
Novel FeOx–polyethylene transparent films: synthesis and mechanism of surface regeneration
 *a
*a
Abstract
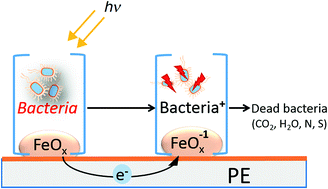
The first evidence for the synthesis of a uniform, adhesive polyethylene–FeOx (PE–FeOx) surface leading efficiently to bacterial inactivation is addressed in this study. PE was loaded with 0.04–0.08% Fe wt/wt PE after RF-plasma pretreatment was required to increase the active sites/polarity and roughness to adhere FeOx on PE. The repetitive bacterial inactivation proceeded in a stable way for several cycles. The oxidative radicals leading to bacterial inactivation under aerobic/anaerobic conditions were investigated by the use of appropriate scavengers. By X-ray photoelectron spectroscopy (XPS) and diffuse reflection spectroscopy (DRS) the changes on the PE–FeOx oxidation states and spectroscopic features during bacterial inactivation were monitored. The regeneration of the initial Fe-oxidation state and consequently of the initial Fe-oxidation state in the PE–FeOx was possible and followed by DRS. Inductive plasma coupled mass spectrometry (ICP-MS) indicated that only sub-ppb levels of Fe were released from the PE–FeOx surface within the reaction time.
 Please wait while we load your content...
Please wait while we load your content...

