, or CdSnO3.21,22 Until recent period, Kelkar et al. synthesized pure phase nanoparticles of Cd2SnO4 by the solution combustion method and obtained a photocurrent of ∼250 μA cm−2 using Cd2SnO4 as a photoanode for solar water splitting.23 When orthorhombic/cubic Cd2SnO4 nanojunctions and CdS QDs coupled with Cd2SnO4 were used as photoanodes, solar water splitting efficiency was improved 10-fold and 40-fold, respectively.24,25 These results indicated CdS/Cd2SnO4 composite can enhance photoelectrochemical efficiency more remarkably. In Kelkar's experiment, CdS nanocrystals were deposited on Cd2SnO4 photo-electrode by chemical bath deposition SILAR route. Only one layer of CdS was absorbed on the surface of Cd2SnO4. There was no sufficient contact between CdS and Cd2SnO4. The contact between the two component is an important factor affecting the photocatalytic activity because the presence of interface is needed to allow the highly efficient interparticle charge transfer. In order to improve further photocatalytic efficiency, it is necessary to synthesize a novel CdS/Cd2SnO4 composite in which there are high interfaces between the two components.
In our work, we first synthesized novel microspheres, which were composed of CdS QDs absorbed on Cd2SnO4 nanosheets by an in situ assembly process via a simple one-step solvothermal reaction. The formation mechanism of the CdS/Cd2SnO4 composite was proposed. The photocatalytic property of as-prepared CdS/Cd2SnO4 composite was evaluated by the degradation of RhB in aqueous solution under simulated visible-light irradiation. The results show that CdS/Cd2SnO4 exhibit enhanced photocatalytic performance compared to pure CdS due to high exciton generation in the CdS QDs, efficient transfer of photogenerated electrons, and lower electron–hole recombination in the CdS/Cd2SnO4 composite. To the best of our knowledge, this is the first examination of the novel hierarchical nanostructure of CdS/Cd2SnO4 composite with enhanced visible-light photocatalytic degradation of organic contaminants.
2 Experimental
2.1 Synthesis procedure
2.1.1 Materials and reagents. All chemicals were analytical grade and were used directly without any treatment.
2.1.2 Synthesis of CdS/Cd2SnO4 composite via one-step reaction. In a typical procedure, 1 mmol of tin(II) chloride dihydrate (SnCl2·2H2O), 2 mmol of cadmium acetate dihydrate (Cd(Ac)2·2H2O), and 4 mmol of L-cysteine (C3H7NO2S) were dissolved in 80 mL of water/diethanolamine (DEA) (3/2 by v/v) solution by vigorous stirring at 40 °C. After being stirred for 0.5 h, the resulting mixture was transferred into a 100 mL Teflon-lined stainless autoclave. The autoclave was sealed and maintained at 180 °C for 12 h and then naturally cooled to ambient temperature. The resultant yellow product was separated by centrifugation, washed with absolute alcohol and deionized water with sonication several times, and dried in a vacuum at 60 °C for 2 days for characterizations.
2.1.3 Synthesis of CdS nanoparticles. CdS nanoparticles were synthesized through a similar procedure in which 1 mmol of Cd(Ac)2·2H2O and 2 mmol of L-cysteine were dissolved in 40 mL of water/DEA (3/2 by v/v) solution by vigorous stirring for 0.5 h at room temperature. Then, the solution was transferred into a 50 mL Teflon-lined stainless autoclave and maintained at 180 °C for 12 h. The bright yellow solid product was centrifuged, washed ultrasonically with absolute alcohol several times, and vacuum-dried at 60 °C for 2 days.
2.1.4 Synthesis of orthorhombic Cd2SnO4. Under the same conditions, orthorhombic Cd2SnO4 could not be synthesized in absence of L-cysteine; only CdSnO3·3H2O and CdSnO3 were obtained. In a typical experiment, 1 mmol of SnCl4·5H2O and 2 mmol of Cd(Ac)2·2H2O were dissolved in 30 mL of deionized water at room temperature. Then 3 mol L−1 NaOH solution was added dropwise into the solution with continuous stirring until the pH of the mixture equalled to 14. A white suspension was formed at basic pH. The suspension was transferred into a 50 mL Teflon-lined stainless autoclave and maintained at 220 °C for 12 h. The resulting grass-green powder was collected and washed with deionized water and ethanol several times and then dried at 60 °C overnight. The synthesized powder was identified as orthorhombic phase Cd2SnO4 (PCPDF # 34-0928) by XRD. The bright yellow orthorhombic Cd2SnO4 corresponding to PCPDF # 31-0242 was obtained after the grass green powder was further annealed at 1050 °C for 4 h.
2.2 Characterization
X-ray diffraction (XRD) patterns were measured with a Rigaku D/MAX-IA diffractometer using Cu, Kα (λ = 1.5406 Å) radiation over a range of 10° ≤ 2θ ≤ 80°. The morphologies and microstructures of the as-prepared samples were observed on a Hitachi S-3400 scanning electron microscopy (SEM) and a Hitachi S-4800 field emission scanning electron microscopy (FESEM). Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were obtained on a JEOL JEM-2100F TEM with an acceleration voltage of 200 kV. Ultraviolet-visible (UV-vis) absorption spectra were recorded for the dry-pressed disk samples using a Shimadzu UV-2550 UV-vis spectrometer. BaSO4 was used as the reflectance standard in UV-vis diffuse reflectance experiment. The Brunauer–Emmett–Teller (BET) surface area and pore size measurement were performed using nitrogen adsorption via a Micromeritics ASAP 2020 (USA) surface area analyzer. X-ray photoelectron spectroscopy (XPS) was performed on an ESCALABMK II X-ray photoelectron spectrometer with Al KR radiation as the X-ray source. The photoluminescence (PL) spectra of the samples were obtained at room temperature using Fluorescence Spectrophotometer (F-7000, Hitachi, Japan) equipped with a Xe lamp.
2.3 Photocatalytic activity test
The photocatalytic experiments of the as-prepared samples for decomposition of RhB were performed in aqueous solution. First, 20 mg of sample was suspended into 100 mL RhB aqueous solution (the dye concentration was 8 mg L−1). The suspension was stirred in the dark for a certain time to reach an adsorption–desorption equilibrium, and then the light was turned on. A 350 W Xe lamp equipped with a cut-off filter (λ > 420 nm) was used as the light source and set about 20 cm apart from the reactor. The light irradiance of visible lights is about 0.180 mW cm−2. At given irradiation time intervals, 6 mL suspension was collected and centrifuged to remove the photocatalyst particles. As a comparison, the photocatalytic activity of commercial TiO2 (Degussa P25, with a surface area of ca. 45 m2 g−1), CdS, and Cd2SnO4 were tested under the same experimental conditions. The UV-vis absorption spectra of the RhB solutions were measured using the UV-2550 spectrophotometer. The concentration change of RhB was judged by the relative absorption at 553 nm compared to the baseline.
3 Results and discussion
3.1 Structural characterization
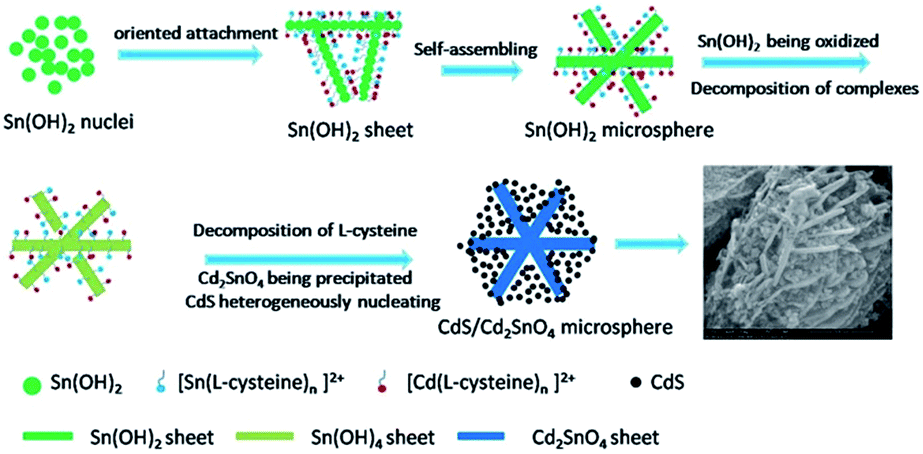
The morphology and microstructure of the final product were characterized by FESEM, as shown in Fig. 1. Interestingly, the composite self-organized into spherical assemblies with an average diameter of ∼2 μm. The high magnification FESEM images show that the CdS/Cd2SnO4 microspheres are in fact built from nanosheets with a thickness of 20–50 nm, with nanoparticles stuffed between the nanosheets. Although the surfaces of these nanosheets are smooth, these nanoparticles still remain in the microspheres after vigorous ultrasonic treatment, demonstrating strong affinity between the nanosheets and nanoparticles.
 |
| | Fig. 1 FESEM images of CdS/Cd2SnO4 microspheres: (a) overall product morphology; (b and c) detailed views of average-sized spheres; and (d) a magnified view of an individual sphere. | |
The microstructure of the CdS/Cd2SnO4 composite was further observed by TEM (HRTEM). Fig. 2a shows nanoparticles aggregated together on the surface of the nanosheets with a size of about 5 nm. Fig. 2b shows clear lattice fringes, suggesting the crystalline nature of the sample. The fringe interval of 0.310 nm shows good agreement with the d-spacing of the (101) planes of hexagonal CdS; and the fringe interval of 0.282 nm corresponds to the d-spacing of the (130) planes of orthorhombic Cd2SnO4. The HRTEM images illustrate that the fringe areas attributed to CdS (circled area) are only several nanometers in size, while the larger fringe regions are consistent with Cd2SnO4. These results indicate that the CdS/Cd2SnO4 microspheres are composed of Cd2SnO4 nanosheets and CdS quantum dots. The size of the CdS QDs is about 3–10 nm.
 |
| | Fig. 2 (a) TEM images of the edge of CdS/Cd2SnO4 microspheres; and (b) HRTEM images of the circled area in (a). | |
The phase and composition of the as-prepared CdS/Cd2SnO4 composite were characterized by XRD, as shown in Fig. 3. For comparison, the XRD patterns of pure CdS and Cd2SnO4 are also shown. The X-ray reflections of CdS and Cd2SnO4 correspond to the hexagonal phase of CdS (JCPDS no. 41-1049) and the orthorhombic phase of Cd2SnO4 (JCPDS no. 31-0242), respectively. The diffraction profiles of the CdS/Cd2SnO4 composite show a mixture of hexagonal CdS and orthorhombic Cd2SnO4. The peaks at 18.3° (110), 31.5° (130), and 33.6° (111) indicate the existence of orthorhombic Cd2SnO4. The peaks in 24.8, 26.5, 28.2, and 43.7° correspond to the (100), (002), (101) and (110) planes of hexagonal CdS. However, the crystallinity of the composite clearly decreases, as indicated by the width of the reflection peaks and the relative decrease in the intensity. The results are consistent with the SEM observations (Fig. S1†), which show that the grain sizes of pure CdS and Cd2SnO4 are about 50 and 100 nm, respectively, which are both larger than that of the composite. In particular, the crystallinity of orthorhombic Cd2SnO4 (JCPDS no. 31-0242) is very high because it comes from the phase change of Cd2SnO4 (JCPDS no. 34-0928) at a high temperature of 1050 °C. The relative intensity of the peak corresponding to the (002) planes compared to these of the (100), (101), (110) planes of CdS increases significantly, which indicates the growth of the partial planes are suppressed.
 |
| | Fig. 3 Powder XRD patterns of as-prepared CdS, Cd2SnO4, and CdS/Cd2SnO4 composite. | |
Fig. 4a and b display the nitrogen adsorption–desorption isotherm and pore-size distribution curves for CdS and CdS/Cd2SnO4. Both of the isotherms are characteristic of type IV with a H3-type hysteresis loops. CdS/Cd2SnO4 composite show larger BET surface areas (74.99 m2 g−1) than CdS (32.44 m2 g−1). From the pore-size distribution (inset of Fig. 4a and b), the total pore volume and the average pore diameter of CdS are 0.325 m2 g−1 and 38.26 nm, respectively. CdS/Cd2SnO4 has larger pore volume (0.418 m2 g−1) and smaller pore diameters (22.27 nm) than CdS. These results agree well with the aforementioned microstructures. The small CdS grain sizes in the CdS/Cd2SnO4 composite lead to small pore diameters, and the hierarchical microspheres structure of the composite induce its large BET surface and pore volume.
 |
| | Fig. 4 Nitrogen adsorption–desorption isotherms and pore-size distribution (inset) for (a) CdS; (b) CdS/Cd2SnO4 composite. | |
The chemical composition and valence state of CdS/Cd2SnO4 composite were analyzed further by XPS, recorded here with Al (K-α) (Fig. 5). The complete survey spectrum is shown in Fig. 5a; it indicates the presence of Cd, Sn, S, O, and C elements, with no other elements being detected. The C element comes from gaseous molecules in the atmosphere and the adventitious carbon-based contaminant, and the binding energy for the C 1s peak at 284.60 eV was used as the reference for calibration. Fig. 4b shows the high-resolution Cd 3d spectrum. The Cd 3d5/2 and Cd 3d3/2 peaks are located at 404.70 and 411.35 eV, respectively. The peak at 404.70 eV could be fitted by two nearly Gaussian functions, centred at 404.37 and 404.99 eV, which correspond well to the binding energies for the Cd–O bond of Cd2SnO4 (ref. 16) and the Cd–S bond of CdS8,13,26–29 in the literature, respectively. As deduced from the intensities of both peaks, the ratio of CdS/Cd2SnO4 is 1.44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The peak energies of 486.20 eV (Sn 3p5/2) and 494.55 eV (Sn 3p3/2) shown in Fig. 4c are in good agreement with previously reported values for pure Cd2SnO4.16,30 The values are slightly higher than the bonding energies of Sn 3p5/2 and Sn 3p3/2 of Sn2+ due to the extra Coulomb interaction between the ion core and the photo-emitted electron in atoms with higher oxidation states.31 The fact that the bonding energies of the Sn(IV) compound are higher than those of the Sn(II) analogue is consistent with previous findings regarding tin sulfides.32,33 Otherwise, the splitting of the 3d doublet of Sn for the above composites is 8.35 eV, which also lies within the acceptable range of the spin–orbit energy splitting of 8.41 ± 0.02 eV for Sn4+.30,34 These results all confirm the presence of only Sn4+ in composites; Sn2+ is entirely oxidized to Sn4+ in the reaction. The position of the S 2p peak (Fig. 4d) is at 161.37 eV; this confirms that the S element exists in the form of S2− chemical state, which accords well with the reference values for CdS.8,27–29 Importantly, no +2, +4, or +6 oxidation states of S were observed.
:1. The peak energies of 486.20 eV (Sn 3p5/2) and 494.55 eV (Sn 3p3/2) shown in Fig. 4c are in good agreement with previously reported values for pure Cd2SnO4.16,30 The values are slightly higher than the bonding energies of Sn 3p5/2 and Sn 3p3/2 of Sn2+ due to the extra Coulomb interaction between the ion core and the photo-emitted electron in atoms with higher oxidation states.31 The fact that the bonding energies of the Sn(IV) compound are higher than those of the Sn(II) analogue is consistent with previous findings regarding tin sulfides.32,33 Otherwise, the splitting of the 3d doublet of Sn for the above composites is 8.35 eV, which also lies within the acceptable range of the spin–orbit energy splitting of 8.41 ± 0.02 eV for Sn4+.30,34 These results all confirm the presence of only Sn4+ in composites; Sn2+ is entirely oxidized to Sn4+ in the reaction. The position of the S 2p peak (Fig. 4d) is at 161.37 eV; this confirms that the S element exists in the form of S2− chemical state, which accords well with the reference values for CdS.8,27–29 Importantly, no +2, +4, or +6 oxidation states of S were observed.
 |
| | Fig. 5 XPS spectra of the CdS/Cd2SnO4 composite: (a) survey; (b) Cd 3d; (c) Sn 3d; and (d) S 2p. | |
UV-visible absorption spectra were employed to compare the optical properties of CdS, Cd2SnO4 and CdS/Cd2SnO4 composite. It can be seen in Fig. 6a that CdS and the CdS/Cd2SnO4 composite have similar absorption spectra and show strong absorption in the visible-light region, with an absorption edge at about 550 nm. Cd2SnO4 show weak absorption with the same absorption edge due to its high transmittance,16,30,31 and the absorption intensity of the composite was lower than that of pure phase CdS. The band gaps of the samples were estimated by the formula αhv = A(hv − Eg)n/2, where α, hv, A, and Eg are the absorption coefficient, photo energy, proportionality constant, and band gap, respectively. Here, n takes the value of 1, and 4, for direct or indirect allowed transition respectively. Both CdS and Cd2SnO4 are semiconductors with a direct band gap, and the value of n is 1 for the direct transition. Fig. 6b presents the (αhv)2 vs. photo energy (hv) curve for the three samples. The band gaps (Eg values) are estimated to be 2.42, 2.31, and 2.46 eV for CdS, Cd2SnO4 (inset), and the CdS/Cd2SnO4 composite, respectively. The Eg value of CdS shows good agreement with the reference values in the literatures (∼2.4 eV).35 The Eg value of Cd2SnO4 is within the range of values of orthorhombic Cd2SnO4 powder (2.3–2.44 eV),20,24,25 although it is lower than that of Cd2SnO4 films (2.7–3.3 eV).19,30 The higher band gaps of the Cd2SnO4 films resulted from weaker crystallinity and the presence of the impurity SnO2 with a higher Eg (3.6 eV). Compared with pure phase CdS and Cd2SnO4, an increase in band gap was observed in the CdS/Cd2SnO4 composite; this can be ascribed to the blue-shift induced by the quantum size effect from CdS QDs. The experimental results suggest that the composite has the potential as a photocatalyst to decompose organic pollutant under visible light irradiation.
 |
| | Fig. 6 (a) UV-vis absorption spectra for CdS, Cd2SnO4, and CdS/Cd2SnO4 composite; (b) plots of (αhv)2 vs. photo energy (hv) of CdS/Cd2SnO4, CdS, and Cd2SnO4 (inset). | |
3.2 Formation mechanism of CdS/Cd2SnO4 microspheres
In order to investigate the formation process and mechanism of CdS/Cd2SnO4 microspheres, we separated initial precipitation from the reaction mixture before the solvothermal process. The XRD patterns and SEM images of the precipitation are shown in Fig. 7. All peaks of precipitation are indexed to the pure phase of Sn6O4(OH)4 (JCPDS no. 46-1486) with low crystallinity due to reaction at a low temperature (40 °C) in the basic solution (pH ≈ 11, experimental result), with no other impurities such as hydroxide of Cd2+ or metal sulfide being found. The corresponding SEM images show that an irregular porous sheet-like architecture was obtained. We can deduce the precipitation of Sn6O4(OH)4 acts as a template for CdS/Cd2SnO4 microspheres. Although the sheet-like structure is irregular at low temperature, these nanosheets are favorable to aggregating into microspheres driven by the minimization of surface energy in the condition of energy provided by the solvothermal process. In the system, L-cysteine plays the critical role of structural direction for the formation of microspheres. It is widely known that there are three main functional groups in the L-cysteine molecule; namely, –NH2, –COOH, and –SH, which all have a strong tendency to coordinate with metal cations.36 It has been reported that L-cysteine can react with Sn2+ and Cd2+ to form complexes in aqueous solution.37,38 In our experiment, we observed that the amount of precipitation in the system decreases significantly due to the formation of soluble complexes, compared to a similar reaction system without L-cysteine. These complexes can preferentially attach on a specific lattice plane of Sn6O4(OH)4 nuclei through hydrogen bonds, which selectively lower the surface energy of the plane and suppress the growth of the plane, resulting in the sheet-like architecture. These sheets further aggregate into microspheres to minimize the surface energy in the solvothermal process. In fact, such a hydrogen bond mediated self-assembly has been found in the cysteine-assisted hydrothermal synthesis of various 3D metal sulfides semiconductor nanostructures.36 In addition, the Ksp of Sn(OH)4 is far smaller than that of Sn(OH)2 (Ksp(Sn(OH)4) = 1.0 × 10−56, Ksp(Sn(OH)2) = 5.0 × 10−27), which leads φ(Sn4+/Sn2+) to decrease further. As a result, the reducing capacity of Sn2+ is enhanced further, and Sn2+ is more easily oxidized to Sn4+ by O2 in the atmosphere in the basic solution. It is worth mentioning that Sn6O4(OH)4 is the dehydration product of Sn(OH)2 after being dried in a vacuum for simple XRD measurements, it is not possible for it to be the real form of precipitation in the solution. However, Sn(OH)2 can exist in the solution. After Sn(OH)2 is oxidized to Sn(OH)4, partial Cd2+ decomposed from the complexes combines with Sn(OH)4 to form Cd2SnO4 in the basic solution. All these reactions happen in situ with the structure only adjusting lightly, i.e., not undergoing the process of the precipitation being dissolved and leading to a second precipitation. Therefore, the microsphere architecture is still retained after Sn(OH)2 transforms into Cd2SnO4. The reactions in situ and the appropriate basicity of the solution are necessary for the formation of Cd2SnO4, which is examined in the following section. Simultaneously, L-cysteine is slowly released from the complexes and decomposes to form S2− in the solvothermal process. Comparing Ksp of CdS with that of SnS and SnS2 (Ksp(CdS) = 8.0 × 10−27, Ksp(SnS) = 1.0 × 10−25, and Ksp(SnS2) = 2.0 × 10−27), we can infer that CdS would preferentially generate and be favorable to forming nuclei heterogeneously on the surface of the nanosheets. In the system, L-cysteine is a weak acid; the basicity of the solution is provided by DEA. The reactions can be described as follows:| | |
Sn2+ + nL-cysteine → [Sn(L-cysteine)n]2+
| (1) |
| | |
Cd2+ + nL-cysteine → [Cd(L-cysteine)n]2+
| (2) |
| | |
Sn2+ + 2OH− → 2Sn(OH)2
| (3) |
| | |
HSCH2CHNH2COOH + 2OH− → CH3COCOO− + NH3 + S2− + H2O
| (4) |
| | |
2Sn(OH)2 + O2 + 2H2O → 2Sn(OH)4
| (5) |
| | |
Sn(OH)4 + 2Cd2+ + 4OH− → Cd2SnO4 + 4H2O
| (6) |
 |
| | Fig. 7 (a) X-ray diffraction patterns; and (b) SEM image of precipitation from the reaction mixture before the solvothermal process. | |
To further illustrate the formation of cadmium stannate in situ and the effect of L-cysteine on the structure and morphology of the product, we compared precipitation from the reaction system without L-cysteine before and after the solvothermal process under similar conditions as the afore mentioned system. The XRD patterns of the precipitation before the solvothermal is still assigned to Sn6O4(OH)4 (not shown here), while the precipitation after the solvothermal consists mainly of cubic CdSnO3·3H2O (JCPDS no. 28-0202) and a small amount of CdSnO3 (JCPDS no. 34-0758), without any other impurities of divalent tin compounds (Fig. S2a†). SEM observations indicate that the precipitation before the solvothermal consists of nanoparticles with a uniform diameter of about 30 nm (Fig. S2b†), and the precipitation after the solvothermal is composed of nanoparticles and micrometer scale cubes (Fig. S2c†). The XRD results further confirm that Sn2+ can be fully oxidized to Sn4+ and form cadmium stannate in DEA basic aqueous solution. The fact that nanoparticles still partially exist after solvothermal indicates that Sn6O4(OH)4 can possibly be converted into cadmium stannate in situ with the structure being retained. These nanoparticles continue to grow and become cubes according to the intrinsic crystalline structure of CdSnO3·3H2O through the Ostwald ripening process, which is different from the oriented attachment mechanism performed in the afore mentioned solution including L-cysteine. Therefore, it can be concluded that L-cysteine serves as a structural direction coordination reagent for hierarchical microspheres.
It is worth noting that both CdSnO3·3H2O and CdSnO3 are all polymorphs of Cd2SnO4. According to previous reports, CdSnO3·3H2O can lose crystal water at about 300 °C, CdSnO3 can transform into cubic Cd2SnO4 at about 500 °C and into orthorhombic Cd2SnO4 at about 1050 °C with the formation of the impurity SnO2.21 In our experiment, to compare the properties of CdS/Cd2SnO4 with those of the same pure Cd2SnO4, we directly synthesized orthorhombic Cd2SnO4 corresponding to JCPDS no. 34-0928 (Fig. S3†) in strong basic NaOH aqueous solution (pH > 14) to avoid the impurity SnO2 resulting from the decomposition of CdSnO3. The orthorhombic Cd2SnO4 corresponding to JCPDS no. 34-0928 can transform into the orthorhombic Cd2SnO4 corresponding to JCPDS no. 31-0242 with thermal treatment at 1050 °C; the latter is the same phase as Cd2SnO4 of the CdS/Cd2SnO4 microspheres (Fig. 3). The results indicate that the special morphology and composition of the CdS/Cd2SnO4 microspheres can be attributed to the synergistic assistance of all the reaction reagents, demonstrating the uniqueness of the synthesis route. The initial template can determine the structure of cadmium stannate; namely, nanosheets lead to orthorhombic Cd2SnO4, while nanoparticles tend to form CdSnO3·3H2O and CdSnO3.
In addition, when SnCl4·5H2O is used instead of SnCl2·2H2O, CdS/SnO2 nanoparticles with a diameter of about 100 nm are obtained instead of CdS/Cd2SnO4 microspheres under similar conditions (Fig. S4†). Cd2SnO4 and CdSnO3 cannot be produced because the acidity of Sn4+ is stronger than that of Sn2+; this decreases the basicity of the reaction system, which is unfavorable for the formation of Cd2SnO4, leading to only SnO2 being obtained. On the other hand, Sn4+ has a stronger ability to coordinate with L-cysteine than Sn2+ does, so no hydroxide of quadrivalent tin is precipitated as a template for heterogeneous nucleation, as the solution is clear before the solvothermal. Subsequently, it is possible that CdS and SnO2 simultaneously form and merge into a nanoparticle, with no hierarchical microspheres being assembled. These results demonstrate further that the hydroxide of tin(II) serves as a template for hierarchical microspheres. Scheme 1 illustrates the formation process of hierarchical CdS/Cd2SnO4 microspheres based on the previous experimental results and analyses.
 |
| | Scheme 1 A schematic illustration of the proposed formation mechanism of CdS/Cd2SnO4 microspheres. | |
3.3 Photocatalytic activity
Fig. 8a shows the photodegradation process of the RhB aqueous solution in the presence of CdS/Cd2SnO4 microspheres (20 mg). The results show that the composite can adsorb about 34% of RhB within 30 min. The dye concentration adsorbed onto the surface of the catalyst increase further with time until an adsorption–desorption equilibrium in the solution is reached at about 90 min. When the light is turned on, the absorption peaks at 554 nm decrease rapidly with an absorption band shift to shorter wavelengths due to the de-ethylation of RhB. The results indicate that the composite can accomplish good adsorption in the dark and degradation to RhB under visible-light irradiation. Fig. 8b shows the adsorption and degradation of RhB in the presence of different catalysts, where c is the absorption of RhB at a wavelength of 554 nm after adsorption or irradiation for t min and c0 is the original absorption before the catalyst is put in the solution. Commercial TiO2 (P25), a standard material, was used for comparison with our samples. After adsorption–desorption equilibriums were reached, about 7.2, 3.2 and 38.3% of RhB was adsorbed on the surfaces of CdS, Cd2SnO4, and CdS/Cd2SnO4, respectively; for P25, only 4% of RhB was adsorbed. CdS has slightly better adsorption ability than P25 due to its nanometer size (Fig. S1(a)†), and the effective adsorption activity of the composite can be ascribed to its high surface area and large pore volume. After visible-light irradiation for 60 min, 16.5, 1.8, and 72.4% of RhB remained in solution for CdS, CdS/Cd2SnO4, and P-25, respectively. Cd2SnO4 showed no photocatalytic performance at all. To eliminate the effect of adsorption, we calculated the rate constants of different catalysts by linear fitted plots of ln(A0/A) vs. irradiation time (t). As for RhB aqueous solution with low concentration, the photocatalytic reaction follows a pseudo-first-order reaction and its kinetics can be express as ln(c/c0) = −kt, where k is the rate constant, c0 and c are the RhB concentrations at the initial state after the adsorption equilibrium and after irradiation for t min, respectively. As seen from Fig. 8c, the rate constant (k) of the RhB decomposition over CdS/Cd2SnO4 is estimated to be ca. 0.05748 min−1, which is 2.42 times larger than that of pure CdS (0.02954 min−1), and 11.73 larger than of P-25. The results indicated that CdS/Cd2SnO4 has obvious enhanced photocatalytic activity.
 |
| | Fig. 8 (a) UV-vis spectral changes of RhB aqueous solution in the presence of CdS/Cd2SnO4 microspheres (20 mg); (b) comparison of photocatalytic degradation of RhB over CdS, Cd2SnO4, CdS/Cd2SnO4 composite, and P25 (TiO2); and (c) plot of ln(A0/A) vs. visible irradiation time over various catalysts (RhB aqueous solution: 100 mL, 8 mg L−1, catalyst: 20 mg, 500 W Xe-lamp, λ > 420 nm). (d) Photo-stability tests over CdS/Cd2SnO4 composite for the cycling photodegradation of RhB. | |
To demonstrate the potential applicability of the CdS/Cd2SnO4 composite in photocatalysis, we further investigated its stability. The recycling experiments were carried out five times under the same reaction conditions (as shown in Fig. 8d). It is widely reported that photocorrosion of sulfide in the photocatalytic reaction can result in the deactivation of photocatalysts. However, in this case there was hardly decrease in the photocatalytic activity after five cycling runs, indicating its superior stability during photocatalysis. Therefore, the CdS/Cd2SnO4 composite has a promising practical application in the treatment of organic wastewater originating from its high activity and stability.
Cd2SnO4 shows no visible-light photocatalytic activity, even though its band gap is within the scope of visible light. This can be attributed to its weak absorption and high transmittance (Fig. 6a). Although the CdS nanoparticles shows superior photocatalytic activity compared to P25, the CdS/Cd2SnO4 composite exhibit enhanced photocatalytic efficiency, which can be attributed to two key factor: high surface area and the improved separation of charge carriers. The as-prepared CdS/Cd2SnO4 composite has higher surface area with smaller pore sizes and larger pore volume than CdS. Since the photocatalytic decomposition of organic compounds takes place on the surface of a photocatalyst, the enrichment of the organic compounds close to the photocatalyst is an important contributing factor for achieving higher photocatalytic performance.39 The higher surface areas could increase the contact area between the composite and the RhB solution. It is also believed that the holes in photocatalyst can transfer to the organic compounds more easily to accomplish rapid degradation. Based on the reasons, the CdS/Cd2SnO4 composite shows superior adsorption ability and photocatalytic activity.
The recombination of photo-generated charge carriers is another important cause due to the high density of surface states/defects where photo-generated electrons and/or holes may get trapped.24,25 The improved separation of charge carriers in the CdS/Cd2SnO4 composite can be testified by photoluminescence (PL) emission spectra. Since PL emission spectra mainly results from the combination of excited electrons and holes, PL emission spectra are useful in determining the efficiency of charge carrier trapping, migration and transfer, and are helpful in understanding the fate of electron–hole pairs in semiconductor particles. A low PL intensity implies a low recombination rate of the electron–hole pair under light irradiation.40 Fig. 9 shows a comparison of PL spectra of pure CdS and CdS/Cd2SnO4 composite when the excitation wavelength is 370 nm. It can be seen that CdS give a broad emission around 586 nm, which is attributed to excitation emission.41,42 The emission intensity of CdS/Cd2SnO4 composite is lower than that of pure CdS, suggesting the improved charge carrier separation in the composite. In fact, the charge carrier separation mechanism is usually used to project different combinations of heterojunctions with type II alignments for enhanced photocatalytic efficiency.43–45 In addition, high exciton generation in CdS QDs is also important factor that enhances photocatalytic activity.
 |
| | Fig. 9 Room temperature PL spectra of pure CdS and CdS/Cd2SnO4 composite. | |
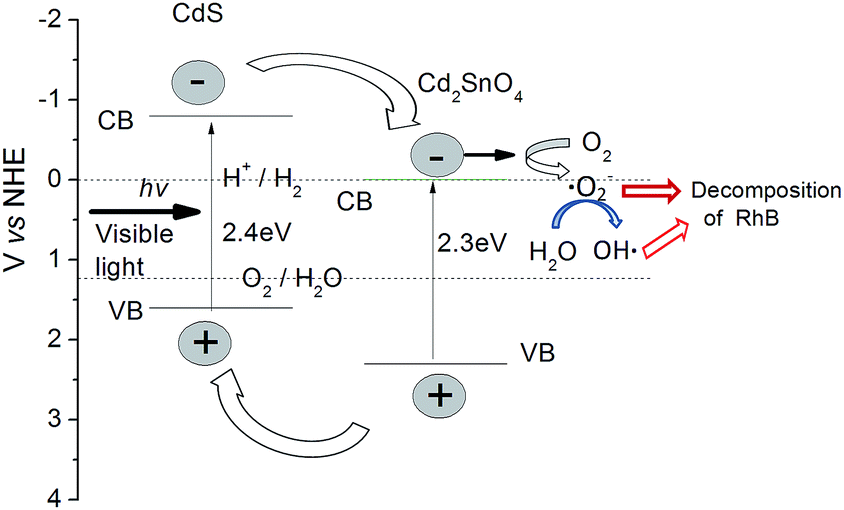
To clarify the photocatalytic mechanism of CdS/Cd2SnO4 composite, it is necessary to detect the main oxidative species in the photocatalytic reaction. The can be achieved by trapping oxidative species. In this experiment, tert-butanol (t-BuOH), 1,4-benzoquinone (BZQ) and disodium ethylene diamine tetra acetic acid (Na2-EDTA) were selected to detect the hydroxyl radicals (OH˙), superoxide radical (˙O2−) and hole (h+), respectively.46 As show in Fig. 10, the photo-degradation of RhB for CdS/Cd2SnO4 composite is significantly suppressed after the addition of a scavenger for ˙O2− (BZQ), however, only a slight decrease is observed with the presence of a scavenger for OH˙ (t-BuOH). In contrast, the injection of hole scavenger (Na2-EDTA) increase the photocatalytic activity of the composite, suggesting the trapping of hole increase the number of photo-generated electrons. These results indicate that the ˙O2− radical play a dominant role in the CdS/Cd2SnO4 composite system, the OH˙ radical play an assistant role and holes are not involved.
 |
| | Fig. 10 Trapping experiment of photocatalytic degradation of RhB over CdS/Cd2SnO4 composite with/without the presence of scavengers. | |
On the basis of experimental results, a proposed photocatalytic mechanism of the CdS/Cd2SnO4 composite is revealed in Fig. 11. The conduction band (CB) of CdS (−0.8 V vs. NHE)25,43 is more negative than that of Cd2SnO4 (−0.1 V vs. NHE).24 Under visible light irradiation, both of the semiconductors can be excited, the photo-generated electrons can only be transferred from CdS to Cd2SnO4, while the holes can only be transferred to the VB of CdS due to their intimate contact. Therefore, the electrons and holes will accumulate in the CB of Cd2SnO4 and the VB of CdS, respectively, leading to effective charge separation in the composite. The electrons retained at Cd2SnO4 can react with absorbed oxygen molecules (O2), which are then reduced to form superoxide radical (˙O2−). The unstable ˙O2− can react with water quickly and produce a few hydroxyl radicals (OH˙), to simultaneously decompose RhB.
 |
| | Fig. 11 Schematic diagrams illustrating the possible photocatalytic mechanism of the CdS/Cd2SnO4 microspheres. | |
Compared with the other nano-scale powdered photocatalysts, the as-prepared CdS/Cd2SnO4 microsphere photocatalyst has several advantages. It is widely known that for practical wastewater applications, a good photocatalyst should be able to be reclaimed and re-used easily, in addition to having high efficiency. The hierarchical structures of the CdS/Cd2SnO4 microspheres provide the composite a larger specific surface area and superior sedimentation capacity. Strong affinities between the Cd2SnO4 nanosheets and CdS nanoparticles prevent the CdS nanoparticles from individually dispersing in the aqueous solution in photocatalytic reactions. Our experiments also found the composite can be separated from the aqueous solution more easily than pure CdS nanoparticles can. Therefore, the as-prepared CdS/Cd2SnO4 microspheres can be regarded as novel photocatalysts that are ideal for the industrial application, many of which have been seriously impeded by the high cost of separating nanocrystal catalysts.
4 Conclusions
In summary, for the first time, CdS QDs decorated hierarchical Cd2SnO4 microspheres were fabricated via a in situ assembly in a one-pot solvothermal process. The formation process of the hierarchical CdS/Cd2SnO4 microspheres was studied, and possible mechanisms were proposed. The initial precipitation of Sn(OH)2 serves as a template, and the complexes of L-cysteine coordinated with metal ion provided structural direction for the hierarchical microspheres. The oxidization of Sn2+ and the precipitation of Cd2SnO4 occurred in situ and CdS heterogeneously nucleate on the surface of Cd2SnO4. The CdS/Cd2SnO4 microspheres exhibited superior photocatalytic efficiency compared to the CdS nanoparticles, Cd2SnO4, and Degussa P-25 in the degradation of RhB under visible light irradiation due to the efficient charge separation at the interface of the two semiconductors, higher adsorption ability to the organic compounds, and quantum effect of CdS. This synthetic method can be extended to the preparation of a variety of composite photocatalysts.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51521001) and the Ministry of Science and Technology of China (2015DFR50650).
Notes and references
- A. Kubacka, G. M. Fernandez and G. Colon, Chem. Rev., 2012, 112, 1555 CrossRef CAS PubMed.
- K. Ramasamy, M. A. Malik, N. Revaprasadu and P. O'Brien, Chem. Mater., 2013, 25, 3551 CrossRef CAS.
- F. Fresno, R. Portela, S. Suarez and J. M. Coronado, J. Mater. Chem. A, 2014, 2, 2863 CAS.
- T. Gao and T. H. Wang, Cryst. Growth Des., 2010, 10, 4995 CAS.
- J. Jin, J. G. Yu, G. Liu and P. K. Wong, J. Mater. Chem. A, 2013, 1, 10927 CAS.
- X. L. Wang, Z. C. Feng, D. Y. Fan, F. T. Fan and C. Li, Cryst. Growth Des., 2010, 10, 5312 CAS.
- Y. Kim, Y. H. B. Kim and D. J. Jang, J. Mater. Chem. A, 2014, 2, 5791 CAS.
- Z. Yu, X. Wu, J. Wang, W. Jia, G. Zhu and F. Qu, Dalton Trans., 2013, 42, 4633 RSC.
- S. Xiong, B. Xi and Y. Qian, J. Phys. Chem. C, 2010, 114, 14029 CAS.
- Z. X. Yang, W. Zhong, Y. Deng, C. T. Au and Y. W. Du, Cryst. Growth Des., 2011, 11, 2172 CAS.
- W. T. Sun, Y. Yu, H. Y. Pan, X. F. Gao, Q. Chen and L. M. Peng, J. Am. Chem. Soc., 2008, 130, 1124 CrossRef CAS PubMed.
- J. E. Murphy, M. C. Beard, A. G. Norman, S. P. Ahrenkiel, J. C. Johnson and P. Yu, et al., J. Am. Chem. Soc., 2006, 128, 3241 CrossRef CAS PubMed.
- P. Chang, H. Cheng, W. Li, L. Zhuo, L. He, Y. Yu and Z. Zhao, Phys. Chem. Chem. Phys., 2014, 16, 16606 RSC.
- F. Su, J. Lu, Y. Tian, X. Ma and J. Gong, Phys. Chem. Chem. Phys., 2013, 15, 12026 RSC.
- Y. Liu, L. Ren, X. Qi, Y. Wang, X. Liu and J. Zhong, RSC Adv., 2014, 4, 8772 RSC.
- D. A. Cristaldi, G. Impellizzeri, F. Priolo, T. Gupta and A. Gulino, J. Phys. Chem. C, 2012, 116, 3363 CAS.
- M. M. El-Nahass, A. A. Atta, M. M. A. El-Raheemb and A. M. Hassanien, J. Alloys Compd., 2014, 585, 1 CrossRef CAS PubMed.
- J. R. Mamazza, D. L. Morel and C. S. Ferekides, Thin Solid Films, 2005, 484, 26 CrossRef PubMed.
- T. Meng, B. McCandless, W. Buchanan, E. Kimberly and R. Birkmire, J. Alloys Compd., 2013, 556, 39 CrossRef CAS PubMed.
- X. Huang, J. Lv, Z. Li and Z. Zou, J. Alloys Compd., 2010, 507, 341 CrossRef CAS PubMed.
- Y. Tang, Y. Jiang, Z. Jia, B. Li, L. Luo and L. Xu, Inorg. Chem., 2006, 45, 10773 Search PubMed.
- W. Wang, Y. Xiao, X. Zhao, B. Liu and M. Cao, CrystEngComm, 2014, 16, 922 RSC.
- S. A. Kelkar, P. A. Shaikh, P. Pachfule and S. B. Ogale, Energy Environ. Sci., 2012, 5, 5681 CAS.
- A. Deshpande, S. Kelkar, S. Rayaluc and S. Ogale, J. Mater. Chem. A, 2014, 2, 492 CAS.
- S. Kelkar, C. Ballal, A. Deshpande, S. Warule and S. Ogale, J. Mater. Chem. A, 2013, 1, 12426 CAS.
- A. Veamatahau, B. Jiang, T. Seifert, S. Makuta, K. Latham and M. Kanehara, et al., Phys. Chem. Chem. Phys., 2015, 17, 2850 RSC.
- P. Kundu, P. A. Deshpande, G. Madrasb and N. Ravishankar, J. Mater. Chem., 2011, 21, 4209 RSC.
- F. Xu, Y. Yuan, H. Han, D. Wu, Z. Gao and K. Jiang, CrystEngComm, 2012, 14, 3615 RSC.
- F. X. Xiao, J. Miao and B. Liu, J. Am. Chem. Soc., 2014, 136, 1559 CrossRef CAS PubMed.
- H. Khallaf, C. T. Chen, L. B. Chang, O. Lupan, A. Dutta and H. Heinrich, et al., Appl. Surf. Sci., 2012, 258, 6069 CrossRef CAS PubMed.
- K. Jeyadheepan, M. Thamilselvan, K. Kim, J. Yi and C. Sanjeeviraja, J. Alloys Compd., 2015, 620, 185 CrossRef CAS PubMed.
- H. Su, Y. Xie, Y. Xiong, P. Gao and Y. Qian, J. Solid State Chem., 2001, 161, 190 CrossRef CAS.
- A. M. Tripathi and S. Mitra, RSC Adv., 2014, 4, 10358 RSC.
- Z. Zhang, C. Shao, X. Li, Y. Sun, M. Zhang, J. Mu, P. Zhang, Z. Guo and Y. Liu, Nanoscale, 2013, 5, 606 RSC.
- L. A. Silva, S. Y. Ryu, J. Choi, W. Choi and M. R. Hoffmann, J. Phys. Chem. C, 2008, 112, 12069 CAS.
- W. Shi, S. Song and H. Zhang, Chem. Soc. Rev., 2013, 42, 5714 RSC.
- W. Cai, J. Hu, Y. Zhao, H. Yang, J. Wang and W. Xiang, Adv. Powder Technol., 2012, 23, 850 CrossRef CAS PubMed.
- P. Thakur, S. Joshi, S. S. Kapoor and T. Mukherjee, Langmuir, 2009, 25, 6334 CrossRef CAS PubMed.
- P. Wang, J. Wang, X. Wang, H. Yu, J. Yu, M. Lei and Y. Wang, Appl. Catal., B, 2013, 132–133, 452 CrossRef CAS PubMed.
- K. Fujihara, S. Izumi, T. Ohno and M. Matsumura, J. Photochem. Photobiol., A, 2000, 132, 99 CrossRef CAS.
- P. Zhao and K. Huang, Cryst. Growth Des., 2008, 8, 717 CAS.
- L. Wang and W. Wang, CrystEngComm, 2012, 14, 3315 RSC.
- A. Kar, S. Kundu and A. Patra, RSC Adv., 2012, 2, 10222 RSC.
- Z. Fang, Y. Liu, Y. Fan, Y. Ni, X. Wei, K. Tang, J. Shen and Y. Chen, J. Phys. Chem. C, 2011, 115, 13968 CAS.
- C. R. Jiao, D. M. Chen and J. F. Tong, Adv. Mater. Res., 2011, 412, 473 CrossRef.
- M. Guo, Y. Wang, Q. He, W. Wang, W. Wang, Z. Fu and H. Wang, RSC Adv., 2015, 5, 58633 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17679d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.