DOI:
10.1039/C5RA16859G
(Paper)
RSC Adv., 2015,
5, 91829-91835
Preparation of Rh/C and its high electro-catalytic activity for ethanol oxidation in alkaline media
Received
20th August 2015
, Accepted 18th October 2015
First published on 19th October 2015
Abstract
The ethanol electrooxidation behaviors on Rh/C in alkaline media were studied compared with that on Pd/C. The Rh/C and Pd/C catalysts were synthesized by a microwave heating-glycol reduction method and characterized by X-ray diffraction (XRD) and transmission electron microscopy (TEM). The electrocatalytic activities of the catalysts were investigated by cyclic voltammetry (CV), chronoamperometry (CA), electrochemical impedance spectroscopy (EIS) and single cell discharge testing methods. The results showed that Rh/C presented higher activity and better stability toward ethanol oxidation than Pd/C. On the CV plots, it was observed that the onset potential of oxidation on Rh/C shifted negatively and the backward peak current on Rh/C decreased as compared to that of Pd/C. Intermediate species are produced during the oxidation process, as revealed by the complex impedance spectra. Rh/C presented higher tolerance for surface poisoning than Pd/C, which can be reflected by the negative impedance on the Pd/C electrode. The selectivity for ethanol oxidation to CO2 (existing as CO32− in alkaline media) on Rh/C and Pd/C electrodes were determined to be as low as 10.07% and 2.18%, respectively. Nevertheless, the ability of Rh for breaking the C–C bond in ethanol is still slightly higher than that of Pd under the same conditions.
1. Introduction
The Direct Alcohol Fuel Cells (DAFCs) are considered as a promising technology for transportation and portable electronic devices.1 Ethanol is environmental friendly and may serve as a potential alternative due to its higher energy density than methanol.2 Pt and Pt-based catalysts have been extensively investigated as electro-catalysts for oxidation of methanol and ethanol,3–10 and significant progress has been made in their development. For example, Xin et al.4,5 developed a highly active PtSn catalyst for the ethanol oxidation reaction (EOR). However, the high price and limited supply of Pt constitute a major barrier to the development of DAFCs. Therefore, there is a desire to develop low cost non-platinum electro-catalysts with comparable or improved kinetics towards the EOR.11,12 Among the non-platinum catalysts, Pd and Rh are suitable low-cost transition metals and shown marked superiority over Pt in terms of oxidation rate and electrode stability.13–21 Shen et al.13–15 prepared a series of Pd-based catalysts and found that Pd catalysts exhibited higher activity and stability than did Pt catalysts in the EOR in alkaline solutions. However, Pd is easily poisoning by intermediate products and difficult to break C–C bond.22,23 Therefore, a significant challenge in the development of DAFC technology is the need for highly active catalysts for the EOR that involve complete oxidation per ethanol molecule to CO2 releasing 12 electrons and the cleavage of the C–C bond.24 In recent years, Rh based catalysts have been investigated by several research groups. For example, de Souza et al.21 found that PtRh combined in bimetallic surfaces for ethanol oxidation decreases the acetaldehyde yield, compared to pure platinum on Pt73Rh27 and Pt55Rh45. Kowal et al.,19,20 synthesized PtRhSnO2/C electrocatalysts by depositing Pt and Rh atoms on Sn oxides nanoparticles. The authors argue that these catalysts are efficient to cleave the C–C bond of ethanol at room temperature and cause its predominant oxidation to CO2 at very low overpotentials compared to platinum. In another study, Q. He et al.25 reported that RhCeO2/C has better activity for ethanol oxidation and Stephen K. Murphy et al.26 reported that a Rhodium (Xantphos) (benzoate) catalyst activates aldehyde carbon–hydrogen (C–H) bonds with high chemoselectivity to trigger carbon–carbon (C–C) bond cleavage. However, the information and the understanding of the activity of Rh-based electrocatalysts for ethanol electro-oxidation are still very scarce. Also, little is known about the mechanism of the ethanol oxidation reaction on Rh catalysts in alkaline media. The present work investigates the electro-oxidation behaviors of Rh/C by compared to Pd/C. The differences of EIS patterns of the ethanol oxidation on Rh/C and Pd/C and the selectivity for ethanol oxidation to CO2 will be discussed.
2. Experimental
2.1 Catalyst preparation and characterization
All chemicals used were of analytical grade, and all solutions were freshly prepared in deionized water. Rh/C catalyst was synthesized by the intermittent microwave heating-glycol reduction method. Firstly, 2.5 mL of 0.05 M RhCl3 aqueous solution was added to the 100 mL reactor containing 50 mL glycol, 2 mL of 1.0 M KOH solution, and 0.24 g Vulcan XC-72 carbon powder (Cabot Corp. USA). After that, the mixed solution was changed into carbon paste by ultrasonic concussion and moderately heated in a microwave oven (900 W, 2.45 GHz, Galanz, WD900ASL23-2, China). The reaction product was filtered and washed extensively with water and ethanol. The residue was dried at 90 °C for 24 h. Pd/C was prepared by using the same method for comparing the performance of different catalysts. The electrodes containing different catalysts were prepared using 60% PTFE emulsion as bonder and foamed nickel as the electric collector. The loading of each electrode was 0.1 mg cm−2.
Structure and morphology of the catalyst was investigated using XRD and TEM. The XRD patterns were generated by the 2θ scan with the scanning rate of 8° min−1 and the angular resolution of 10–80°. TEM was taken through a JEOL-1230 microscope, operating at the applied voltage of 200 kV.
2.2 Electrochemical measurements
Electrochemical measurements were carried out using CHI 760D (Chen Hua, Shanghai, China) electrochemistry station in a conventional three-electrode cell with a Pt coil (counter electrode), a saturated Hg/HgO electrode (reference electrode) and a prepared electrode (working electrode). The CV, CA and EIS were collected in 1.0 M KOH and 1.0 M C2H5OH solution. The CV tests were conducted at a 10 mV s−1 with potential ranging from −0.8 to 0.4 V. The EIS were recorded in the constant potential mode with the amplitude of 50 mV by scanning frequency from 105 to 0.1 Hz.
2.3 Single cell discharge testing
The discharge testing of single cell was carried out to further elucidate the performance of Pd/C and Rh/C (Pd and Rh loading of 0.1 mg cm−2) on the ethanol oxidation in an anion-exchange membrane DEFC, using a Neware charge–discharge device (Shenzhen Neware Corporation, China) at room temperature. The anode was Pd/C and Rh/C electrodes, while the cathode was the gas diffusion electrode which involves diffusion layer (acetylene black) and activation layer (MnO2/C). The redox reaction area on both electrodes was 2 × 2 cm2, the current density of discharge was 15, 20, and 30 mA cm−2. The anodic electrolyte was the mixed solution of 3.0 M C2H5OH and 6.0 M KOH, accordingly, the cathodic electrolyte was 6.0 M KOH solution.
2.4 Estimation of reaction product
The CO2 ion selective electrode was used to detect CO2 concentration in the solution after single cell discharge testing. CO2 ion selective electrode was only responsive to CO2 and different concentration of CO2 corresponds to different potential values (E). In this work, NaHCO3 was used as a standard solution. First, 10−5, 10−4, 10−3, 10−2, 10−1 M NaHCO3 solutions were prepared. Secondly, adding hydrochloric acid to the solutions and adjusting pH < 3.8 respectively, CO32− was transformed to CO2 completely and detected by CO2 ion selective electrode, thus E can be obtained. Then obtained E and −lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C(CO2) can be used to draw a standard curve. Finally, under the same condition, E of the sample solutions, i.e., Pd/C electrolyte solution, Rh/C electrolyte solution and blank group, was measured to obtain the CO2 concentrations from the standard curve. To be specific, Pd/C electrolyte solution was obtained by the mixed solution of 3.0 M C2H5OH and 6.0 M KOH after the single cell discharge of AEM DEFC using Pd/C electrolyte for 3 h, while Rh/C electrolyte solution was gotten in the same way. Blank group was achieved by laying the mixed solution of 3.0 M C2H5OH and 6.0 M KOH in electrolytic cell for 3 h. All the text was carried out at room temperature.
C(CO2) can be used to draw a standard curve. Finally, under the same condition, E of the sample solutions, i.e., Pd/C electrolyte solution, Rh/C electrolyte solution and blank group, was measured to obtain the CO2 concentrations from the standard curve. To be specific, Pd/C electrolyte solution was obtained by the mixed solution of 3.0 M C2H5OH and 6.0 M KOH after the single cell discharge of AEM DEFC using Pd/C electrolyte for 3 h, while Rh/C electrolyte solution was gotten in the same way. Blank group was achieved by laying the mixed solution of 3.0 M C2H5OH and 6.0 M KOH in electrolytic cell for 3 h. All the text was carried out at room temperature.
3. Results and discussion
3.1 XRD and TEM
XRD pattern of the Rh/C is shown in Fig. 1. The broad diffraction peak at about 25° is ascribed to Vulcan XC-72 carbon. The XRD reflections showed face-centered cubic lattice structures with diffraction peaks at Bragg angles of 2θ = 42, 46.5, 72.121°, corresponding to the (111), (200), (220) planes of the Rh metallic phase, respectively. The average crystal size of Rh is about 3.8 nm, which is calculated by using the Scherrer equation from XRD patter, indicating that Rh nanoparticles dispersed on Vulcan XC-72 could be very small. TEM image was obtained to provide more information on particle size and distribution. Fig. 2 shows the TEM image of Rh/C particle. As seen clearly in Fig. 2, the Rh/C particles are uniform and well distributed on the carbon support, while the average particle size of Rh/C from TEM image is 3–5 nm, which is consistent with the XRD results.
 |
| | Fig. 1 XRD pattern of Rh/C. | |
 |
| | Fig. 2 TEM image of Rh/C. | |
3.2 Cyclic voltammetry
Fig. 3(a) shows CV curves of Rh/C catalyst in 1.0 M KOH and 1.0 M KOH + 1.0 M C2H5OH solutions at room temperature, respectively. In absence of ethanol, three potential peaks can be observed in the forward scan, which correspond to different electrochemical processes occurring over the electrode surface. The peak on Rh/C that appears at potentials range between −0.8 and −0.6 V is associated with the oxidation of absorbed and desorbed hydrogen. The second peak on Rh/C, which emerges in −0.45 V, can be attributed to the adsorption of OH−. The third peak occurs at potentials above 0.3 V on Rh/C catalyst. It can be attributed to the formation of the rhodium oxide layer on the surface of the catalyst. The negative current observed during the backward scan suggests a reduction process, probably reduction of oxygen. When add ethanol to the solution, the hydrogen absorbed/desorbed region is significantly suppressed in the presence of ethanol in the solution. The ethanol oxidation reaction starts at −0.78 V and a current peak centered at −0.20 V is observed during the forward scan. In the reverse scan, another current peak is found centered at −0.59 V. Fig. 3(b) shows the CV curves of ethanol oxidation on Pd/C and Rh/C catalysts. In the forward scan, peaks current are related to the oxidation of freshly chemisorbed species derived from ethanol adsorption. In the reverse scan, peaks are associated with the removal of carbonaceous species that are not completely oxidized in the positive scan, rather than the oxidation of freshly chemisorbed species.27–29 It is worth noting that, in the reverse scan, on the Pd/C at −0.18 V, a sharp increasing current (about 90 mA) is seen, while the current of Rh/C (about 22 mA) increases slowly and its current density is much smaller than Pd/C, indicating that the adsorption degree is less serious than Pd/C. The onset potential on Rh/C (about −0.78 V) is shifted negatively as much as 0.28 V as compared to that on Pd/C (about −0.5 V), indicating better activity of the Rh/C catalyst for ethanol oxidation. The anodic onset potential is important parameter for evaluating the activity of catalysts during the electro-oxidation process. In addition, one oxidation peak along with a shoulder for Rh/C can be seen (around −0.08 V), indicating that the products may not be the same in the case of Pd/C. According to the published articles that Rh presents excellent selectivity for complete conversion of ethanol to CO2.25,26 Therefore, the shoulder oxidation peak may be due to the oxidation of intermediates from decomposition of ethanol.
 |
| | Fig. 3 Cyclic voltammograms for (a) Rh/C electrode in 1.0 M KOH and 1.0 M KOH + 1.0 M C2H5OH; (b) Pd/C and Rh/C electrodes in 1.0 M KOH + 1.0 M C2H5OH. The catalysts loading are all 20 μg cm−2, and the potential sweep rate is 10 mV s−1. | |
3.3 Chronoamperometry
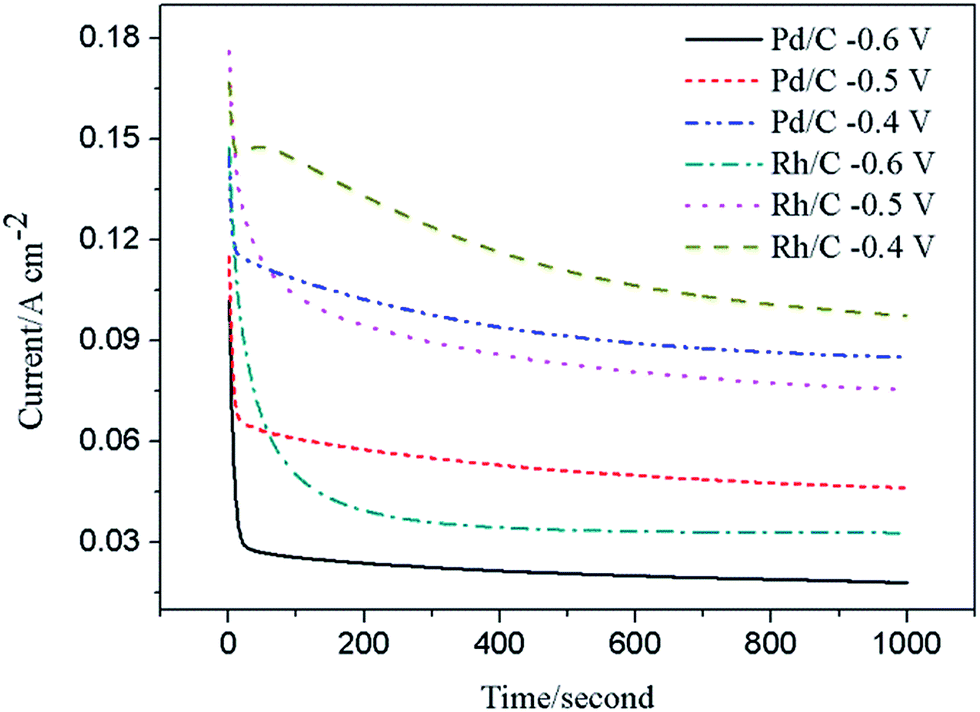
Chronoamperometric experiment was carried out to observe the stability and possible poisoning of the catalyst under short-time continuous operation. Fig. 4 provides the CA curves under various potentials for Rh/C and Pd/C catalysts in a 1.0 M KOH + 1.0 M C2H5OH solution after 1000 s. It can be clearly seen that, at low potentials from −0.6 to −0.4 V, the current densities on Rh/C electrode are higher than that on Pd/C electrode, indicating a higher catalytic activity of Rh/C for ethanol oxidation compared with Pd/C, which is consists with our CV results (Fig. 3(b)).
 |
| | Fig. 4 Chronoamperograms for Rh/C and Pd/C electrodes in 1.0 M KOH + 1.0 M C2H5OH at the potentials from −0.6 to −0.4 V. | |
3.4 Mechanism investigation of ethanol oxidation by EIS
To provide more insight into the ethanol oxidation behavior on the Rh/C electrodes, EIS is used in this study to investigate the kinetics of ethanol oxidation. EIS has been shown to be a sensitive electrochemical technique for the studies on the electro-oxidation kinetics of formic acid oxidation on Pd/C30 and methanol oxidation on Pt/C.31 Complex impedance plots of the formic acid oxidation on Pd/C catalysts at different applied potentials are shown in Fig. 4. At potentials below −0.1 V, as shown in Fig. 5(a), impedance plots occur in the first quadrant and the diameters of the arcs decrease with the increase of potentials, indicating a decreasing polarization resistance with the increase of potential. At −0.40 V, the arc become very small, indicating faster electron-transfer kinetics at higher potentials. However, with the increase of potentials above −0.30 V, the impedance plot starts becoming larger than −0.4 V in Fig. 5(b), suggesting the surface with little activity for ethanol oxidation. Here in the ethanol oxidation system, the phenomenon could be caused by the inhibiting effect of carbonaceous species that are not completely oxidized. At potential between 0 and 0.3 V (Fig. 5(c)), the spectra become small again and the diameter of the arc decreases with the increases of potential, indicating a new electrochemical reaction kicks in at this higher potential regime.30
 |
| | Fig. 5 Complex impedance plots for 1.0 M KOH + 1.0 M C2H5OH oxidation on Rh/C catalyst at room temperature as a function of potential (a) between −0.6 and −0.4 V; (b) between −0.3 and −0.1 V; (c) between 0 and 0.3 V. | |
In order to discuss the ethanol oxidation mechanism on different catalytic surfaces, the impedance plots of ethanol oxidation on Pd/C were compared with that on Rh/C. Fig. 6 shows the complex impedance plots of ethanol oxidation on Pd/C. As shown in Fig. 6(a), the impedance patterns on Pd/C are similar to those on Rh/C at potentials below −0.1 V. However, at −0.1 V, the impedance plot starts transiting from the first quadrant to the second quadrant in Fig. 6(b). Impedance plot in the second quadrant indicates the polarization resistance is negative and that is to say the steady state oxidation current decreases with the increase of polarization potential. This phenomenon observed previously in the electro-oxidation of methanol on Pt32 and formic acid on Pd/C.30 In their study, the negative impedance behaviors are usually attributed to the adsorption of reaction intermediates on the catalyst surface. In this work, the negative resistance could be caused by the heavy intermediates adsorption on the Pd surface and inhibits the reaction of ethanol oxidation by blocking the active surface of Pd/C electrode with the associated loss of surface active sites. It should be noted that all the impedance spectra obtained on Rh/C electrode are located within the first quadrant, suggesting the ability of resist poisoning of Rh/C is stronger than Pd/C. In addition, the reaction mechanism on both catalysts may different and it will be discussed in detail later.
 |
| | Fig. 6 Complex impedance plots for 1.0 M KOH + 1.0 M C2H5OH oxidation on Pd/C catalyst at room temperature as a function of potential (a) between −0.6 and −0.2 V; (b) between −0.1 and 0.05 V; (c) between 0.1 and 0.4 V. | |
Based on the voltammetric and impedance results, the equivalent circuit shown in Fig. 7 was used to fit the EIS data. Fig. 7(a) depicts the equivalent circuit for the electrodes that exhibit normal impedance behaviors on Rh/C and Pd/C electrodes. Constant-phase element (CPE) is the double-layer capacitance. RS and RCT are the solution resistance and charge-transfer resistance, respectively. For the special impedance behaviors observed on Rh/C electrode at −0.3 to −0.1 V and −0.1 to −0.05 V on Pd/C electrode, the equivalent circuit is shown in Fig. 7(b). C1 and R1 represent the capacitance and resistance of the electrooxidation of adsorbed intermediates. RCT, an indicative of the rate of electrochemical reaction, is always a positive number and its value can be determined from an impedance arc at a low frequency domain. From the fitting, the variation of the RCT with electrode potentials was evaluated, which was depicted in Fig. 8. First, negative RCT was only obtained on the Pd/C electrode at potentials between from −0.1 to 0.05 V, due to the strong surface-adsorbed carbonaceous species. It can be noted that RCT on Rh/C electrode increases with increasing potential between from −0.3 to −0.1 V, and not obtained negative RCT, indicating that Rh/C has excellent carbonaceous species tolerance. Second, RCT for ethanol oxidation on Pd/C electrode is much larger than that on the Rh/C electrode, suggesting that Rh/C display better catalytic activity because of its fast reaction kinetics.
 |
| | Fig. 7 Equivalent circuits for electrooxidayion of ethanol: (a) for normal impedance, and (b) for special impedance. | |
 |
| | Fig. 8
R
CT of ethanol electrooxidation at Rh/C and Pd/C electrodes. | |
3.5 Single cell performance
Fig. 9 shows the constant-current discharging curve of the oxidation of ethanol in an anion-exchange membrane direct ethanol fuel cell (AEM DEFC) that consists of Rh/C (or Pd/C) anode, an AEM, and a MnO2/C cathode at room temperature. The constant-current discharging tests are carried out by varying the current density at 15, 20 and 30 mA cm−2. It can be observed that the cell voltage of the fuel cell decreases with the current density; for instance, in Rh/C electrode, when the current density increases from 15, 20, to 30 mA cm−2, the cell voltage decreases from 0.57, 0.55 to 0.45 V, respectively. However, the cell voltages for Pd/C are 0.41, 0.37 and 0.32 V respectively, which are much lower than Rh/C under the same current density. In addition, it is clear from Fig. 6 that the cell voltage for Rh/C is more stable than that of Pd/C under the same current density, which further shows that Rh/C electrode exhibits much better catalytic activity and stability than Pd/C.
 |
| | Fig. 9 Comparison of the single AEM DEFC constant-current discharging curves respectively using Pd/C and Rh/C electrodes as the anode at room temperature. | |
3.6 Oxidation product analysis through CO2 ion selective electrode
The CO2 ion selective electrode is merely responsive and very sensitive to CO2 and it displays different potential values in the solution with different CO2 concentrations. As shown in Table 1, different concentrations of NaHCO3 solution ranging from 10−5 to 10−1 M correspond to different E. Then fitting E and −lgC(CO2) of different concentrations, a standard curve can be obtained as shown in Fig. 10. It can be observed that E and −lgC(CO2) are in linear relationship, thus a standard eqn (1) can be achieved:| | | E = −199.8 + 48.2lgC | (1) |
Table 1 The E value of NaHCO3 standard solution
|
C
(NaHCO3) (mol L−1) |
−lgC(CO2) |
E (mV) |
| 10−5 |
5 |
−443 |
| 10−4 |
4 |
−390 |
| 10−3 |
3 |
−344 |
| 10−2 |
2 |
−296 |
| 10−1 |
1 |
−249 |
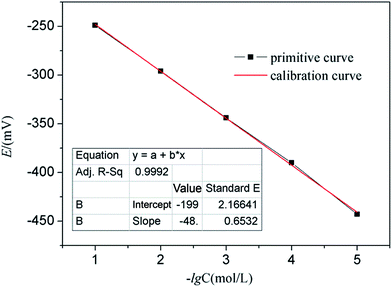 |
| | Fig. 10 Standard curve of NaHCO3 solution. | |
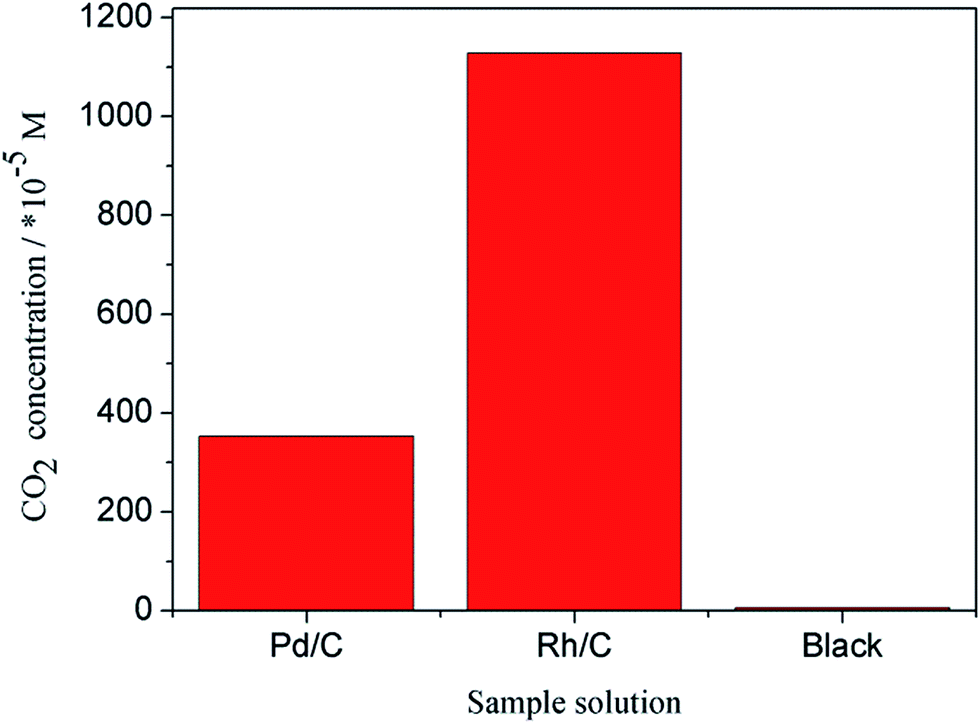
In this investigation, CO2 ion selective electrode is used to detect CO2 concentration in the three sample solutions and the data are tabulated in Table 2. As shown in Table 2, the E of Pd/C, Rh/C and blank group is −308, −278 and −402, respectively. Substituting E into eqn (1), the C(CO2) can be obtained. And n(CO2) can be calculated by the volume of the three sample solutions. As shown in Table 2 and Fig. 11, the CO2 concentration in black group is very low, while that in the group Pd/C and Rh/C are higher than black group. The reason is that ethanol is converted to CO2 by catalytic oxidation. It also be noted that the CO2 concentration in Rh/C group (1.628 × 10−2 M) is higher than that of Pd/C (3.529 × 10−3 M), indicating that Rh/C has better catalytic ability. By further calculation, the selectivity for ethanol oxidation to CO2 on the Pd/C and Rh/C are 2.18% and 10.07%, respectively.
Table 2 The data of sample solutions
| Sample solution |
Volume (mL) |
E (mV) |
C
(CO2) (mol L−1) |
n
(CO2) (mol) |
| Pd/C |
2.5 |
−318 |
3.529 × 10−3 |
8.824 × 10−6 |
| Rh/C |
2.5 |
−286 |
1.628 × 10−2 |
4.069 × 10−5 |
| Blank group |
2.5 |
−402 |
6.382 × 10−5 |
1.596 × 10−7 |
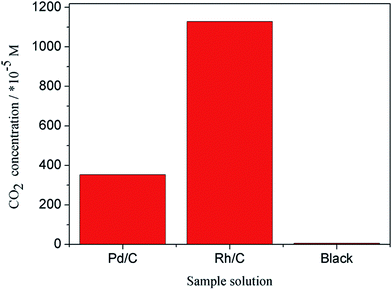 |
| | Fig. 11 CO2 concentration in sample solutions. | |
A dual-path mechanism for ethanol oxidation in alkaline media was proposed.23,33 In the C2-pathway, the C–C bond of ethanol remains intact upon oxidation. Ethanol is converted to acetaldehyde and eventually to acetic acid/acetate. In the C1-pathway, the C–C bond can be broken in ethanol or acetaldehyde, leading eventually to carbon dioxide (or (bi)carbonate). For the Pd/C, the selectivity for ethanol oxidation to CO2 on the Pd/C is only 2.18% and much less than that on the Rh/C (10.07%). This suggests that the ability of Pd for breaking C–C bond in ethanol to produce CO2 is very limited. Therefore, the C2-pathway will be considered on the Pd/C. This assumption is reasonable considering that the selectivity for ethanol oxidation to CO2 on the Pd/C was determined to be as low as 2.5% by the FTIR experiments.22 The selectivity for ethanol oxidation to CO2 on the on the Rh/C is five times as much as Pd/C, indicating Rh/C catalyst has a better capability for breaking the C–C bond cleavage. The oxidation ethanol on the Rh/C can be contributed via both the C1-pathway and the C2-pathway.
4. Conclusions
In this work, Rh/C and Pd/C were synthesized by microwave heating-glycol reduction method. Electrochemical evaluation shows that Rh/C catalyst has a higher activity and stability towards ethanol oxidation in alkaline media at room temperature. As compared to Pd/C electrode, the onset potential on Rh/C is shifted negatively as much as 0.28 V and the backward peak current on Rh/C was smaller 78 mA. Moreover, in a direct fuel cell, Rh/C has higher activity and stability than Pd/C. The Rh/C catalyst is more resistant to the poise caused by adsorbed oxygen intermediates than the Pd/C catalyst. Finally, the selectivity for ethanol oxidation to CO2 on the Rh/C is five times as much as Pd/C. This is mainly due to the relatively inert nature of Rh/C on C–C bond cleavage and the different reaction pathways on Pd/C and Rh/C.
References
- A. L. West, M. H. Griep, D. P. Cole and S. P. Karna, Anal. Chem., 2014, 86, 7377–7382 CrossRef CAS PubMed
 .
.
- Y. H. Qin, H. C. Li, H. H. Yang, X. S. Zhang, X. G. Zhou, L. Niu and W. K. Yuan, J. Power Sources, 2011, 196, 159–163 CrossRef CAS PubMed .
- S. Song, W. Zhou, Z. Liang, R. Cai, G. Sun, Q. Xin, V. Stergiopoulos and P. Tsiakaras, Appl. Catal., B, 2005, 50, 65–72 CrossRef PubMed .
- C. Coutanceau, L. Demarconnay, C. Lamy and J.-M. Léger, J. Power Sources, 2006, 156, 14–19 CrossRef CAS PubMed .
- W. J. Zhou, S. Q. Song, W. Z. Li, Z. H. Zhou, G. Q. Sun, Q. Xin, S. Douvartzides and P. Tsiakaras, J. Power Sources, 2005, 140, 50–58 CrossRef CAS PubMed .
- Q. He, S. Mukerjee, B. Shyam, D. Ramaker, S. Parres-Esclapez, M. J. Illán-Gómez and A. Bueno-López, J. Power Sources, 2009, 193, 408–415 CrossRef CAS PubMed .
- L. Jiang, A. Hsu, D. Chu and R. Chen, Int. J. Hydrogen Energy, 2010, 35, 365–372 CrossRef CAS PubMed .
- J. Anderson, A. Karakoti, D. J. Díaz and S. Seal, J. Phys. Chem. C, 2010, 114, 4595–4602 CAS .
- J. Melke, A. Schoekel, D. Gerteisen, D. Dixon, F. Ettingshausen, C. Cremers, C. Roth and D. E. Ramaker, J. Phys. Chem. C, 2012, 116, 2838–2849 CAS .
- M. Subhramannia, K. Ramaiyan and V. K. Pillai, Langmuir, 2008, 24, 3576–3583 CrossRef CAS PubMed .
- T. Maiyalagan, B. Viswanathan and U. Varadaraju, J. Nanosci. Nanotechnol., 2006, 6, 2067–2071 CrossRef CAS PubMed .
- K. Chen, P. Shen and A. Tseung, J. Electrochem. Soc., 1995, 142, L185–L187 CrossRef CAS PubMed .
- P. K. Shen and C. Xu, Electrochem. Commun., 2006, 8, 184–188 CrossRef CAS PubMed .
- C. Xu, P. K. Shen and Y. Liu, J. Power Sources, 2007, 164, 527–531 CrossRef CAS PubMed .
- F. Hu, C. Chen, Z. Wang, K. S. Pei and G. Wei, Electrochim. Acta, 2006, 52, 1087–1091 CrossRef CAS PubMed .
- Z. Liang, T. Zhao, J. Xu and L. Zhu, Electrochim. Acta, 2009, 54, 2203–2208 CrossRef CAS PubMed .
- R. Singh and A. Singh, Carbon, 2009, 47, 271–278 CrossRef CAS PubMed .
- Q. He, W. Chen, S. Mukerjee, S. Chen and F. Laufek, J. Power Sources, 2009, 187, 298–304 CrossRef CAS PubMed .
- M. Li, N. S. Marinkovic and K. Sasaki, Electrocatalysis, 2012, 3, 376–385 CrossRef CAS .
- A. Kowal, S. L. Gojković, K. S. Lee, P. Olszewski and Y. E. Sung, Electrochem. Commun., 2009, 11, 724–727 CrossRef CAS PubMed .
- J. P. I. d. Souza, S. L. Queiroz, K. Bergamaski, E. R. Gonzalez and F. C. Nart, J. Phys. Chem. B, 2002, 106, 9825–9830 CrossRef .
- Z. Y. Zhou, Q. Wang, J. L. Lin, N. Tian and S. G. Sun, Electrochim. Acta, 2010, 55, 7995–7999 CrossRef CAS PubMed .
- L. Ma, D. Chu and R. Chen, Int. J. Hydrogen Energy, 2012, 37, 11185–11194 CrossRef CAS PubMed .
- A. Dutta and J. Datta, J. Phys. Chem. C, 2012, 116, 25677–25688 CAS .
- Q. He, S. Mukerjee, B. Shyam, D. Ramaker, S. Parres-Esclapez, M. J. Illán-Gómez and A. Bueno-López, J. Power Sources, 2009, 193, 408–415 CrossRef CAS PubMed .
- S. K. Murphy, J. W. Park, F. A. Cruz and V. M. Dong, Science, 2015, 347, 56–60 CrossRef CAS PubMed .
- Y. Wang, X. Wang and C. M. Li, Appl. Catal., B, 2010, 99, 229–234 CrossRef CAS PubMed .
- J. Liu, J. Ye, C. Xu, S. P. Jiang and Y. Tong, Electrochem. Commun., 2007, 9, 2334–2339 CrossRef CAS PubMed .
- M. W. Xu, G. Y. Gao, W. J. Zhou, K. F. Zhang and H. L. Li, J. Power Sources, 2008, 175, 217–220 CrossRef CAS PubMed .
- Y. Suo and I. M. Hsing, Electrochim. Acta, 2009, 55, 210–217 CrossRef CAS PubMed .
- I. M. Hsing, X. Wang and Y. J. Leng, J. Electrochem. Soc., 2002, 149, A615–A621 CrossRef CAS PubMed .
- G. X. Cai, J. W. Guo, J. Wang and S. Li, J. Power Sources, 2015, 276, 279–290 CrossRef CAS PubMed .
- S. C. Lai, S. E. Kleijn, F. T. Öztürk, V. C. van Rees Vellinga, J. Koning, P. Rodriguez and M. T. Koper, Catal. Today, 2010, 154, 92–104 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.