DOI:
10.1039/C5RA15905A
(Paper)
RSC Adv., 2015,
5, 96281-96289
New reduction mechanism of CO dimer by hydrogenation to C2H4 on a Cu(100) surface: theoretical insight into the kinetics of the elementary steps†
Received
8th August 2015
, Accepted 27th October 2015
First published on 27th October 2015
Abstract
A systematic DFT study that examines the role of the kinetics of the elementary reaction steps during the course of the reduction of a CO dimer, OCCO*, to C2H4 on Cu(100) is presented for the first time in the present study, and a new mechanism is introduced. Kinetic analysis of the elementary reaction steps has suggested that the further reduction of CO is the key selectivity-determining step for the formation of C2H4 and CH4 on Cu(100) and Cu(111), respectively. The main reaction pathway on Cu(111) proceeds through the reduction of CO to a CHO* intermediate, which may eventually result in CHx species by the breaking of a C–O bond and production of CH4. On Cu(100), OCCO* is first formed by CO dimerization, which is the first step and a more favorable pathway than the further hydrogenation of CO. This explains why only C2 species and not C1 species are observed experimentally on Cu(100). For the formation of C2H4 on Cu(100), the results suggest that the hydrogenation of OCCO* to the OCCHO* intermediate is the most likely reaction path, followed by the formation of intermediate OHCCHO* through further hydrogenation of the OCCHO* intermediate. The formation of OCCO* may be the rate-determining step in the reduction mechanism of the CO dimer. Kinetic analysis of the elementary steps gives a different mechanistic explanation for the selectivity of C2H4 production, which is in contrast to a previous suggested thermodynamic theoretical study on the reduction mechanisms of a CO dimer to C2H4. This present reduction pathway is consistent with the latest experimental results and explains the experimental uncertainty regarding the reaction intermediates. At present, it appears that the mechanism proposed in this study is most agreeable with the present experimental results.
1. Introduction
Reducing carbon dioxide (CO2) to a carbon-neutral fuel and a renewable feedstock of carbon-based fine chemicals would affect the global carbon balance and offer a novel solution to the dilemma of the growing energy demand and global warming.1,2 In this area, a landmark discovery was made in 1985 by Hori, who found that CO2 can be reduced to ethylene (C2H4) and methane (CH4) on Cu electrodes,3 and the extent of C2H4 and CH4 formation depends sensitively on the surface orientation of the Cu electrode.4 On the (100) facet of a Cu fcc crystal, the formation of C2H4 is dominant, whereas the formation of CH4 is favored on the (111) facet. However, an overpotential of almost 1 V on Cu electrodes is required, which hinders the global spread of this technology. Thus, the study of reduction mechanism of CO2 is extremely urgent on a Cu electrode. At present, most studies devoted to understanding the reduction mechanism on Cu electrodes have been experimental,5–10 but a theoretical insight was provided recently for the formation of C1 species such as CH4.11–16 Despite the extensive literature, the molecular-level details of the mechanism of the CO2 reduction on Cu electrodes are still not completely understood, particularly in relation to the C–C coupling step leading to the formation of C2H4 on Cu(100). However, all these studies showed that CO appears to be an important intermediate during the course of reduction. The electroreduction of CO leads to a similar product distribution, as observed for CO2 reduction,17 producing both C2H4 and CH4 through different reduction pathways. Similar hydrocarbon products yielded in experiments on CO electroreduction on Cu electrodes have established that adsorbed CO is a key intermediate in the formation of both C2H4 and CH4.17–19 Thus, the study of CO reduction mechanisms on copper electrodes will lead to a deeper understanding of the reaction chemistry and can eventually lead to the design of more efficient and selective catalysts, which can explain the observed product distribution and the effects of different electrode facets on the catalytic activity and selectivity of the reaction.
Recent experimental and theoretical studies focusing on the CO reduction mechanism have mostly showed that adsorbed CHO* is a key reaction intermediate and the hydrogenative reduction of CO to an adsorbed CHO* as the rate-determining step (rds) eventually results in the formation of CH4 by successive hydrogenation and cleavage of the C–O bond on Cu electrodes.5–16 However, the precise reduction mechanism of CO to C2H4 is still debated. Studies by Schouten and coworkers8–10 suggested that a mechanism may exist whose rds does not depend on proton-coupled electron transfer from an experimental standpoint because they found that the yields of C2 species are pH dependent on an reversible hydrogen electrode (RHE) scale in experiment. To explain this phenomenon, a CO dimerization mechanism in both CO2 and CO electroreduction on Cu(100) was proposed by Schouten and coworkers, and the electroreduction of various potential intermediates to hydrocarbons suggests that the dimerization of CO is the rate-determining step in C2 species formation, followed by either the formation of an enediol-like species or an oxalometallocycle.8 Further work by Schouten and coworkers demonstrated a shift in the onset of C2 formation to potentials around −0.4 V vs. RHE, providing further evidence that CO dimerization may occur.9 Li et al. and Kas et al. also suggested a C–C coupling mechanism based on the yields of C2H4 on oxide-derived Cu electrodes at potentials less negative than −0.5 V vs. RHE.20,21 Previous theoretical studies performed by Montoya and coworkers have shown that the kinetic barriers to the formation of a C–C bond between unprotonated CO adsorbates on Cu(211) surfaces are too high at a vacuum–metal interface for the turnover of production C2 species at reasonable rates, the qualitative trends would not be changed even in the presence of applied electric fields likely to exist in electrochemical environments,22 suggesting that CO* dimerization is kinetically unfavorable on Cu(211), and therefore, the protonation of CO* to form the CHO* intermediate must be proceeded before favorable kinetics can be achieved in the formation of C2H4. Calle-Vallejo and coworkers23 showed that with concerted proton–electron transfer, pH has no impact on the formation of C2H4 on Cu(100), suggesting that the rds occurs at an early stage of the mechanism and that there should be no proton transfer involved in that step or in the one before it from a theoretical standpoint. Thus, it can be concluded that C–C coupling should occur before the first proton transfer for the lowest-overpotential reduction pathways. Simultaneously, their studies also suggested that the activation energies to CO dimerization with an Eley–Rideal mechanism (i.e., only one of the CO molecules adsorbs on the surface and the other one from the gas phase reacts directly with the adsorbed CO molecule, without adsorbing) are not as high on Cu(100) compared with that with an Langmuir–Hinshelwood mechanism, i.e., two CO molecules adsorb at neighboring sites on Cu(111). However, the exergonic binding energy of CO on (211) steps and (100) terraces of Cu15 suggest that the adsorbed, rather than gas-phase CO, is the relevant precursor to the kinetics of C–C coupling between two CO molecules. In addition, previous work from Calle-Vallejo and coworkers also proposed stabilization of the CO dimer and its transition state by scaling the energy using the number of excess electrons in the adsorbed species, as determined from Bader charge analysis. Most recently, Montoya and coworkers presented DFT simulations for the CO dimerization mechanism and demonstrated that CO dimerization should have a lower activation barrier on Cu(100) than Cu(111),24 suggesting that the Cu(100) surface has a high activity for C–C coupling. However, the theoretical study by Montoya and coworkers only considered the C–C coupling mechanism of the Cu(211) surface,22 whereas the Cu(100) facet has been reported to be particularly selective towards C2H4 production. C2 products can be formed at low overpotentials without the formation of C1 products on Cu(100).3,4,8,9,17–19 Although they also considered CO dimer as the intermediate during the course of C2H4 formation, no detailed DFT-based mechanism for the formation of C2H4 on Cu(100) was suggested.24 In the theoretical study of Cu(100) by Calle-Vallejo and coworkers, the model used was purely thermodynamic and assumed that the kinetic barriers of uphill processes are not much larger than their reaction energies and those downhill processes have easily surmountable barriers.23 Despite the extensive experimental and theoretical studies, it is still not known whether adsorbed CO or a more reduced C1 species is coupled to produce C2H4, thus leaving the main reaction path, reaction intermediates and selectivity-determining and rate-determining steps for C2H4 production on Cu(100) electrocatalysts unclear. The C–C bonding formation is important as it would aid in the design of new catalysts that are active at a lower overpotential and open up routes to the production of high-energy fuels by the electrochemical reduction of CO2.
In the present study, we carried out a systematic DFT study of the reduction mechanism of CO dimer to form C2H4 in CO2 electroreduction on the Cu(100) electrode, for the first time examining the kinetic energy of various possible elementary reaction steps during the course of the reduction of the CO dimer to form C2H4 and determining the selectivity-determining and rate-determining steps. As electrocatalytic reactions on catalysts that include the electrolyte environment are far too complex for a complete theoretical description of the kinetics of the elementary steps, the complexity is first reduced. Consequently, in the present study, we restrict our calculations to close packed surfaces and a periodic gas-phase environment. However, theoretical evidence of the reduction mechanism of the CO dimer of a Cu(100) surface can still be presented qualitatively in this work. The results will explain the experimental observations and provide theoretical support for the reduction mechanism of CO dimer previously proposed by Schouten and coworkers.
2. Computational method and modeling
Calculations were performed in the framework of DFT using the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE)25 and employing ultrasoft pseudopotentials26 for the nuclei and core electrons of all atoms. The calculations of the most stable geometry structure of various adsorption and co-adsorption intermediates were performed using periodic super-cells with the Cu electrodes modeled by four-layer (100) slabs with a 3 × 3 surface. A vacuum space of 16 Å was placed above the slabs and adsorption was allowed on only one of the two surfaces exposed. The Kohn–Sham orbitals were expanded in a plane-wave basis set with a kinetic energy cutoff of 26 Ry and the charge-density cutoff of 260 Ry. The Fermi-surface effects were treated by the smearing technique of Methfessel and Paxton, using a smearing parameter27 of 0.02 Ry. The calculations were carried out with spin-polarization, which is essential to properly represent the electronic structure of various adsorbed reaction intermediates. The PWSCF codes contained in the Quantum ESPRESSO distribution28 were used to implement all calculations, while figures of the chemical structures were produced with the XCRYSDEN29–31 graphical package. BZ integrations were performed using a (3 × 3 × 1) uniformly shifted k-mesh for the (3 × 3) supercell. The calculated equilibrium lattice constant for Cu was 3.66 Å, which agreed well with theoretical and experimental values (3.66 and 3.62 Å, respectively).32,33 During the calculations, the structure of the bottom two layers was fixed at the theoretical bulk positions, whereas the top two layers and the adsorbates were allowed to relax and all the other structural parameters were optimized to minimize the total energy of the system. Structural optimization was performed until the Cartesian force components acting on each atom were brought below 10−3 Ry per Bohr and the total energy converged to within 10−5 Ry.
The climbing-image nudged elastic band (CI-NEB) method was used to determine the minimum energy paths (MEPs) for all the elementary steps.34,35 The transition state of the optimized reaction coordinate was approximated by the image of highest energy. The transition state images from the CI-NEB calculations were optimized using the quasi-Newton method, which minimizes the forces to determine the saddle point. Geometry optimization was performed for each intermediate point in the MEPs, in which considering the high cost of CI-NEB calculations, the most stable adsorption configurations of adsorbents obtained on 3 × 3 surface and a three-layer Cu(100) slab with a 2 × 3 surface unit cell were used, the bottom two layers of metal atoms were fixed, while the top layer of metal atoms and all other nonmetal atoms were allowed to relax.
3. Results
The adsorption, co-adsorption energies and geometry structures of the preferred adsorption configurations for various possible reaction intermediates on Cu(100) are given in Tables S1, S2, Fig. S1 and S2 in the ESI.† The most stable adsorption configurations on Cu(100) were used to perform the following MEP analysis. The hydrogenation agent in the elementary reaction steps of reduction mechanism of the CO dimer is the adsorbed hydrogen atoms (H*) formed through H2 dissociation. A Langmuir–Hinshelwood mechanism is involved in the reduction mechanism of the CO dimer because the adsorbents are co-adsorbed structures on Cu(100). The DFT-calculated activation barrier for H2 dissociation is 0.45 eV on Cu(100), which are small and easily surmountable. Therefore, the H2 dissociation should play a relatively minor role in the kinetics of the reduction of the CO dimer.
3.1 Selectivity-determining steps of CO reduction on Cu(111) and Cu(100)
Schouten and coworkers8 suggested that there are two separate pathways for the formation of C2H4: one that shares an intermediate with the pathway to CH4, as observed on Cu(111) and below −0.8 V on the Cu(100) surface at pH 7; and a second pathway that occurs only on the Cu(100) surface. Our recent theoretical study16 reported the formation pathways of CH4 and C2H4 by the chemical and electrochemical reduction of CO2 on Cu(111) and Cu(100) that shared key intermediates COads, CHOads, CH2Oads, and CH2OHads. The key intermediate, CH2ads, can be formed by the hydrogenative dissociation of the CH2OHads intermediate and finally, the CH2 intermediate leads to the formation of CH4 and C2H4 by serial hydrogenation and dimerization, respectively. For this second pathway, they suggested that the CO dimer, OCCO*, is the key intermediate in the formation of C2H4. To date, the selectivity-determining steps are still unclear, namely, CO hydrogenation or CO dimerization is more favorable on Cu(111) and Cu(100). Therefore, MEP analysis for CO further reduction is performed in this section to determine the selectivity-determining steps. On Cu(111) and Cu(100), there are two possible reactions for CO further hydrogenation, namely, the hydrogenation to form an adsorbed formyl (CHO*) and hydroxymethylidyne (COH*). In addition, the direct dissociation to adsorbed C* and O* is also considered. The calculated MEPs (Fig. 1 and 2) suggest that direct dissociation is particularly energetically demanding; the activation barrier is 3.96 and 3.01 eV on Cu(111) and Cu(100), respectively. The two hydrogenation reactions have relatively lower activation barriers, and the formation of CHO* requires an activation barrier nearly 1.6 eV lower than the formation COH* (∼1.0 eV vs. ∼2.6 eV) on Cu(111). The activation barriers for the hydrogenation of CO to form COH* and CHO* are 2.43 and 2.36 eV on Cu(100), respectively, as shown in Fig. 2. These results indicated that CO hydrogenation requires higher activation barriers on Cu(100) than on Cu(111), which is easier to achieve on Cu(111). On this basis, it can be concluded that further CO hydrogenation to CHO* is more favorable on Cu(111).
 |
| | Fig. 1 Minimum energy path of CO dissociation and hydrogenation to form C, COH and CHO on Cu(111). Oxygen atoms are red, hydrogen atoms are white, carbon atoms are gray, and copper atoms are blue. | |
 |
| | Fig. 2 Minimum energy path of CO dissociation and hydrogenation on Cu(100) to form C, COH and CHO. | |
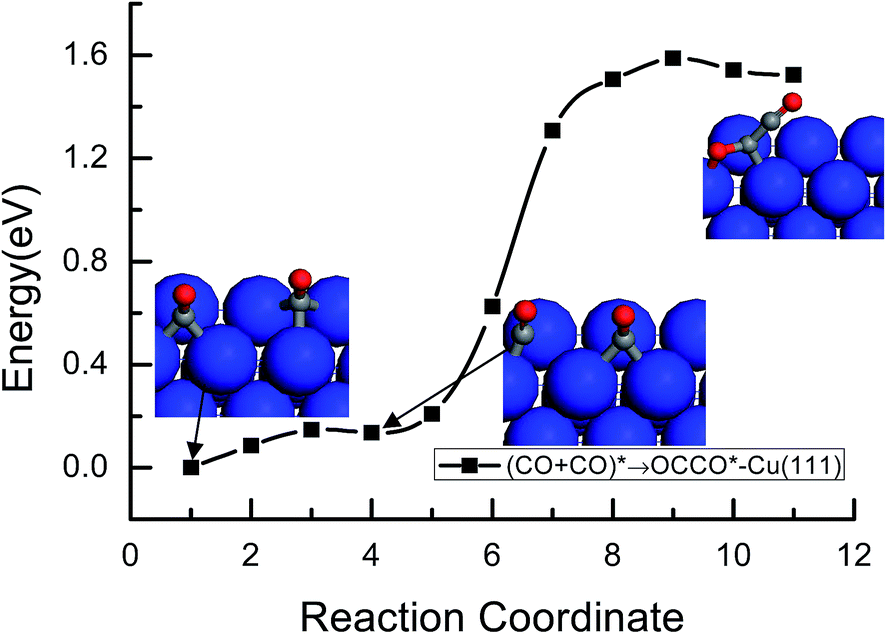
As shown in Fig. 3 and 4, the activation barrier for the formation of OCCO* intermediate by CO dimerization on Cu(111) and Cu(100) is 1.59 and 1.26 eV, respectively. Our present results indicated that CO dimerization to OCCO* intermediate requires a higher activation barrier than CO hydrogenation to CHO* intermediate on Cu(111), whereas on Cu(100), lower activation barrier is required for CO dimerization compared to the formation of the CHO* intermediate by CO hydrogenation. Therefore, CO hydrogenation to CHO* during the course of further CO reduction occurs more easily on Cu(111), whereas CO dimerization to OCCO* is more favorable on Cu(100). A relationship between the surface structure of the Cu electrodes and the mechanism of CO reduction is suggested by our present theoretical study and selectivity-determining steps are revealed on these different Cu single-crystal electrode surfaces. The previous experiment study by Hori et al.3,4,17–19 on Cu single electrode surfaces and the thermodynamic and kinetic theoretical study conducted by Calle-Vallejo et al. and Nørskov et al.23,36 also showed that more C2H4 is formed on Cu(100), whereas more CH4 is produced on Cu(111). Therefore, we can speculate that the formation of CH4 is more favorable on the atomically flat (111) parts of the Cu electrode surface through CO hydrogenation into CHO* intermediate, whereas C2H4 will be formed more easily at the (100) sites by CO dimerization. Simultaneously, previous DFT calculations by Peterson et al. and Nie et al.11,14 also suggested that CHO* is an intermediate in the formation of CH4, and no experimental evidence indicated that CHO* is the precursor to C2H4.8,9 A second key aspect of our present result is that the activation barrier for CO dimerization on the Cu(100) facet is significantly lower than that on the Cu(111) facet. The structure sensitively described by the lowering of the activation barrier on Cu(100) relative to Cu(111) implies that the Cu(100) facet has more activity for the reduction of the CO dimer, as indicated in previous experimental studies. Thus, the unique selectivity for the formation of C2H4 on (100) can be explained by the reduction of CO dimer (for detailed arguments, see ref. 37) and is consistent with Gattrell and coworkers,38 who proposed that this CO dimer would be more stable on Cu(100). In addition, our present results are also in agreement with Schouten and coworkers' experimental study. Currently, the mechanism of C2H4 formation initiated by CO dimerization was studied in more detail on Cu(100) for the first time from the point of view of kinetics in our present study as follows.
 |
| | Fig. 3 Minimum energy paths of CO dimerization to the OCCO* intermediate on Cu(111). | |
 |
| | Fig. 4 Minimum energy paths of CO dimerization to the OCCO* intermediate on Cu(100). | |
3.2 The serial reduction pathways of CO dimer into C2H4 on Cu(100)
To find intermediates in the reduction of CO dimer, OCCO*, to C2H4, we investigated the reduction of C2 species. The kinetic energetics of the OCCO* intermediate for further hydrogenation or dissociation were firstly examined on Cu(100). There are three possibilities for further reduction of the OCCO* intermediate: one is the dissociation of OCCO* by breaking the C–O bond to form the CCO* intermediate; the other two are hydrogenation of OCCO* at C and O atom site to form OCCHO* and OCCOH* intermediates, respectively. To compare the degree of difficulty of these two possibilities, minimum energy path (MEP) analysis was performed. As shown in Fig. 5, the activation barriers for the dissociation of OCCO* to CCO* is 0.91 eV, and further hydrogenation of OCCO* to OCCHO* and OCCOH* on Cu(100) are 0.21 and 1.16 eV, respectively, in which the activation barrier for the hydrogenation of OCCO* on the C atom site is significantly lower and can be overcome easily at an ambient temperature. Although a recent theoretical study conducted by Calle-Vallejo and coworkers23 based on thermodynamics analysis suggested that CCO* is a possible reaction intermediate in the electroreduction of CO to C2 species on Cu(100), cleavage of the C–O bond in OCCO* is unfavorable kinetically on Cu(100). Therefore, it can be concluded that the further reduction of OCCO* to the OCCHO* intermediate should be more kinetically favorable on Cu(100).
 |
| | Fig. 5 Minimum energy paths of OCCO* hydrogenation to OCCHO* and OCCOH*, and dissociation to CCO* by cleavage of the C–O bond on Cu(100). | |
For further reduction of the OCCHO* intermediate, there may be three possibilities on Cu(100). One is the hydrogenation of OCCHO* at the C atom site to glyoxal (OHCCHO*); the other two is the hydrogenation of OCCHO* at the O atom site to HOCCHO* and OCCHOH* intermediate, respectively. As shown in Fig. 6, the activation barriers for the further hydrogenation of OCCHO* to OHCCHO*, HOCCHO* and OCCHOH* intermediate on Cu(100) based on the MEP analysis are 0.09, 0.22 and 0.62 eV, respectively, in which the formation of the OHCCHO* intermediate requires a significantly lower activation barrier, which is an almost non-activated process. Therefore, the hydrogenation of OCCHO* to the OHCCHO* intermediate at the C atom site occurs more easily on Cu(100). Our present theoretical calculations are also agreeable with a recent experimental study by Schouten and coworkers,8 in which OHCCHO* intermediate had been observed experimentally as a reaction intermediate during the course of CO reduction on Cu(100).
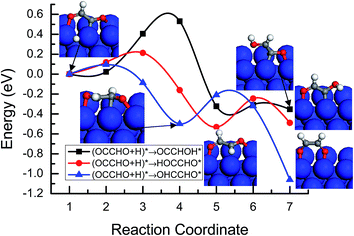 |
| | Fig. 6 Minimum energy paths of OCCHO* intermediate hydrogenation to OCCHOH*, HOCCHO* and OHCCHO* on Cu(100). | |
Similarly, three possibilities are also considered for the further reduction of the OHCCHO* intermediate. One is deoxygenation of OHCCHO* intermediate to form adsorbed OHCCH* and O* atom by cleavage of C–O bond; the other two is the hydrogenation of OHCCHO* at the C and O atom sites to form the OCHCH2O* and OHCCHOH* intermediate, respectively. MEP analysis indicated that the activation barriers for the deoxygenation of OHCCHO* to OHCCH* and O* atom is 1.02 eV and the activation barriers for the further hydrogenation of OHCCHO* to OHCCH2O and OHCCHOH* on Cu(100) are 0.63 and 1.23 eV, respectively, see Fig. 7. The hydrogenation of OHCCHO* at the C atom site to OHCCH2O* has a relatively lower activation barrier on Cu(100). Therefore, the formation of OHCCH2O is the most favorable kinetically reaction pathway on Cu(100) for the further reduction of OHCCHO* intermediate.
 |
| | Fig. 7 Minimum energy paths of the further reduction of OHCCHO* intermediate to OHCCH2O*, OHCCHOH* and OHCCH* on Cu(100). | |
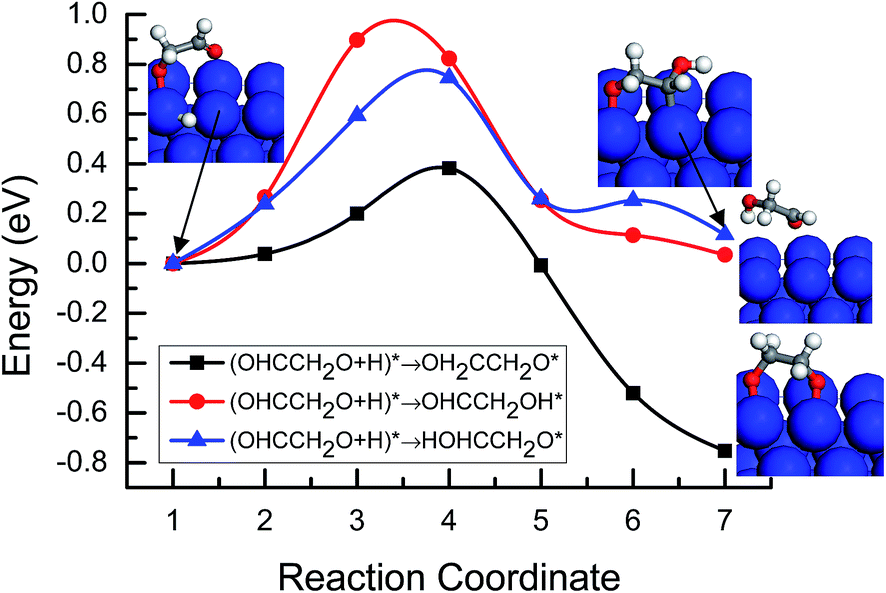
Based on the abovementioned analysis, we can speculate that OHCCHO* and OHCCH2O* are possible reaction intermediates in the reduction mechanism of CO dimer on Cu(100). There are also three possibilities for the further reduction of OHCCH2O*. One is the hydrogenation of OHCCH2O* at the C atom site in the CHO part into OH2CCH2O* intermediate. The other two is the hydrogenation of OHCCH2O* at the O atom site in CH2O and the CHO part to the OHCCH2OH* and HOHCCH2O* intermediate, respectively. For these three possibilities, MEP analysis was also performed. As shown in Fig. 8, the activation barriers for the further reduction of OHCCH2O* intermediate to OH2CCH2O*, OHCCH2OH* and HOHCCH2O* on Cu(100) are 0.38, 0.98 and 0.78 eV, respectively, in which the formation of OH2CCH2O* has a relatively lower activation barrier on Cu(100). Therefore, it can be predicted that OH2CCH2O* should be the most kinetically favorable state derived from the hydrogenation of OHCCH2O* at the C atom site in the CHO part on Cu(100).
 |
| | Fig. 8 Minimum energy paths of further hydrogenation of the OHCCH2O* intermediate to OH2CCH2O*, OHCCH2OH* and HOHCCH2O* on Cu(100). | |
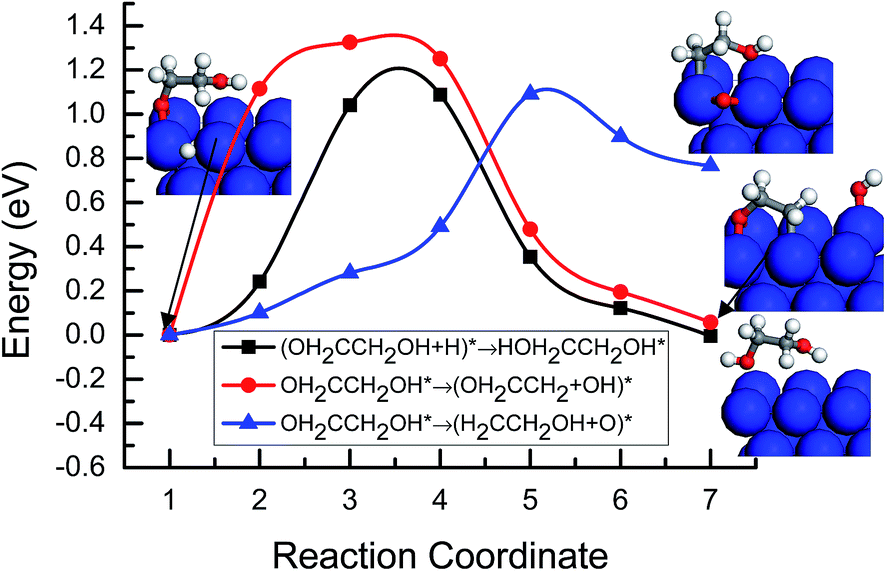
For further reduction of the OH2CCH2O* intermediate, there may be two possibilities. One is the hydrogenation of OH2CCH2O* at the O atom site to the OH2CCH2OH* intermediate; another is deoxygenation of OH2CCH2O* to form the OH2CCH*2 intermediate by cleavage of the C–O bond. MEP analysis of these two possibilities was performed for comparison. As shown in Fig. 9, the activation barriers for the hydrogenation of OH2CCH2O* to the OH2CCH2OH* intermediate and deoxygenation to OH2CCH*2 on Cu(100) are 1.21 and 2.71 eV, respectively. By comparing the activation barriers, the formation of OH2CCH2OH* has a relatively lower barrier, and it is significantly lower than that of the formation of OH2CCH*2. Therefore, we can conclude that OH2CCH2OH* should be a more kinetically favorable intermediate derived from the hydrogenation of the OH2CCH2O* intermediate than the other OH2CCH*2 intermediate formed by cleavage of the C–O bond in OH2CCH2O*.
 |
| | Fig. 9 Minimum energy paths of the further reduction of OH2CCH2O* intermediate to OH2CCH2OH* by hydrogenation and OH2CCH*2 on Cu(100) by cleavage of the C–O bond. | |
Three possibilities are considered for further reduction of the OH2CCH2OH* intermediate. One is the hydrogenation of OH2CCH2OH* at the O atom site in the CH2O part to form HOH2CCH2OH* intermediate; the other two is dehydroxylation and deoxygenation of OH2CCH2OH* to form OH2CCH*2 and H2CCH2OH* intermediate, respectively. MEP analysis of these three possibilities showed that the activation barriers for the further hydrogenation of OH2CCH2OH* to HOH2CCH2OH*, and the dehydroxylation and deoxygenation of OH2CCH2OH* to OH2CCH*2 and H2CCH2OH* on Cu(100) are 1.21, 1.36 and 1.12 eV, respectively, see Fig. 10. The hydrogenation of OH2CCH2OH* to the HOH2CCH2OH* intermediate and the deoxygenation of OH2CCH2OH* to the H2CCH2OH* intermediate have almost identical and relatively lower activation barriers on Cu(100), indicating that these two pathways may be parallel and may occur simultaneously during the course of C2H4 formation.
 |
| | Fig. 10 Minimum energy paths of the further reduction of the OH2CCH2OH* intermediate to HOH2CCH2OH*, OH2CCH*2 and H2CCH2OH* by hydrogenation, deoxygenation and dehydroxylation, respectively, on Cu(100). | |
HOH2CCH2OH* and H2CCH2OH* intermediates may be formed simultaneously on Cu(100) based on the abovementioned analysis. Therefore, the final product, C2H4, will be formed possibly by the dehydroxylation of HOH2CCH2OH* and H2CCH2OH* intermediates. For these two possibilities, MEP analysis indicated that the activation barriers for the dehydroxylation of HOH2CCH2OH* and H2CCH2OH* intermediates to C2H4 on Cu(100) are 0.45 and 0.28 eV, respectively, as shown in Fig. 11. The results showed that the dehydroxylation of H2CCH2OH* to the final product, C2H4, has a relatively lower activation barrier on Cu(100), indicating that the reaction may occur more easily on Cu(100). However, the activation barriers of the dehydroxylation of HOH2CCH2OH* to C2H4 is also very low, which is also surmountable at ambient temperature. Therefore, these two pathways may occur simultaneously on Cu(100).
 |
| | Fig. 11 Minimum energy paths of the further reduction of the HOH2CCH2OH* and H2CCH2OH* intermediates into C2H4 by dehydroxylation on Cu(100). | |
4. Discussion
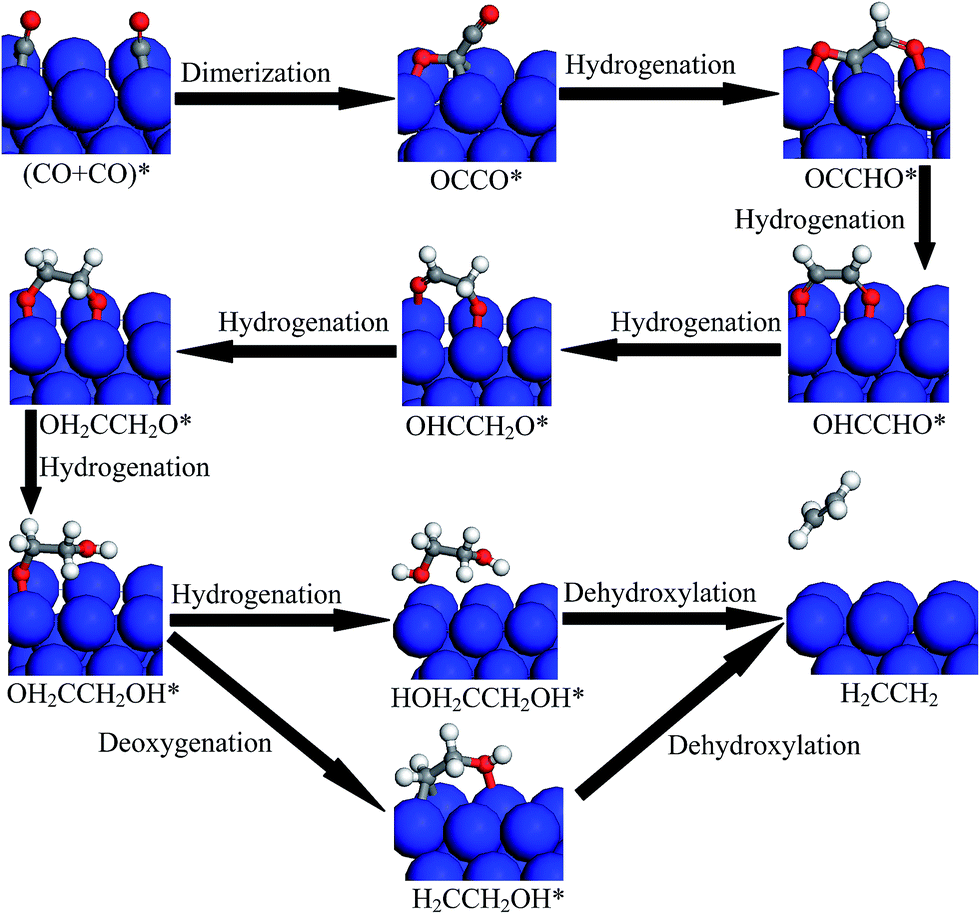
In the present study, a systematic determination of the CO reduction mechanism to the production C2H4 is presented by CO dimerization on Cu(100), including the selectivity-determining steps, the optimal reduction pathways and rate-determining steps. We present the first DFT study that examines the role of the kinetics of elementary reaction steps in the reduction mechanism of the CO dimer to the production of C2H4 on Cu(100), by MEP analysis. Based on the abovementioned results from MEP analysis, the optimal reaction pathways by CO dimerization to the production C2H4 on Cu(100) can be summarized in Table 1. The CO dimer, OCCO*, is first formed by CO dimerization, and this reaction is more favorable on Cu(100) than the further hydrogenation of CO. Based on the activation barriers, the OH2CCH2OH* intermediate either forms the HOH2CCH2OH* intermediate by direct hydrogenation or forms the H2CCH2OH* intermediate by cleavage of the C–O bond, which maybe parallel pathways in the reduction mechanism of the CO dimer on Cu(100). Finally, the production of C2H4 can be formed by the dehydroxylation of HOH2CCH2OH* and H2CCH2OH* intermediates. Based on the calculated activation barriers, the relatively slow steps on Cu(100) include (CO + CO)* → OCCO*, (OH2CCH2O + H)* → OH2CCH2OH*, (OH2CCH2OH + H)* → HOH2CCH2OH*, and OH2CCH2OH* → (H2CCH2OH + O)*, in which (CO + CO)* → OCCO* has the highest activation barrier, and it may be the rate-determining step of the reduction mechanism of the CO dimer to C2H4 on Cu(100). Our present mechanistic study explains the experimental observations by Schouten and coworkers in which the rate-determining step consists of the coupling of two CO molecules mediated by electron transfer to form the OCCO* intermediate on Cu(100). Simultaneously, coupling of the C–C bond, hydrogenation at the O atom site, and cleavage of the C–O bond is more difficult on Cu(100) for CO reduction to the production of C2H4. The possible reduction mechanism of CO dimer to C2H4 is proposed in Fig. 12 based on our DFT calculated results on Cu(100).
Table 1 Optimal reduction pathways for CO dimerization to the production of C2H4 and the activation barriers (Eact) of each elementary reaction step on Cu(100)
| Reaction paths |
Eact (eV) |
| (CO + CO)* → OCCO* |
1.26 |
| (OCCO + H)* → OCCHO* |
0.21 |
| (OCCHO + H)* → OHCCHO* |
0.09 |
| (OHCCHO + H)* → OHCCH2O* |
0.63 |
| (OHCCH2O + H)* → OH2CCH2O* |
0.38 |
| (OH2CCH2O + H)* → OH2CCH2OH* |
1.21 |
| (OH2CCH2OH + H)* → HOH2CCH2OH* |
1.21 |
| OH2CCH2OH* → (H2CCH2OH + O)* |
1.12 |
| HOH2CCH2OH* + * → H2CCH2 + 2OH* |
0.45 |
| H2CCH2OH* → H2CCH2 + OH* |
0.28 |
 |
| | Fig. 12 Proposed reduction pathways for the production of C2H4 in the reduction mechanism of CO dimer on Cu(100). | |
In a previous mechanistic study on CO reduction to C2H4, the combination of two adsorbed CH*2 species and a CO-insertion type mechanism, namely, coupling between CHx and CO leading to the formation of CHxCO species, were proposed by Hori and coworkers3,4,17–19,37 as alternative pathways for the production of C2H4, but these two mechanisms cannot explain why the main C–C coupling product C2H4 is observed experimentally on Cu(100). From a thermodynamic viewpoint, recent theoretical considerations on the reduction of CO by dimerization to the production of C2H4 on the Cu(100) electrode were performed by Calle-Vallejo and coworkers, and the possible reduction pathways were proposed. Based on a thermodynamic study, the OCCHO* intermediate is less stable than the OCCOH* intermediate, suggesting that the initial hydrogenation of the O atom in the OCCO* intermediate is a more favorable pathway on Cu(100), and the CCO* intermediate was then formed by dehydroxylation of OCCOH* intermediate (i.e., cleavage of the C–O bond). The next favorable step is the hydrogenation of CCO* at the C atom site to form HCCO* intermediate, followed by the hydrogenation of HCCO* in the carbonyl group to the HCCHO* intermediate. In the next step, another hydrogenation takes place at the C atom site of HCCHO*, producing the H2CCHO* intermediate. The further reduction pathway of H2CCHO* proceeds with the hydrogenation of the C atom bonded to the O atom, leading to the formation of the H2CCH2O* intermediate. Finally, the production of C2H4 was formed by cleavage of the C–O bond on Cu(100). Therefore, previous thermodynamic studies showed that CCO*, HCCO*, HCCHO*, H2CCHO*, and H2CCH2O are intermediates of the reduction mechanism of the CO dimer. However, our present mechanistic studies showed that the initial hydrogenation of OCCO* at the C atom site to OCCHO* should be a more favorable pathway on Cu(100) than forming the OCCOH* intermediate by hydrogenation of the O atom; the corresponding activation barriers are 0.21 and 1.16 eV. OCCHO*, OHCCHO*, OHCCH2O*, OH2CCH2O*, OH2CCH2OH*, HOH2CCH2OH*, and H2CCH2OH* are key reduction intermediates in the reduction mechanism of the CO dimer on Cu(100), which are not in agreement with a previous thermodynamic study. Furthermore, the formation of HCCHO* is relatively more difficult during the course of the further reduction of OHCCHO*, which is not a kinetically favorable pathway, and an activation barrier of 1.02 eV is required by deoxygenation of OHCCHO* (i.e. the cleavage of C–O bond). In our present mechanistic study, the H2CCH2O* intermediate can be formed by the deoxygenation of OH2CCH2O* and dehydroxylation of OH2CCH2OH*. However, the activation barriers of H2CCH2O* formation in these two pathways are very high (2.71 and 1.36 eV, respectively) compared to the further hydrogenation of OH2CCH2O* and OH2CCH2OH*. Therefore, the formation of the H2CCH2O* intermediate may not be a kinetically favorable step during the course of CO reduction by dimerization. Considering the difficulty of cleavage of the C–O bond, we can infer that H2CCHO* is not a reduction intermediate the reduction mechanism of the CO dimer. The present mechanistic study results showed that the preferred pathway on Cu(100) based solely on the reaction free energies may be misleading, and the reaction kinetics of elementary reaction steps provide a different mechanistic explanation for selective production C2H4 compared to CH4 on Cu electrodes. An alternative reaction pathway that suggests the production of C2H4 through the OCCO* intermediate from CO dimerization is provided in our present mechanistic study. This reduction pathway is consistent with the latest experimental results and explains the puzzle in the experiment from Schouten and coworkers,8,9 who suggested that OHCCHO*, HOCCOH* and H2CCH2O* are possible reduction intermediates in the reduction mechanism of the CO dimer to C2H4, but this is still uncertain. Our present research results provide an explanation that OHCCHO* is a key reaction intermediate in the reduction mechanism of the CO dimer other than HOCCOH* and H2CCH2O* on Cu(100). At present, it appears that the mechanism proposed in Fig. 12 is most agreeable with the experimental results. In reality, the electrode potential should be considered in our present MEP analysis. However, the kinetic model of coupling with the electrode potential still needs to be explored further because of the uncertainty of the electrode potential, in which the work function differs among these adsorbed states when kinetically modeling electrochemical reaction systems, leading to differences in the electrode potentials during the reaction. Although the relationship between the kinetic electrochemical reduction pathways of CO and electrode potential from our present data cannot be deduced, the conclusions in the trends are expected to be reasonably accurate.
5. Conclusions
A systematic DFT study that examines the role of kinetics of the elementary steps in reduction mechanism of the CO dimer to the production C2H4 on Cu(100) is presented for the first time in the present study, and a new reduction mechanism is introduced. Kinetic analysis of elementary reaction steps has suggested that further reduction of CO is the key selectivity-determining step for the formation of C2H4 and CH4 on Cu(100) and Cu(111), respectively. The main reaction pathway on Cu(111) proceeds through the reduction of CO to the CHO* intermediate, which may eventually result in CHx species by breaking of the C–O bond and the production of CH4. On Cu(100), CO dimer, OCCO* is first formed by CO dimerization, which is the first step and a more favorable pathway than the further hydrogenation of CO. This explains why only C2 species and not C1 species are observed experimentally on Cu(100). For the formation of C2H4 on Cu(100), the results suggest that the hydrogenation of OCCO* to the OCCHO* intermediate is the most possible reaction path, followed by the formation of the OHCCHO* intermediate through further hydrogenation of the OCCHO* intermediate. The OH2CCH2OH* intermediate is formed by the serial hydrogenation of OHCCHO*. The OH2CCH2OH* intermediate can form HOH2CCH2OH* by direct hydrogenation and H2CCH2OH* by breaking the C–O bond, which might be a parallel pathway in the reduction mechanism of the CO dimer on Cu(100). Finally, the production C2H4 is formed by dehydroxylation of the HOH2CCH2OH* and H2CCH2OH* intermediates. The formation of a dimer, OCCO* may be the rate-determining step in the reduction of the CO dimer on Cu(100). In contrast to the previous suggested thermodynamic theoretical study on the reduction mechanisms of the CO dimer into the production of C2H4, our present reaction kinetics of elementary steps provide a different mechanistic explanation for the selectivity of C2H4 production. This reduction pathway is consistent with the latest experimental results and explains the puzzle in the experiment on the uncertainty of the reaction intermediates. Our present research results also suggest that OHCCHO* is a key reaction intermediate in reduction mechanism of the CO dimer on Cu(100) other than HOCCOH* and H2CCH2O* suggested in a previous thermodynamic study. At present, it appears that the mechanism proposed in this study is most agreeable with the present experimental results.
Acknowledgements
This study is financially supported by the National Natural Science Foundation of China (Grant No. 21303048), the Hunan Provincial Natural Science Foundation of China (Grant No. 13JJ4101), the Construct Program of the Key Discipline in Hunan Province (Applied Chemistry) and the Doctoral Start-up Fund of Hunan University of Arts and Science.
References
- F. Hasegawa, S. H. Yokoyama and K. Imou, Bioresour. Technol., 2010, 101, S109–S111 CrossRef CAS PubMed.
- M. Abu-zahra, L. H. Schneiders, J. P. M. Niederer, P. H. Feron and G. F. Versteeg, Int. J. Greenhouse Gas Control, 2007, 1, 37–46 CrossRef CAS.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 14, 1695–1698 CrossRef.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Nørskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 CAS.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- K. J. P. Schouten, Z. Qin, E. P. Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- K. J. P. Schouten, E. P. Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- P. Hirunsit, W. Soodsawang and J. Limtrakul, J. Phys. Chem. C, 2015, 119, 8238–8249 CAS.
- A. A. Peterson, F. Abild-Pederson, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 CAS.
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pederson and J. K. Nørskov, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS.
- L. H. Ou, RSC Adv., 2015, 5, 57361–57371 RSC.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS.
- Y. Hori, A. Murata, T. Tsukamoto, H. Wakebe, O. Koga and H. Yamazaki, Electrochim. Acta, 1994, 39, 2495–2500 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- R. Kas, R. Kortlever, A. Milbrat, M. T. M. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B, 1990, 41, 7892–7895 CrossRef.
- M. Methfessel and A. T. Paxton, Phys. Rev. B, 1989, 40, 3616–3621 CrossRef CAS.
- S. Baroni, A. Dal Corso, S. de Gironcoli and P. Giannozzi, PWSCF and PHONON: Plane-Wave Pseudo-Potential Codes. http://www.pwscf.org, 2001 Search PubMed.
- A. Kokalj, J. Mol. Graphics Modell., 1999, 17, 176–179 CrossRef CAS PubMed.
- A. Kokalj and M. Causà, Scientific Visualization in Computational Quantum Chemistry. In Proceedings of High Performance Graphics Systems and Applications European Workshop, CINECA-Interuniversity Consortium, Bologna, Italy, 2000 Search PubMed.
- A. Kokalj and M. Causà, XCrySDen: X-Window CRYstalline Structures and DENsities. http://www.xcrysden.org/, 2001 Search PubMed.
- J. Greeley, A. A. Gokhale, J. Kreuser, J. A. Dumesic, H. Topsøe, N. Y. Topsøe and M. Mavrikakis, J. Catal., 2003, 213, 63–72 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, CRC Press/Taylor and Francis, Boca Raton, FL, 91st edn, 2011 (Internet Version 2011) Search PubMed.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 15, 897–898 CrossRef.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra15905a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.