
A multifunctional hierarchically assembled magnetic nanostructure towards cancer nano-theranostics
Hajar Mousavia,
Behrooz Movahedi*a,
Ali Zarrabi*b and
Marzieh Jahandarb
aDepartment of Nanotechnology Engineering, Faculty of Advanced Sciences and Technologies, University of Isfahan, Isfahan, 81746-73441, Iran. E-mail: b.movahedi@ast.ui.ac.ir; Fax: +98 31 37932342; Tel: +98 31 37934404
bDepartment of Biotechnology, Faculty of Advanced Sciences and Technologies, University of Isfahan, Isfahan, 81746-73441, Iran. E-mail: a.zarrabi@ast.ui.ac.ir
First published on 3rd September 2015
Abstract
Nowadays, iron oxide nanoparticles are among the most interesting carriers in simultaneous drug delivery and magnetic resonance imaging applications (theranostics). In this study, Fe3O4 magnetic nanoparticles were synthesized by a co-precipitation method followed by coating with an active-bioglass layer. This nanostructure is functionalized with hyperbranched polyglycerol through ring-opening polymerization of glycidol. The carrier was characterized using TEM, FT-IR, XRD, TGA, and elemental analysis. The results showed that the diameter of the carrier was between 20–30 nm. The cytotoxicity and cellular uptake results indicated that this nanostructure did not induce any cytotoxicity while expressing good potential as a contrast agent for magnetic resonance imaging. Moreover, curcumin was loaded on the carrier as a hydrophobic sample drug. The results showed a significant increase in curcumin solubility, which revealed the potential of this nanostructure in simultaneous cancer diagnosis and therapy.
Introduction
Magnetic particles have been used for over five decades. These particles were originally applied as a hyperthermia treatment against cancer cells. Owing to the special features of magnetic nanoparticles (MNPs) like their super-paramagnetic properties, great interest has recently been developed among researchers to carry out more studies in this field.1,2 Among the variety of MNPs, iron oxide nanoparticles are more interesting because of their intrinsic biocompatibility. Up to now, the fabrication of magnetic nanoparticles has been widely studied, and different methods such as co-precipitation,3–5 reverse micelles,6,7 thermal decomposition,8–12 sol–gel13–15 polyol production,16–18 and hydrothermal methods19–21 have been reported.Due to the high surface area of magnetic nanoparticles, they have a tendency to aggregate. Hence, their re-dispersion is almost impossible. In addition, Fe3O4 nanoparticles are not stable in the presence of oxygen, and they turn into Fe2O3 phase,22 which could change their properties from super-paramagnetic into paramagnetic. Therefore, many methods such as surface modification with different layers were proposed for their stabilization. These layers provide suitable sites for conjugation of other biological molecules such as drug-targeting and tracing agents especially in medical applications.23–25
Generally, MNPs are coated with inorganic layers specially silica7,11 and gold5,12,13 or organic layers such as different polymers, lipids, micelles and dendrimers. According to the literature, iron oxide nanoparticles have already been coated with various layers, especially polymers. Polymeric materials perform a remarkable role as a coating for MNPs. Polyethylene glycol, polyvinyl alcohol, dextran and chitosan are the most common polymers for theranostics (simultaneous therapy and diagnosis) applications.1,16,22,26–35 Recently, scientists have applied polyglycerol (PG) as a coating of Fe3O4 nanoparticles. According to the literature, polyglycerol is a hyperbranched, biocompatible, biodegradable and a hydrophilic polymer, consisting of ether scaffold with hydroxyl end group functionality.36–41 Because of the biocompatibility features of chitosan, this polymer has frequently been used in drug delivery systems. Nevertheless, chitosan's further surface engineering is limited due to its lower surface functional groups in comparison to hyperbranched polyglycerol. Moreover, chitosan is positively charged,32 thus, preventing the anionic ring opening polymerization of glycidol on its surface hydroxyl groups.
Hyperbranched-PG (HPG) is synthesized through polymerization of glycidol monomer on a hydroxyl epoxide core.42 The most popular method for synthesizing PG is the ring-opening polymerization of glycidol.43 Polymerization of hyperbranched polyglycerol improves the dispersity of MNPs in aqueous media such as phosphate buffer saline (PBS), and decreases the protein absorption compared to naked Fe3O4 MNPs.11,18 In addition, the ether scaffold of PG creates a suitable site for loading hydrophobic drugs such as curcumin.
Curcumin, a hydrophobic polyphenol derived from the rhizome of the herb curcuma lounge, has a wide spectrum of biological and pharmacological activities.44 Curcumin possesses multiple properties including anti-inflammatory, anti-oxidant, anti-proliferative, anti-carcinogenic, anti-angiogenic45 as well as anti-microbial44,46 activities in various cell culture and in vivo studies. Various in vivo studies (animal and human) have proved that curcumin is enormously safe in a high dosage. Poor solubility of curcumin has been a great challenge for medical applications. Thus, enhancing the bioavailability of curcumin needs more researches. Recently, Altunbas et al.47 designed the peptide hydrogel by self-assembly method for localized delivery of curcumin for in vitro models. Sontosh et al.48 used liposome nanoparticles of 2-hydroxypropyl-γ-cyclodextrin to increase the solubility of curcumin for in vivo OS-xenograft models. In 2012, scientists used biodegradable copolymer of mPEG–PLA for delivery of curcumin, which significantly improved the loading efficiency of curcumin and low-potency of intracellular delivery of curcumin.49
In this study, Fe3O4 nanoparticles were synthesized by co-precipitation method and then they were coated with bioactive glass. The core surface charge should be negative in order to polymerize the PG on the core surface through anionic ring opening polymerization. Thus, bioglass interface was used as a cheap, available and biocompatible interface (ligand exchange moiety). According to the literature, bioglass (BG) has great biocompatibility in a biological environment.50–53 To the best of our knowledge, this paper is the first report on bioglass coated Fe3O4 nanoparticles.
In the next step, the MNPs were coated by hyperbranched-PG by the ring-opening polymerization of glycidol in order to enhance the surface functional groups and drug loading cavities. The reasons PG is selected are: (a) PG is one of the most biocompatible polymers,54 (b) PG is an amphiphilic molecule and is appropriate for loading hydrophobic drugs while being circulated in the aqueous media, and (c) PG has several exposed surface functional groups (hydroxyl groups), which make any surface modifications such as conjugating targeting agents and tracing agents possible. This carrier was then characterized in terms of size and size distribution, thermal behaviour, polydispersity, biocompatibility and cellular uptake in detail. Ultimately, curcumin was loaded on the carrier as a model drug, and the loading efficiency as well as drug release was assessed.
Experimental
Materials
Fe(III) chloride hexahydrate (99%), Fe(II) chloride tetrahydrate (99%), ammonium hydroxide (28% w/v in water), ethanol (99.9%), oleic acid, tetra-ethyl-ortho-silicate (TEOS), triethyl phosphate (TEP), calcium nitrate tetrahydrate (CaNO3·4H2O), HCl (37%) and glycidol (96%) were purchased from Sigma Chemical Co. (St Louis, MO, USA). The MTS assay was purchased from Invitrogen Company. All the chemicals and reagents were used without further purification.Synthesis of naked and carboxyl group modified Fe3O4 MNPs
Naked Fe3O4 MNPs were prepared by co-precipitation of Fe2+ and Fe3+ ions in the presence of aqueous ammonia solution under nitrogen atmosphere. Briefly, 45 ml deionized water containing 297 mg FeCl2·4H2O and 810 mg FeCl3·6H2O (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was stirred vigorously for 20 min. To this solution, 3 ml of ammonia was added slowly in order to precipitate uniform magnetic nanoparticles. The resulting precipitate was stirred overnight to evaporate excess ammonia. After several washing cycles with deionized water, the nanoparticles were re-suspended in 25 ml deionized water and were centrifuged at 1000 rpm for 8 min to remove large aggregates.
:2) was stirred vigorously for 20 min. To this solution, 3 ml of ammonia was added slowly in order to precipitate uniform magnetic nanoparticles. The resulting precipitate was stirred overnight to evaporate excess ammonia. After several washing cycles with deionized water, the nanoparticles were re-suspended in 25 ml deionized water and were centrifuged at 1000 rpm for 8 min to remove large aggregates.
0.340 g FeCl2·4H2O and 1.160 g FeCl3·6H2O were dissolved in 35 ml deionized water to produce Fe3O4 nanoparticles with carboxyl functional groups. Then, 3 ml aqueous ammonia was added to the solution dropwise under stirring conditions. After 5 min, 100 μl oleic acid was slowly added to the solution. After 20 min of stirring, 200 μl oleic acid was added to the solution, and then the solution was stirred for 10 minutes until the reaction was complete. The magnetic nanoparticles were separated using a strong neodymium magnet and they were washed three times with deionized water and ethanol. The bilayer MNPs were re-dispersed in 50 ml deionized water for further purposes.
MNP coating with bioglass and polyglycerol
Bioglass sol was prepared by sol–gel method. First, 233 ml of TEP was hydrolysed by deionized water and hydrochloride acid as a catalyst for 4 h. Then, 2.5 ml TEOS was hydrolysed in 840 μl deionized water in the presence of 665 μl ethanol and hydrochloridric acid as a catalyst. These two solutions were mixed and were stirred for 1 h. Then, 1 g calcium nitrate was added to them. The result was a viscous solution after 12 h of stirring. For the synthesis of magnetic nanoparticles coated with bioglass, 30 mg of synthesized Fe3O4was dispersed in 30 ml deionized water. Then, 30 μl of bioglass sol was added to this solution dropwise and the mixture was stirred for 12 h. The pH of the Fe3O4 nanoparticles solution was kept around 5. The coated MNPs precipitate was collected using a neodymium magnet and it was washed 3 times with deionized water prior to being dried at 60 °C in a vacuum oven. Ultimately, the Fe3O4@BG nanostructure was calcined at 600 °C under nitrogen atmosphere.Hyperbranched polyglycerol was coated on the Fe3O4@BG nanostructure with ring-opening polymerization of glycidol. To this end, 30 mg of the Fe3O4@BG nanoparticles were mixed with 6 ml of glycidol for 1 h using an ultrasonic bath. Then, the homogeneous mixture was stirred vigorously at 140 °C for 20 h until a black gel was obtained. The gel was cooled to room temperature, and 6 ml of PBS solution was added so that it would precipitate the Fe3O4@BG nanoparticles grafted with hyperbranched polyglycerol. The nanoparticles were separated using a magnet, and dialyzed (molecular weight cut off: 1200) overnight. The black nanoparticles of Fe3O4@BG@HPG were obtained after vacuum drying.
Cytotoxicity assay
The cytotoxicity of Fe3O4@BG grafted hyperbranched polyglycerol was evaluated by determining the viability of HT-29 cell line of colon cancer cells after incubation in a medium containing 0.05 and 0.2 mg ml−1 of the nanostructure. The Fe3O4@BG@HPG nanoparticles were sterilized with 75% ethanol solution and they were recovered after drying under vacuum before application. Control experiments were performed by a complete growth culture medium without nanoparticles. Cell viability testing was carried out through MTS assay, which is a colorimetric method for determining the number of viable cells in proliferation as described in eqn (1):
 | (1) |
Magnetic resonance imaging (MRI)
The Fe3O4@BG@HPG nanoparticles were suspended in water at different concentrations of 0.20 and 0.05 mg ml−1. The imaging was conducted after cellular uptake during 7 days of treatment. The tubes were placed into the MRI apparatus. The pictures were captured in T2 relaxation time mode from a multi-echo spin-echo sequence (32 echoes, repetition time (TR): 1600 ms, echo time (TE): 15–480 ms.Cellular uptake
Colon cancer cells were utilized to evaluate the intracellular uptake of the Fe3O4@BG@HPG nanoparticles. The colon cancer cells were routinely cultured in their medium and seeded at a density of 105 cells per well in 24-well culture plates for 48 h before the medium was replaced with media containing MNPs at 0.20 mg ml−1. In the control experiment, the cells were seeded and cultured in the same manner without MNPs. After incubation at 37 °C for pre-determined periods, the cells were washed three times with PBS to remove MNPs in the medium. Then, they were collected by centrifugation, and the cell pellets were dissolved in 37% HCl (5 molar) at 80 °C for 3 h. The iron concentration was calculated by inductively coupled plasma-mass spectrometer (ICP-MS). Each experiment was conducted three times. The ICP results (particle/cell) were normalized with DNA content and the results were presented in terms of pg ml−1.Drug loading and release
First, the stock solution of curcumin was prepared in ethanol to make the calibration curve. The absorbance of the solutions was measured at 450 nm using UV-visible spectrophotometry. 0.5 mg of the Fe3O4@BG@HPG nanoparticles was dissolved in 10 ml phosphate buffered saline (PBS) in order to evaluate the drug loading. Different concentrations of drug solution, prepared in ethanol, were added to the solution of MNPs and the mixture was stirred for 24 h at room temperature. The ethanol was removed through 24 h stirring. To remove non-encapsulated drugs, the suspension was centrifuged at 4000 rpm for 20 min. The supernatant of this step is non-encapsulated drugs, which were dissolved in ethanol. Then, the absorbance of this solution was measured at 450 nm. The concentration of non-encapsulated drug was calculated according to the calibration curves of curcumin.0.15 mg of the Fe3O4@BG@HPG nanoparticles was dissolved in 3 ml deionized water to evaluate the drug release. Then, 1 ml of drug solution (0.90 mg ml−1 in ethanol) was added to the solution and the mixture was stirred for 24 h at room temperature. In the next step, non-encapsulated drugs were removed by centrifugation at 4000 rpm for 20 min. The remaining solution was centrifuged again at 14000 rpm for 20 min to separate the drug-loaded carriers followed by drying in a vacuum oven. Then, 5 ml of PBS solution (pH = 7.4) was added to 1 mg of drug loaded carrier. After 1, 2, 4, 8, 12, 24, 48 and 72 h, the drug release was measured at 37 °C. The non-released drug was separated using a strong magnet, and the absorbance of the drug release was measured at 450 nm. The amount of drug was measured according to the calibration curve. The drug loading efficiency and encapsulation efficiency were assessed according to eqn (2) and (3), respectively.55–57
 | (2) |
 | (3) |
Characterization methods
FT-IR spectra was obtained in a transmission mode in nitrogen atmosphere (JASC, FT-IR-6300 (400–4000 cm−1), Japan). For thermogravimetric analysis (TGA), samples weighed from5 to 15 mg. The size of MNPs in water was determined by transmission electron microscopy (TEM, Zeiss-EM10C, 80 kV) and dynamic light scattering (DLS, Molvern, MAL1001767). The elemental analysis (Leco) was utilized for measuring the percentage of organic components of the carrier.Results and discussion
Nanostructure characterizations
In this paper, bioglass layer is considered as a biocompatible ligand exchange moiety enabling the polymerization of hyperbranched polyglycerol on the core surface. There are several other alternatives for bioglass and hyperbranched polyglycerol, among which chitosan has gained the most attention.The advantages of using bioglass coating over chitosan coating are as follows: (a) smaller particles are achieved when coated with bioglass,32,35,58 (b) bioglass acts as an inert coating, while chitosan is active and could interact well with hydrophobic drugs thus interfering in drug release behaviour of PG,32,59 (c) strong covalent bond between bioglass and Fe3O4 may not be cleaved before the nanocarrier has accomplished its mission of drug delivery, while the weak electrostatic interaction between negatively charged Fe3O4 NPs surface and the positively protonated chitosan may be cleaved then.32,33
The advantages of using hyperbranched polyglycerol over chitosan are as follows: (a) hyperbranched polyglycerol is more biocompatible than chitosan,54 (b) hyperbranched polyglycerol with numerous exposed surface functional groups is a better choice for conjugating the targeting agent and the tracing agent rather than chitosan, (c) because of its dendritic structure, a hyperbranched polymer has a higher loading capacity for drug molecules compared to chitosan, (d) hyperbranched polyglycerol coating thickness is significantly less than chitosan coating thickness, and (e) there is a significant decrease in paramagnetic behaviour of Fe3O4 nanoparticles after coating with chitosan in comparison to PG-coated Fe3O4 nanoparticles.32,34,35,60
It is worth mentioning that using chitosan as a drug carrier does not guarantee avoiding the use of other polymeric shells since:
(1) Plasma proteins could recognize chitosan surface easily and could remove them from the circulation within seconds to minutes through the reticuloendothelial system (RES). Thus, a stealth shielding should be imparted on the surface of chitosan delivery systems to increase the systemic circulation time.61
(2) High cationic charge density on the chitosan surface may lead to cell membrane disruption. This necessitates covering the chitosan nanoparticle with some biocompatible coatings.62
Fig. 1a shows that the average hydrodynamic size of carrier is 36.4 nm. The poly dispersity index is 0.163, confirming that the carrier has a relatively narrow size distribution around 36 nm. Fig. 1b shows that the size of Fe3O4@BG@HPG is almost 20–30 nm. Based on these results, it is proposed that the polymerization of glycidol has separated the aggregated nanoparticles (NPs) besides the functionalization of MNPs with carboxyl group, which inhibited the growth of hydroxyl-functionalized nucleation. The applied method makes the reaction follow the LaMer mechanism,22 which forms monodispersed NPs by separating the nucleation and growth steps.
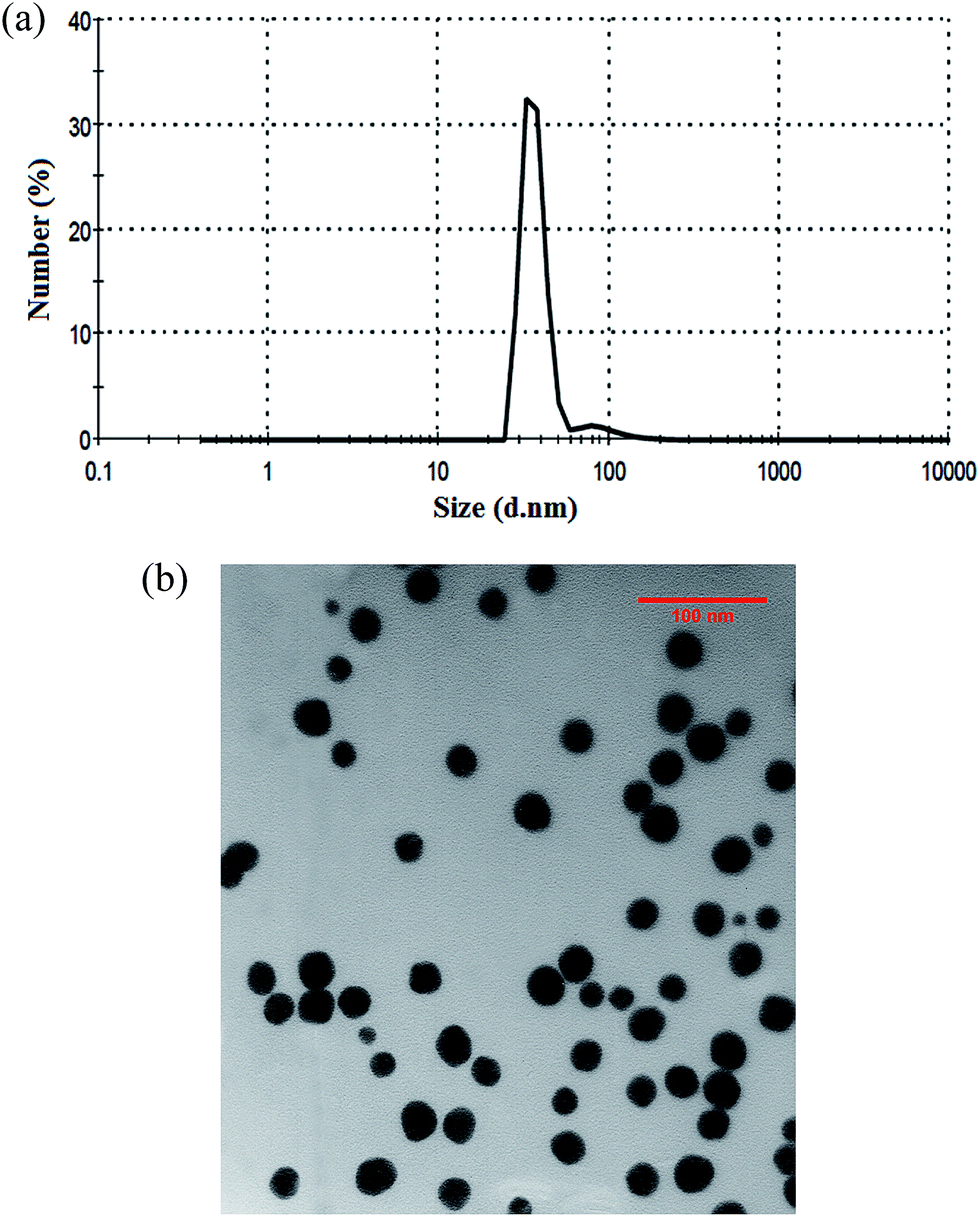 | ||
| Fig. 1 (a) Dynamic light scattering of bioglass@Fe3O4-grafted-HPG and (b) TEM image of bioGlass@Fe3O4-grafted-HPG. | ||
Fig. 2a shows the FT-IR spectra of physical/chemical interaction of oleic acid and Fe3O4 NPs. The characteristic peaks at 2851 and 2922 (cm−1), attributed to the free oleic acid in physical interaction, are symmetric and asymmetric stretches of the methylene groups, respectively. These peaks are shifted to 2852 and 2923.56 (cm−1) after the chemical reaction of MNPs and oleic acid. However, these changes are negligible and not reliable. These peaks could only prove the presence of oleic acid. However, the symmetric (sym, 1441 cm−1) and asymmetric (asym, 1593 cm−1) vibration peaks of carboxylate groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) could also prove the attachment of oleic acid to MNPs surfaces.53 Fig. 2b shows the FT-IR spectra of MNPs coated with bioglass before and after calcination, as well as Fe3O4@BG-grafted-HPG. The peak at 2955 (cm−1) is related to the carboxylic group of oleic acid on the surface of bare MNPs. The peak exists after coating MNPs with bioactive-glass (before calcination), but after the calcination process, the peak is completely removed because the calcination process removes a high percentage of oleic acid carbon chain. According to Fig. 2b, the peaks at 880 and 638 (cm−1) refer to Si–O and Si–H bonds, which confirm the successful coating of bioglass on MNPs. The intense peak at 1070 (cm−1) refers to the C–O–C ether stretch, which confirms the formation of etheric scaffold of polyglycerol. The heightened peaks at 2862 and 3666 (cm−1) are related to the stretches and vibration of CH2 groups as well as surface carboxylic groups, respectively. These peaks in addition to C–O–C peak confirm the formation of polyglycerol on the surface of Fe3O4@BG.53 The bioglass plays the role of a ligand exchange agent since its layer on the MNPs surfaces transforms the particle's end functional groups from the carboxyl to hydroxyl, which is critical in the polymerization process.
O) could also prove the attachment of oleic acid to MNPs surfaces.53 Fig. 2b shows the FT-IR spectra of MNPs coated with bioglass before and after calcination, as well as Fe3O4@BG-grafted-HPG. The peak at 2955 (cm−1) is related to the carboxylic group of oleic acid on the surface of bare MNPs. The peak exists after coating MNPs with bioactive-glass (before calcination), but after the calcination process, the peak is completely removed because the calcination process removes a high percentage of oleic acid carbon chain. According to Fig. 2b, the peaks at 880 and 638 (cm−1) refer to Si–O and Si–H bonds, which confirm the successful coating of bioglass on MNPs. The intense peak at 1070 (cm−1) refers to the C–O–C ether stretch, which confirms the formation of etheric scaffold of polyglycerol. The heightened peaks at 2862 and 3666 (cm−1) are related to the stretches and vibration of CH2 groups as well as surface carboxylic groups, respectively. These peaks in addition to C–O–C peak confirm the formation of polyglycerol on the surface of Fe3O4@BG.53 The bioglass plays the role of a ligand exchange agent since its layer on the MNPs surfaces transforms the particle's end functional groups from the carboxyl to hydroxyl, which is critical in the polymerization process.
 | ||
| Fig. 2 The FT-IR spectra of (a) MNPs coated with carboxyl group and (b) Fe3O4@BG-grafted HPG. | ||
Fig. 3 shows the X-ray diffraction pattern of Fe3O4@BG. The main typical peaks appeared in (311), (400), (511) and (440). These peaks belong to the Fe3O4magnetite phase. According to FT-IR spectra and XRD pattern, the MNPs were coated with bioglass and polyglycerol without putting the magnetite characteristics at risk.
 | ||
| Fig. 3 The XRD pattern of Fe3O4@BG after calcination. | ||
Fig. 4 shows the TGA diagrams of naked Fe3O4 MNPs, Fe3O4@BG and Fe3O4@BG@HPG. The weight loss of naked Fe3O4 MNPs and Fe3O4@BG was found to be around 3–5%, which is attributed to the moisture content of the nanoparticles. The TGA curve of Fe3O4@BG@HPG has a weight loss around 57%. This greater weight loss is due to the degradation of hyperbranched polyglycerol layer on the MNPs surfaces. The result is in agreement with the previous literature. Wang et al.53 reported a weight loss of around 63% for HPG grafted with Fe3O4. Elemental analysis could also prove the TGA results. As described in Table 1, there was no organic component in naked MNPs and Fe3O4@BG, while the organic component of Fe3O4@BG@HPG was significantly increased.
 | ||
| Fig. 4 The TGA analysis of naked Fe3O4, Fe3O4@BG and Fe3O4@BG@HPG. | ||
| Sample | Carbon percentage (wt%) | Hydrogen percentage (wt%) |
|---|---|---|
| Fe3O4 | 0.01 | 1.45 |
| Fe3O4@BG | 0.01 | 1.85 |
| Fe3O4@BG@HPG | 3.07 | 1.90 |
In vitro assessments
 | ||
| Fig. 5 Cytotoxicity assessment of Fe3O4@BG@HPG with 0.20 mg ml−1 concentration in terms of cell morphology (a) control sample (b) after 24 h and (c) after 48 h. | ||
Fig. 6a shows that for both concentrations in the desired interval, there is a remarkable correlation between the nanoparticles concentration and the cell viabilities. In addition, in all cases, the viabilities are above 90% and there are no significant viability changes as the concentration increases. The SPSS results indicated that the p-value was less than 0.05 for all samples.
 | ||
| Fig. 6 Cell viability results for Fe3O4@BG@HPG: (a) effect on HT-29 colon cancer cells and (b) uptake of Fe3O4@BG@HPG by HT-29 colon cancer cells. | ||
Since Fe3O4@BG@HPG demonstrated no cytotoxicity in high concentrations (0.20 mg ml−1), the 7 day uptake tests were conducted in this concentration and the results are shown in Fig. 6b. As it is anticipated, longer incubation time caused an increase in cellular uptake during 3 days from 1.4 to 2.13 pg ml−1. It is noted that these parameters did not change much with longer incubation time from 3 days to 7 days. This may be due to cell saturation with Fe3O4@BG@HPG after 3 days. The nearly high cellular uptake is proposed to be attributed to the high amount of hydroxyl functional groups of polyglycerol, which makes the nanoparticles highly hydrophilic and biocompatible. Wang et al.26,53 got similar amount of cellular uptake for Fe3O4@SiO2@HPG and MNPs@HPG.
Fig. 7a and b show the magnetic resonance images of different concentrations of Fe3O4@BG@HPG in PBS and colon cancer cell line. According to cytotoxicity assessment, the MR images were darker for higher concentrations of MNPs. The results showed an enhancement in contrast to both samples, attributed to the nature of iron oxide NPs that work as a negative contrast agent.53 This result is in good agreement with cellular uptake results.
 | ||
| Fig. 7 (a) MR images of Fe3O4@BG@HPG at various concentrations in PBS (0.20, 0.15, 0.10 mg ml−1 carrier concentration and control sample, left to right respectively) and (b) colon cancer cell with 0.20 mg ml−1 concentration of carrier during 7 days with clockwise navigation from the control. | ||
Drug investigations
The main aim of such studies is focused on increasing the solubility of hydrophobic drugs like curcumin as an herbal medicine. It is clear that curcumin could be encapsulated in Fe3O4@BG@HPG because of hydrophobic interactions between ether scaffold of polyglycerol and curcumin molecules.According to Fig. 8a, the drug loading and encapsulation efficiencies were increased with the increase in stirring time and the initial amount of drug. The statistical evaluations proved that the time, the ratio of drug to the carrier, and the correlation of these two parameters have affected the efficiency of drug encapsulation and drug loading. These results revealed that the solubility of curcumin increased from 6 μg ml−1 to 187 μg ml−1. A 31-fold increase in drug solubility showed that the nanostructured carrier has great potential as a delivery system for hydrophobic drugs such as curcumin.
 | ||
| Fig. 8 (a) Curcumin loading efficiency and (b) curcumin in vitro release curve. | ||
The results of drug release investigations are shown in Fig. 8b. It is obvious that with longer stirring time, the drug release was gradually increased. In the early hours, this rate was slow and did not show any burst effect. According to Fig. 8b, 50% of the drug content was released during the first 24 h. This rate was fixed after 132 h, and approximately 90% of the loaded drug was released.21,29,30
Conclusions
The Fe3O4 nanoparticles were synthesized and coated with bioglass layer. This nanostructure was grafted by hyperbranched polyglycerol through ring-opening polymerization. The carboxyl group of MNPs transformed to the bioglass hydroxyl group to support the attachment of hyperbranched polyglycerol. This nanostructure has a great potential as a theranostic agent. The non-toxic carrier showed a great affinity towards the colon cancer cell. This carrier greatly increased the solubility of curcumin as a hydrophobic and anticancer herbal drug. Moreover, drug release did not show any burst effect. Thus, the contrast enhancement and high capacity of drug loading confirm the potential of Fe3O4@BG@HPG as theranostic agents.References
- K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef PubMed.
- S. Laurent, S. Dutz, U. O. Hafell and M. Mahmoudi, Adv. Colloid Interface Sci., 2011, 166, 8 CAS.
- M. Yallapu, S. F. Othman, E. T. Curtis, B. K. Gupta, M. Jaggi and S. C. Chauhan, Biomaterials, 2011, 32, 1890 CrossRef CAS PubMed.
- A. Bumb, M. W. Brechbiel, P. L. Choyke, L. Fugger, A. Eggeman, D. Prabhalaran, J. Hutchinson and P. J. Dobson, Nanotechnology, 2008, 19, 335601 CrossRef CAS PubMed.
- J. P. Jolivet, C. Chanéac and E. Tronc, Chem. Commun., 2004, 45, 481 RSC.
- G. Ding, Y. Guo, Y. Lv, X. Liu, L. Xu and X. Zhang, Colloids Surf., B, 2012, 91, 68 CrossRef CAS PubMed.
- H. Zeng and S. Sun, Adv. Funct. Mater., 2008, 18, 391 CrossRef CAS PubMed.
- A. E. Fard, A. Zarepour, A. Zarrabi, A. Shanei and H. Salehi, J. Magn. Magn. Mater., 2015, 394, 44 CrossRef PubMed.
- N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575 RSC.
- W. Wu, Æ. Q. He and Æ. C. Jiang, J. Phys. Chem. C, 2008, 12, 9174 Search PubMed.
- S. Yoffe, T. Leshuk, P. Everett and F. Gu, Curr. Pharm. Des., 2013, 19, 493 CrossRef CAS.
- J. Daou, G. Pourroy and S. B. Colin, Chem. Mater., 2006, 18, 4399 CrossRef.
- Z. Li, M. Kawashita, T. Kudo and H. Kanetaka, J. Mater. Sci.: Mater. Med., 2012, 23, 2461 CrossRef CAS PubMed.
- Y. Deng, C. Deng, D. Qi, C. Liu, J. Liu and X. Zhang, Adv. Mater., 2009, 21, 1377 CrossRef CAS PubMed.
- H. Cui, Y. Liu and W. Ren, Adv. Powder Technol., 2013, 24, 93 CrossRef CAS PubMed.
- M. Muthiah, I. K. Park and C. S. Cho, Adv. Drug Delivery Rev., 2013, 31, 1224 CAS.
- Y. Qui, S. Barkhordari, M. Yadollahi and H. Namazi, Adv. Drug Delivery Rev., 2012, 64, 49 CrossRef PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866 CrossRef CAS PubMed.
- W. F. Ma, K. Y. Wu, J. Tang, D. Li, C. Wei, J. Gua, S. L. Wang and C. C. Wang, J. Mater. Chem., 2012, 22, 15206 RSC.
- P. Russo, D. Acierno, M. Palomba, G. Carotenuto, R. Rosa, A. Rizzuti and C. Leonelli, J. Nanotechnol., 2012, 24, 1 CrossRef PubMed.
- Q. Yuan, W. Geng, Y. Chi and X. Li, Mater. Res. Bull., 2012, 46, 2396 CrossRef PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elset and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS PubMed.
- J. Chomoucka, J. Drbohlavova, D. Huska, V. Adam, R. Kizek and J. Hubalek, Pharmacol. Res., 2010, 62, 144 CrossRef CAS PubMed.
- J. R. McCarthy, Adv. Drug Delivery Rev., 2010, 62, 1023 CrossRef CAS PubMed.
- J. E. Kim, J. Y. Shin and M. H. Cho, Arch. Toxicol., 2012, 86, 685 CrossRef CAS PubMed.
- L. Wang, K. G. Neoh, E. T. Kang and B. Shuter, Biomaterials, 2011, 32, 2166 Search PubMed.
- W. Wu, J. Shen, Z. Gai, K. Hong, P. Banerjee and S. Zhou, Biomaterials, 2011, 32, 9876 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 13, 36 CrossRef PubMed.
- F. Y. Cheng, C. H. Su, Y. S. Yang, C. S. Yeh, C. Y. Tsai, C. L. Wu, M. T. Wu and D. B. Shiej, Biomaterials, 2005, 26, 729 CrossRef CAS PubMed.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064 CrossRef CAS PubMed.
- A. Xu and S. Sun, Adv. Drug Delivery, 2013, 65, 729 Search PubMed.
- S. Mohammadi-Samani, R. Miri, M. Salmanpour, N. Khalighian, S. Sotoudeh and N. Erfani, Res. Pharm. Sci., 2013, 8, 25 CAS.
- J. Safari and L. Javadian, RSC Adv., 2014, 4, 48973 RSC.
- M. Shen, Y. Yu, G. Fan, G. Chen, Y. M. Jin, W. Tang and W. Jia, Nanoscale Res. Lett., 2014, 9, 296 CrossRef PubMed.
- Y. Arum, Y. O. Oh, H. W. Kang, S. H. Ahn and J. Oh, Fish. Aquat. Sci., 2015, 18, 89 Search PubMed.
- A. Zarrabi and M. Vossoughi, J. Mol. Liq., 2015, 208, 145 CrossRef CAS PubMed.
- A. Zarrabi, M. Shokrgozar, M. Vosoughi and M. Farokhi, J. Mater. Sci.: Mater. Med., 2014, 25, 499 CrossRef CAS PubMed.
- S. Gupta, R. Tyagi, V. S. Parmar, S. K. Sharma and R. Haag, Polymer, 2012, 53, 3053 CrossRef CAS PubMed.
- A. Zarrabi, M. Adeli, M. Vosoughi and M. A. Shokrgozar, Macromolecular, 2011, 3, 383 Search PubMed.
- M. Adeli, A. K. Fard, F. Abedi, B. K. Chegeni and F. Bani, Nanomedicine: Nanotechnology, Biology and Medicine, 2013, 9, 1203 CrossRef CAS PubMed.
- M. Witting, M. Molina, K. Obst, R. Plank, K. M. Eskl, H. C. Hennies, M. Calderon, W. Frieb and S. Hedtrich, Nanomedicine: Nanotechnology, Biology and Medicine, 2015, 11, 1179 CrossRef CAS PubMed.
- M. Calderon, M. A. Quadir, S. K. Sharma and R. Haag, Chem. Soc. Rev., 2011, 22, 190 Search PubMed.
- A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240 CrossRef CAS.
- V. Jahed, A. Zarrabi, A. Bordbar and S. Hafezi, Food Chem., 2014, 165, 241 CrossRef CAS PubMed.
- S. Prasad, S. C. Gupta, A. K. Tyagi and B. B. Aggrwal, Biotechnol. Adv., 2014, 32, 1053 CrossRef CAS PubMed.
- A. E. Krausz, et al., Nanomedicine: Nanotechnology, Biology and Medicine, 2015, 11, 195 CrossRef CAS PubMed.
- A. Altunbas, S. J. Lee, S. A. Rajasekaran, J. P. Schneider and D. J. Pochan, Biomaterials, 2011, 32, 5906 CrossRef CAS PubMed.
- S. Dhule, P. Penfornis, T. Frazier, R. Walker, J. Feldman, G. Tan, J. He, A. Alb, V. John and R. Pochampally, Nanomedicine: Nanotechnology, Biology and Medicine, 2012, 8, 440 CrossRef CAS PubMed.
- R. Yang, S. Zhang, D. K. Gao, Y. Zhao and Z. Wang, Pharm. Res., 2012, 29, 3512 CrossRef CAS PubMed.
- S. Padilla, J. Roman, S. S. Salcedo and M. V. Regi, Acta Biomater., 2006, 2, 331 CrossRef CAS PubMed.
- C. Jayalekshmi, S. P. Victor and C. P. Sharma, Colloids Surf., B, 2013, 101, 196 CrossRef PubMed.
- B. H. Hou, S. M. Hou, Y. S. Hsueh, J. Lin, H. C. Wu and F. H. Lin, Biomaterials, 2009, 30, 3956 CrossRef PubMed.
- L. Wang, K. G. Neoh, E. T. Kang, B. Shuter and S. C. Wang, Adv. Funct. Mater., 2009, 19, 2615 CrossRef CAS PubMed.
- K. Saatchi, P. Soema, N. Gelder, R. Misri, K. McPhee, J. H. E. Baker, S. A. Reinsberg, D. E. Brooks and U. O. Hafeli, Bioconjugate Chem., 2012, 23, 372 CrossRef CAS PubMed.
- Y. L. Lin, Y. K. Liu, N. M. Tsai, J. H. Hsieh, C. H. Chen, C. M. Lin and K. W. Liao, Nanomedicine, 2012, 8, 318 CrossRef CAS PubMed.
- J. Liu, L. Xu, C. Liu, D. Zhang, S. Wang, Z. Deng, W. Lou, H. Xu, Q. Bai and J. Ma, Polymer, 2012, 90, 16 CAS.
- P. Ranjan, A. Mukerjee, L. Helson and R. Haag, Biomedcentral, 2012, 10, 38 Search PubMed.
- Y. Wu, Y. Wang, G. Luo and Y. Dai, Bioresour. Technol., 2009, 100, 3459 CrossRef CAS PubMed.
- J. L. Arias, L. H. Reddy and P. Couvreur, J. Mater. Chem., 2012, 22, 7622 RSC.
- L. Wang, D. Su, L. Jiang and X. Feng, Soft Mater., 2014, 12, 306 CrossRef CAS PubMed.
- Z. Hou, C. Zhan, Q. Jiang, Q. Hu, L. Li, D. Chang, X. Yang, Y. Wang, Y. Li, S. Ye, L. Xie, Y. Yi and Q. Zhang, Nanoscale Res. Lett., 2011, 6, 563 CrossRef PubMed.
- M. Malhotra, C. T. Duchesneau, S. Saha, I. Kahouli and S. Prakash, Int. J. Nanomed., 2013, 8, 2041 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |