
Preparation, electrochemical responses and sensing application of Au disk nanoelectrodes down to 5 nm†
Abstract
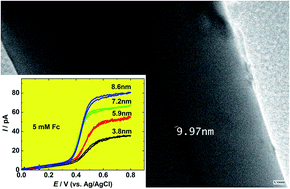
In this work, we report the preparation and electrochemical responses of Au disk nanoelectrodes as small as 5 nm in radii by the improvement of the laser-assisted pulling technique. A Au microwire is sealed into a bilayer capillary and pulled into an ultrasharp Au nanowire sealed in a silica tip using a laser-assisted puller. The ultrasharp tip is then sealed into a piece of glass tube, which is manually polished to expose the Au. Transmission electron microscopy, steady-state voltammetry of small redox species (e.g., ferrocene (Fc), ferrocene methanol (FcH2OH), potassium ferricyanide (Fe(CN)63−), hexaammineruthenium(III)chloride (Ru(NH3)63+)), and cyclic voltammetry in a H2SO4 solution are utilized to characterize the nanoelectrodes. The heterogeneous electron transfer rate constants for the oxidation/reduction of Fc, FcH2OH, Fe(CN)63−, and Ru(NH3)63+ are determined from steady-state voltammetry using the method developed by Mirkin and Bard and found to be ko = 7.9 ± 3.6 cm s−1 and α = 0.41 ± 0.04 for Fc, ko = 7.4 ± 7.1 cm s−1 and α = 0.41 ± 0.23 for FcCH2OH, ko = 4.0 ± 3.6 cm s−1 and α = 0.52 ± 0.17 for Fe(CN)63−, and ko = 7.2 ± 6.9 cm s−1 and α = 0.53 ± 0.22 for Ru(NH3)63+. These Au disk nanoelectrodes have been used to investigate the direct electrochemistry of ferritin molecules, which are immobilized on the Au surface by an electrostatic interaction between the cysteamine monolayer modified on the Au surface and ferritin molecules. From the voltammetric peak of ferritin on the Au nanoelectrode (200 nm in radius), the amount of ∼3900 molecules or 6.1 zmol can be calculated.
 Please wait while we load your content...
Please wait while we load your content...

