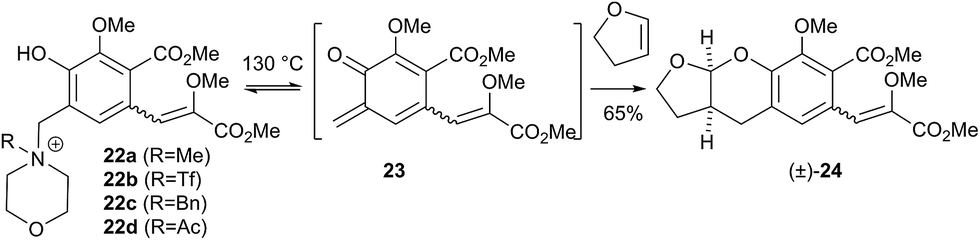
DOI:
10.1039/C5RA17108C
(Communication)
RSC Adv., 2015,
5, 80212-80215
An ortho-quinone methide based strategy towards the rubromycin spiroketal family†
Received
24th August 2015
, Accepted 9th September 2015
First published on 10th September 2015
Abstract
A method for the generation/in situ hetero-Diels–Alder cycloaddition of a trisubstituted ortho-quinone methide (o-QM) is described. Heating of ortho-hydroxybenzylamines was found to be a more effective strategy for the formation of o-QMs than use of the corresponding N-substituted quaternary salts hitherto employed. This method was used to synthesise the spiroketal core of the rubromycins.
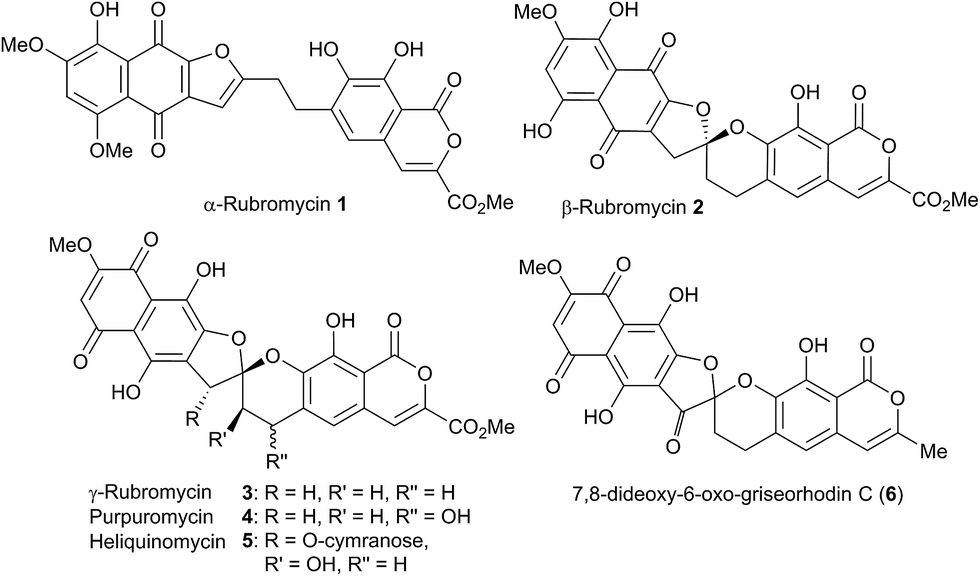
A number of spiroketal containing natural products exist where either one, or both of the rings is benzannelated.1 Amongst these, the rubromycin family 1–6 (Fig. 1) has received considerable interest from the synthetic community.2
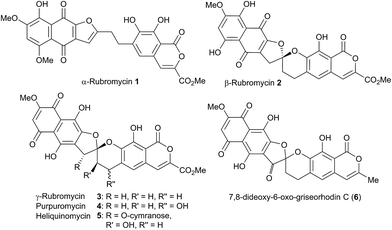 |
| | Fig. 1 Selected members of the rubromycin family. | |
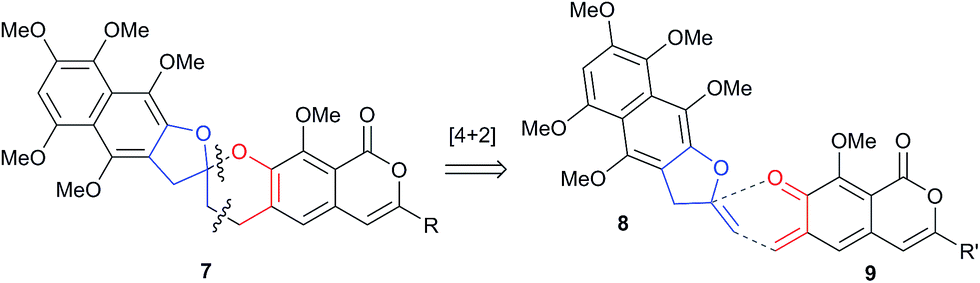
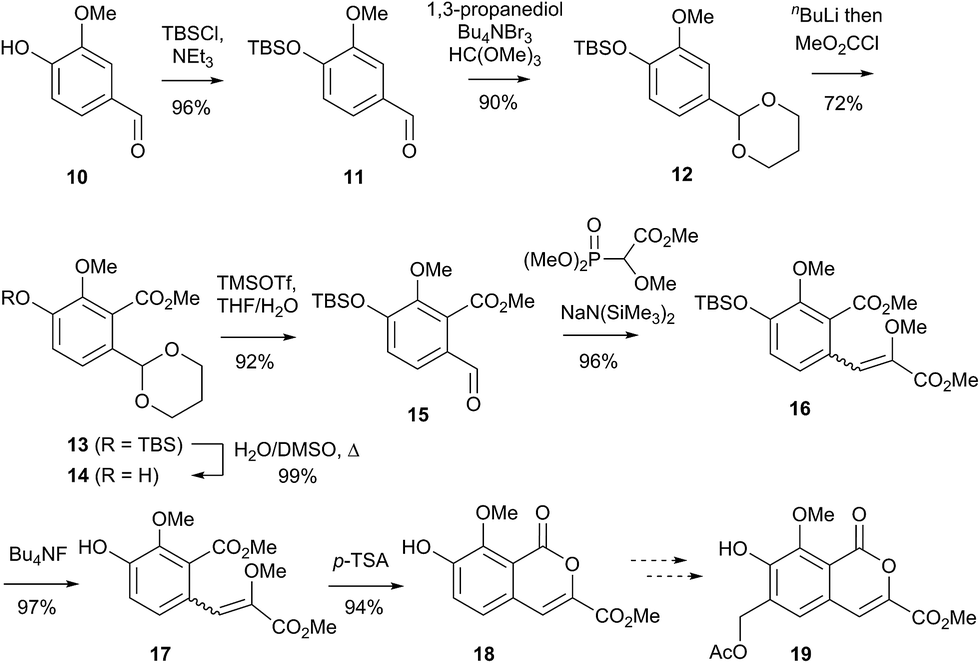
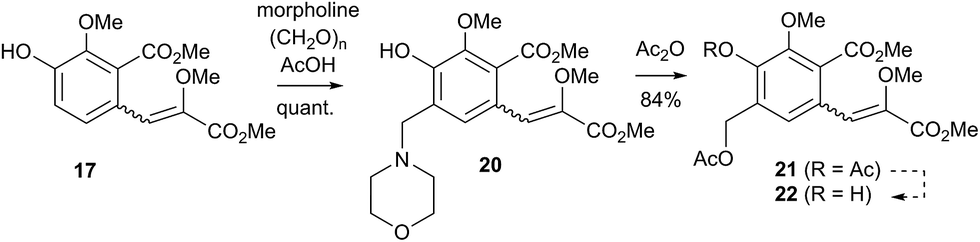
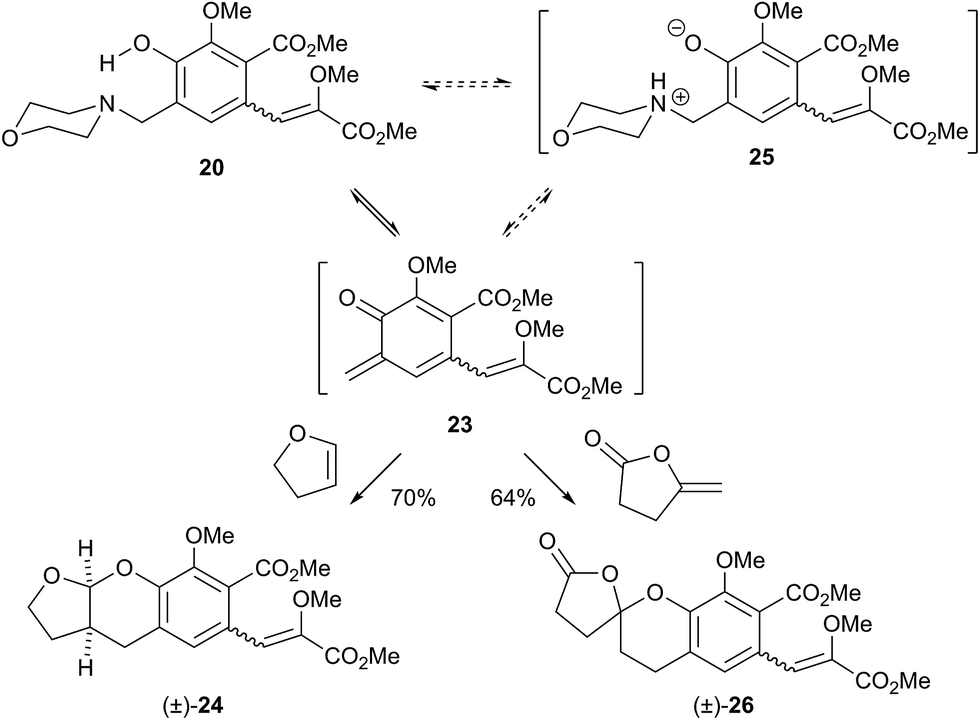
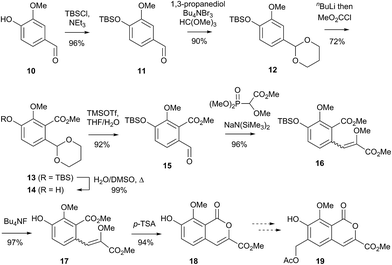
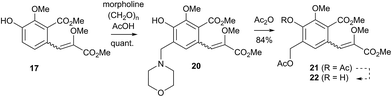
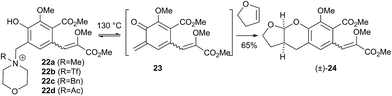
Initial curiosity of these molecules is presumably on account of their attractive biological activities, notably γ-rubromycin 3 is a telomerase and HIV-1 reverse transcriptase inhibitor at micromolar concentrations (IC50 = 2.64 and 19.9 μm respectively).3 In contrast α-rubromycin 1 is far less potent (IC50 > 200 μm in each case), testament to the pharmacological importance of the spirocyclic core. Moreover, the beguiling and challenging molecular architectures of the rubromycins has prompted the development of a variety of innovative cyclisation methods to be developed for the construction of the biologically key spirocyclic core.2 A number of successful total syntheses of γ-rubromycin 3 have been published based upon these.4 For a number of years we have been intrigued by the possibility of a general synthetic route to the rubromycin family via a hetero-Diels–Alder cycloaddition approach that would nominally bring together an enol ether 8 (masked naphthazarin) and an isocoumaryl ortho-quinone methide5 (o-QM) 9 (Scheme 1). Syntheses employing such a union would obviously be highly attractive in terms of overall brevity and subsequent analogue synthesis. To this end, we have previously reported on methods for the construction of benzannelated spirocycles generating an o-QM under basic conditions at low temperature6 as well as via thermolytic extrusion of AcOH7 from the same ortho-hydroxybenzyl acetate starting materials. Herein, we describe our synthesis of a fully elaborated o-QM precursor which has the potential for later conversion to the isocoumarin portion of the rubromycins and our efforts to employ this molecule in hetero-Diels–Alder reactions with enol ethers. Our synthesis began with conversion of vanillin (10) into the ester 13 (Scheme 2) according to the route of Reiβig8 via O-silylation of the phenol with tert-butyldimethylsilyl chloride followed by protection of the aldehyde as an acetal using 1,3-propanediol, HC(OMe)3 and n-Bu4NBr3 as catalyst.9 The subsequent ortho-lithiation of 12 according to the published procedure8 was found to be highly temperature dependant. Optimal conditions were found to require deprotonation at −6 °C followed by subsequent trapping of the aryllithium formed with MeOCOCl and warming to 10 °C to give the arylester 13 in 72% yield on >30 g scale (lit.8 56%). Next a method for the selective deprotection of the aldehyde was needed that retained the silyl-protecting group on the phenol. An exhaustive screen of conditions to achieve acetal deprotection were examined however initially success was in finding a method for selective removal of the silyl protecting group using refluxing DMSO/H2O to give phenol 14. We were subsequently drawn to a report by Mohan et al.10 describing how Bi(OTf)3 could be used as a mild reagent for the deprotection of acetals in THF/H2O. A second screening was therefore carried out using all the available metal triflates, present in the laboratory. Sn(OTf)2 was found to catalyse the desired hydrolysis, leaving the silyl-protecting group intact, to give benzaldehyde 15 in 65%. Conscious that this reaction might be being caused by adventitious TfOH we also examined the use of TMSOTf in THF/H2O. This far cheaper reagent combination led to the desired product in an improved 78% yield. Horner–Wadsworth–Emmons olefination of benzaldehyde 15 with (MeO)2P(O)CH(OMe)CO2Me11 gave the enol ethers 16 in 96% yield as a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diasteromers in favour of the Z-isomer, following which, removal of the silyl-protecting group was achieved with TBAF to give the phenols 17. Cyclisation of the phenols 17 was achieved using p-TSA to give the isocoumarin 18 present in the rubromycins in 94% yield. Our initial aim was then the conversion of isocoumarin 18 into the ortho-hydroxybenzyl acetate 19 which we hoped would be capable of generating an o-QM (cf. 9) as per our previous reported procedures.6,7 Multiple strategies were investigated including O-allylation/Claisen rearrangement and iodination/cross coupling. In each case whilst construction of a new C–C bond at C6 could be achieved, ultimate conversion to the ortho-hydroxybenzyl acetate 19 failed in each case. Attempted direct ortho-formylation via a myriad of known methods returned the starting material 18. Combined, these failures were in large part due to the extreme insolubility of many of the intermediates coupled with the low nucleophilicity of the aromatic ring and fragility of the isocoumarin. Aware that these issues might also prove problematic during any subsequent attempted o-QM formation from 19, we therefore turned our attention to the more soluble and stable phenol 17 and its conversion into a suitable ortho-hydroxybenzyl acetate 22. Once more a range of strategies were explored for the acetoxymethylenation of 17. The low nucleophilicity of the phenolic ring was again apparent with none of the attempted reactions leading to 19 but in contrast to our reactions with isocoumarin 18, Mannich reaction via the 4-methylenemorpholin-4-ium ion led smoothly to the amine 20 (Scheme 3). We then converted this to the diacetylated compound 21 through refluxing in Ac2O according to the procedure of Kozlowski,12 interestingly this process presumably proceeds via an o-QM (23). Selective hydrolysis of the phenolic acetate or methods to preferentially form the mono-acetylated compound 22 e.g. through refluxing in AcOH were sadly not forthcoming. ortho-Hydroxybenzylamines had previously been used as precursors for the generation of ortho-quinone methides via N-alkylation, acylation or oxidation followed either by heating or photolysis.5 In particular we were intrigued by the findings of Wilson and co-workers that N-methylmorpholinium salts can be converted to o-QMs at 80 °C and encouraged by their isolation of a spiroketal byproduct during their synthesis of (±)-xyloketal D.13 The amine 20 reacted with a range of electrophilic reagents (MeI, Tf2O, BnBr, AcCl) to give the quaternary ammonium salts 22a–d (Scheme 4).
:1 mixture of diasteromers in favour of the Z-isomer, following which, removal of the silyl-protecting group was achieved with TBAF to give the phenols 17. Cyclisation of the phenols 17 was achieved using p-TSA to give the isocoumarin 18 present in the rubromycins in 94% yield. Our initial aim was then the conversion of isocoumarin 18 into the ortho-hydroxybenzyl acetate 19 which we hoped would be capable of generating an o-QM (cf. 9) as per our previous reported procedures.6,7 Multiple strategies were investigated including O-allylation/Claisen rearrangement and iodination/cross coupling. In each case whilst construction of a new C–C bond at C6 could be achieved, ultimate conversion to the ortho-hydroxybenzyl acetate 19 failed in each case. Attempted direct ortho-formylation via a myriad of known methods returned the starting material 18. Combined, these failures were in large part due to the extreme insolubility of many of the intermediates coupled with the low nucleophilicity of the aromatic ring and fragility of the isocoumarin. Aware that these issues might also prove problematic during any subsequent attempted o-QM formation from 19, we therefore turned our attention to the more soluble and stable phenol 17 and its conversion into a suitable ortho-hydroxybenzyl acetate 22. Once more a range of strategies were explored for the acetoxymethylenation of 17. The low nucleophilicity of the phenolic ring was again apparent with none of the attempted reactions leading to 19 but in contrast to our reactions with isocoumarin 18, Mannich reaction via the 4-methylenemorpholin-4-ium ion led smoothly to the amine 20 (Scheme 3). We then converted this to the diacetylated compound 21 through refluxing in Ac2O according to the procedure of Kozlowski,12 interestingly this process presumably proceeds via an o-QM (23). Selective hydrolysis of the phenolic acetate or methods to preferentially form the mono-acetylated compound 22 e.g. through refluxing in AcOH were sadly not forthcoming. ortho-Hydroxybenzylamines had previously been used as precursors for the generation of ortho-quinone methides via N-alkylation, acylation or oxidation followed either by heating or photolysis.5 In particular we were intrigued by the findings of Wilson and co-workers that N-methylmorpholinium salts can be converted to o-QMs at 80 °C and encouraged by their isolation of a spiroketal byproduct during their synthesis of (±)-xyloketal D.13 The amine 20 reacted with a range of electrophilic reagents (MeI, Tf2O, BnBr, AcCl) to give the quaternary ammonium salts 22a–d (Scheme 4).
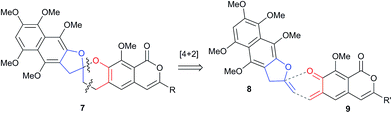 |
| | Scheme 1 Proposed cycloaddition based strategy to the rubromycins. | |
 |
| | Scheme 2 Efforts towards a fully elaborated o-QM precursor. | |
 |
| | Scheme 3 Synthesis of a fully elaborated o-QM precursor. | |
 |
| | Scheme 4 Proof of o-QM formation. | |
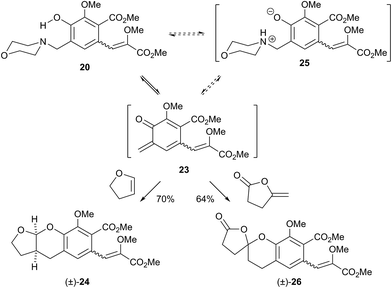
Pleasingly when the iodomethane adduct 22a was heated to 130 °C with 2,3-dihydrofuran (DHF), the fused acetal (±)-24 was formed in 65% yield proving the intermediacy of the desired o-QM 23. However a similar cycloaddition with γ-methylene-γ-butyrolactone did not give any of the expected7 spirocycle. Furthermore we then attempted to induce the formation of 23 under our previously reported low temperature anionic conditions6 in conjunction with DHF, since simple exo-enol ethers (cf. those akin to 8) readily rearrange to their endo-cyclic isomers via thermal or protic conditions. Warming this reaction mixture from −78 → 20 °C after the addition of i-PrMgCl as base did not lead to the expected acetal (±)-24 but instead to complex mixtures. Similar results were obtained with the salts 23b–d even though we examined a large range of bases, temperatures and times. Unalkylated ortho-hydroxybenzylamines had previously been reported as precursors for o-QMs albeit only under photochemical conditions at room temperature or at vastly elevated temperatures.5,14 For completeness we investigated if the amine 20 also formed o-QMs under thermal only conditions. This was indeed found to be the case. When the amine 20 was heated with DHF at 130 °C the fused acetal (±)-24 was formed in an improved 70% yield (Scheme 5). Moreover when γ-methylene-γ-butyrolactone was used as the 2π partner, the desired spirocycle (±)-26 was formed in 64% yield. This suggests that contrary to current thinking, the unalkylated amine is a more effective precursor for forming an o-QM (23) than the corresponding N-alkylated one. This indicates that protonation of the amine by the phenol is sufficient to initiate elimination. It is not clear if protonation is a discrete event (i.e. 20 → 25) in the formation of an o-QM or if the whole process is more concerted, maybe even in a pericyclic like-manner (i.e. 20 → 23).
 |
| | Scheme 5 Synthesis of a spiroketal via o-QM cycloaddition. | |
Conclusions
In conclusion we have synthesised a highly decorated ortho-hydroxybenzylamine 20 that is capable of forming an ortho-quinone methide 23. Notable in the synthesis of 20 is a method for the selective removal of a tert-butyldimethylsilyl group from a phenol in the presence of a 1,3-diol protected aldehyde. Orthogonal removal of this acetal protecting group without deprotection of the phenol was achieved using TMSOTf in THF/H2O. These selective deprotection methods may prove useful in other syntheses. Finally, the ortho-hydroxybenzylamine 20 can be alkylated and heated with DHF to form a fused acetal (±)-24, however of greater note is the fact that in lieu of alkylation, the amine 20 appears to function as a more effective o-QM precursor. Cycloaddition with a model 2π-partner has given a spiroketal (±)-26 similar to that found in the rubromycins. Future work will investigate this o-QM generation method and adapt this cycloaddition protocol to incorporate the entire rubromycin carbon skeleton.
Acknowledgements
We thank the EPSRC for a studentship (to N.W.) under grant EP/G041431/1 and the EPSRC UK National Mass Spectrometry Facility at Swansea University.
Notes and references
-
(a) J. Sperry, Z. E. Wilson, D. C. K. Rathwell and M. A. Brimble, Nat. Prod. Rep., 2010, 27, 1117 RSC;
(b) M. Wilsdorf and H.-U. Reissig, Angew. Chem., Int. Ed., 2012, 51, 9486 CrossRef CAS PubMed.
-
(a) M. Brasholz, S. Sörgel, C. Azap and H. Reiβig, Eur. J. Org. Chem., 2007, 3801 CrossRef CAS PubMed;
(b) D. J. Atkinson and M. A. Brimble, Nat. Prod. Rep., 2015, 32, 811 RSC.
-
(a) M. E. Goldman, G. S. Salituro, J. A. Bowen, J. M. Williamson, D. L. Zink, W. A. Schleif and E. A. Emini, Mol. Pharmacol., 1990, 38, 20 CAS;
(b) M. Chino, K. Nishikawa, A. Yamada, M. Ohsono, T. Sawa, F. Hanaoka, M. Ishizuka and T. Takeuchi, J. Antibiot., 1998, 51, 480 CrossRef CAS;
(c) C. Puder, S. Loya, A. Hizi and A. Zeeck, Eur. J. Org. Chem., 2000, 729 CrossRef CAS;
(d) Y. Mizushina, T. Ueno, M. Oda, T. Yamaguchi, M. Saneyoshi and K. Sakaguchi, Biochim. Biophys. Acta, 2000, 1523, 172 CrossRef CAS;
(e) T. Ueno, H. Takahashi, M. Oda, M. Mizunuma, A. Yokoyama, Y. Goto, Y. Mizushina, K. Sakaguchi and H. Hayashi, Biochemistry, 2000, 39, 5995 CrossRef CAS PubMed;
(f) E. P. M. T. Cohn, K. L. Wu, T. R. R. Pettus and N. O. Reich, J. Med. Chem., 2012, 55, 3678 CrossRef CAS PubMed;
(g) T. Yuen, Y. Ng, F. C. F. Ip, J. L. Chen, D. J. Atkinson, J. Sperry, Y. Nancy and M. A. Brimble, Aust. J. Chem., 2013, 66, 530 CAS;
(h) N. Nishiya, Y. Oku, Y. Kumagai, Y. Sato, E. Yamaguchi, A. Sasaki, M. Shoji, Y. Ohnishi, H. Okamoto and Y. Uehara, Chem. Biol., 2014, 21, 530 CrossRef CAS PubMed;
(i) C. E. P. Bernardo and P. J. Silva, PeerJ, 2014, 2, e470, DOI:10.7717/peerj.470.
-
(a) S. Akai, K. Kakiguchi, Y. Nakamura, I. Kuriwaki, T. Dohi, S. Harada, O. Kubo, N. Morita and Y. Kita, Angew. Chem., Int. Ed., 2007, 46, 7458 CrossRef CAS PubMed;
(b) D. C. K. Rathwell, S. H. Yang, K. Y. Tsang and M. A. Brimble, Angew. Chem., Int. Ed., 2009, 48, 7996 CrossRef CAS PubMed;
(c) K.-L. Wu, E. V. Mercado and T. R. R. Pettus, J. Am. Chem. Soc., 2011, 133, 6114 CrossRef CAS PubMed;
(d) L. Wei, J. Xue, H. Liu, W. Wang and Y. Li, Org. Lett., 2012, 14, 5302 CrossRef CAS PubMed;
(e) M. Wilsdorf and H.-U. Reiβig, Angew. Chem., Int. Ed., 2014, 53, 4332 CrossRef CAS PubMed;
(f) W. Wang, J. Xue, T. Tian, J. Zhang, L. Wei, J. Shao, Z. Xie and Y. Li, Org. Lett., 2013, 15, 2402 CrossRef CAS PubMed.
-
(a) R. W. van de Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367 CrossRef CAS;
(b) S. E. Rokita, Quinone Methides, Wiley, Hoboken, N.J., 2009 Search PubMed;
(c) T. P. Pathak and M. S. Sigman, J. Org. Chem., 2011, 76, 9210 CrossRef CAS PubMed;
(d) N. J. Willis and C. D. Bray, Chem.–Eur. J., 2012, 18, 9160 CrossRef CAS PubMed;
(e) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655 CrossRef CAS PubMed;
(f) M. S. Singh, A. Nagaraju, N. Anand and S. Chowdhury, RSC Adv., 2014, 4, 55924 RSC.
-
(a) C. Bray, Org. Biomol. Chem., 2008, 6, 2815 RSC. See also:
(b) M. A. Marsini, Y. D. Huang, C. C. Lindsey, K. L. Wu and T. R. R. Pettus, Org. Lett., 2008, 10, 1477 CrossRef CAS PubMed.
- C. Bray, Synlett, 2008, 16, 2500 Search PubMed.
- M. Brasholz, X. Luan and H. Reiβig, Synthesis, 2005, 20, 3571 CrossRef.
- R. Gopinath, S. J. Haque and B. K. Patel, J. Org. Chem., 2002, 67, 5842 CrossRef CAS PubMed.
- M. C. Carrigan, D. Sarapa, R. C. Smith, L. C. Wieland and R. S. Mohan, J. Org. Chem., 2002, 67, 1027 CrossRef CAS PubMed.
- V. Guay and P. Brassard, Synthesis, 1987, 3, 294 CrossRef.
- A. N. Lowell, P. D. Wall, S. P. Waters and M. C. Kozlowski, Tetrahedron, 2010, 66, 5573 CrossRef CAS PubMed.
-
(a) J. D. Pettigrew, R. P. Freeman and P. D. Wilson, Can. J. Chem., 2004, 82, 1640 CrossRef CAS;
(b) J. D. Pettigrew, J. A. Bexrud, R. P. Freeman and P. D. Wilson, Heterocycles, 2004, 62, 445 CrossRef CAS.
-
(a) K. Nakatani, N. Higashida and I. Saito, Tetrahedron Lett., 1997, 38, 5005 CrossRef CAS;
(b) W. Chen, Y. Kang, R. G. Wilde and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5179 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1420253. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra17108c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a