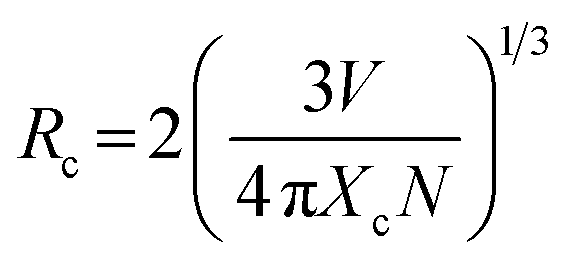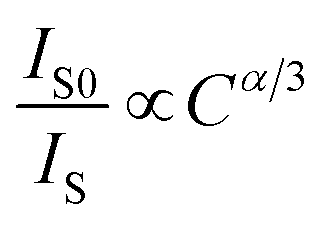DOI:
10.1039/C5RA19345A
(Paper)
RSC Adv., 2015,
5, 96272-96280
Tunable white-light emission via energy transfer in single-phase LiGd(WO4)2:Re3+ (Re = Tm, Tb, Dy, Eu) phosphors for UV-excited WLEDs
Received
19th September 2015
, Accepted 26th October 2015
First published on 28th October 2015
Abstract
A series of novel single-phase color tunable LiGd(WO4)2(LGW):Re3+ (Re = Tm, Tb, Dy, Eu) phosphors have been synthesized by a solid-state reaction. X-ray diffraction (XRD), FT-IR, photoluminescence (PL) and fluorescence decay curves were utilized to characterize the as-prepared samples. Under the excitation of UV light, LGW:Tm3+, LGW:Tb3+, LGW:Dy3+ and LGW:Eu3+ exhibit the characteristic emissions of Tm3+ (blue), Tb3+ (green), Dy3+ (yellow) and Eu3+ (red), respectively. On the one hand, by simply adjusting the doping concentration of Eu3+ ions in the LGW:2% Tm3+, 4% Tb3+, x% Eu3+ system, a white emission in a single-phase was achieved by blending simultaneously the blue, green, and red emissions of Tm3+, Tb3+, and Eu3+ ions in the LGW host, in which the energy transfer from Tb3+ to Eu3+ ions was found to play an important role. On the other hand, a white emission can also be realized based on the energy transfer from Tm3+ to Dy3+ ions in the LGW:2% Tm3+, x% Dy3+ system. The energy transfer mechanism from Tm3+ to Dy3+ ions has been demonstrated to be through dipole–quadrupole interaction and the critical distance (RTm–Dy) calculated by the concentration quenching method is 16.03 Å. The PL properties of as-prepared materials indicate that LGW:Re3+ (Re = Tm, Tb, Dy, Eu) may be potentially applied as single-phase white-light-emitting phosphors for UV-excited WLEDs.
1. Introduction
Nowadays, with the continuous increase of global electricity consumption for artificial lighting, the development of new high-efficiency lighting sources has become particularly important for reducing the world’s energy consumption.1–4 As a hot spot in the field of solid-state lighting applications, white light-emitting diodes (WLEDs) are expected to be the most promising long-term solution due to their attractive merits of energy savings, longer lifetimes (>100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h), faster response, higher luminous efficiency and environmental friendliness compared with conventional incandescent and fluorescence lamps.5–7 The 2014 Nobel Prize in Physics, awarded to Isamu Akasaki, Hiroshi Amano and Shuji Nakamura “for the invention of efficient blue light-emitting diodes which has enabled bright and energy-saving white light sources”, adequately affirmed this technology.8 Currently, the most common method for producing commercial WLEDs is based on a blue-emitting InGaN LED chip with a yellow-emitting phosphor (YAG:Ce3+). Despite their wide applications and high luminous efficacy (>100 lm W−1), the major disadvantage of such a combination is that they are limited to a high correlated color temperature (CCT > 6000 K) and low color rendering index (Ra < 80) due to the lack of a sufficient red spectral component, which restricts their widespread commercialization in the general lighting market.9–11 This limitation can be solved by utilizing an ultraviolet (UV) light-emitting diode (LED) chip coated with multiphased phosphors emitting blue, green, and red light. However, the fatal shortcoming of this method is the relatively low luminescence efficiency owing to the strong reabsorption of the blue light by the red and green phosphors.12–14 Moreover, multi-phosphor systems are difficult to make, because the phosphors have to be prepared separately, the particle sizes of individual phosphor materials have to be adapted to one another in order to avoid agglomeration or sedimentation, and the final product has to be mixed homogeneously in exact ratios.8 To solve the above challenges, recently, single-phase white-light-emitting phosphors pumped by UV or near-UV LED chips have attracted great interest and been considered as an effective approach for phosphor-converted WLEDs, since they show significant advantages compared with the multiphased phosphors, including higher luminescence efficiency, higher color rendering index, better reproducibility, lower manufacturing costs and an easier fabrication process.15–18
000 h), faster response, higher luminous efficiency and environmental friendliness compared with conventional incandescent and fluorescence lamps.5–7 The 2014 Nobel Prize in Physics, awarded to Isamu Akasaki, Hiroshi Amano and Shuji Nakamura “for the invention of efficient blue light-emitting diodes which has enabled bright and energy-saving white light sources”, adequately affirmed this technology.8 Currently, the most common method for producing commercial WLEDs is based on a blue-emitting InGaN LED chip with a yellow-emitting phosphor (YAG:Ce3+). Despite their wide applications and high luminous efficacy (>100 lm W−1), the major disadvantage of such a combination is that they are limited to a high correlated color temperature (CCT > 6000 K) and low color rendering index (Ra < 80) due to the lack of a sufficient red spectral component, which restricts their widespread commercialization in the general lighting market.9–11 This limitation can be solved by utilizing an ultraviolet (UV) light-emitting diode (LED) chip coated with multiphased phosphors emitting blue, green, and red light. However, the fatal shortcoming of this method is the relatively low luminescence efficiency owing to the strong reabsorption of the blue light by the red and green phosphors.12–14 Moreover, multi-phosphor systems are difficult to make, because the phosphors have to be prepared separately, the particle sizes of individual phosphor materials have to be adapted to one another in order to avoid agglomeration or sedimentation, and the final product has to be mixed homogeneously in exact ratios.8 To solve the above challenges, recently, single-phase white-light-emitting phosphors pumped by UV or near-UV LED chips have attracted great interest and been considered as an effective approach for phosphor-converted WLEDs, since they show significant advantages compared with the multiphased phosphors, including higher luminescence efficiency, higher color rendering index, better reproducibility, lower manufacturing costs and an easier fabrication process.15–18
The most widely used method to realize white light in a single-phased phosphor is by codoping a sensitizer and an activator into the same host matrix. It should be pointed out that energy transfer plays a critical role in achieving white light emission in a single-phase phosphor. As a result, single-phase white-light-emitting phosphors based on the mechanism of energy transfer from a sensitizer to an activator have been realized in many inorganic hosts, such as NaCaBO3:Ce3+, Tb3+, Mn2+,19 Na2Ca4Mg2Si4O15:Eu2+, Mn2+,20 Ca4(PO4)2O:Ce3+, Eu2+,21 BaY2Si3O10:Ce3+, Tb3+, Eu3+,22 Sr7La3[(PO4)2.5(SiO4)3(BO4)0.5](BO2):Ce3+, Mn2+,23 and so on. However, some harsh reaction conditions such as high temperature (above 1000 °C) and reducing atmosphere especially for Eu2+ or Ce3+ ion doped phosphors, lead to a high investment and energy costs. So, it will be interesting to find a new single-phase white-light phosphor which can be synthesized at a moderate temperature and in ambient atmosphere. Recently, scheelite-like double tungstate phosphors with the general formula of MRe(WO4)2 (M = alkali metal, Re = rare earth) for WLED applications have gained increasing interest for their promising luminescence properties, excellent physical and chemical stability, low synthetic temperature, and environmentally friendly characteristics.24–27 In addition, the optical properties of these materials can be well improved due to their strong ultraviolet absorption capacity and effective transfer of the absorbed energy to the doping ions.
In this research, a series of novel single-phase LiGd(WO4)2(LGW) phosphors with tunable emission color through singly doping or codoping rare earth ions (Tm3+, Tb3+, Dy3+, and Eu3+) have been successfully synthesized by solid-state reaction. We find that LGW:Tm3+ phosphors show blue emission under the excitation of 360 nm, which match well with the emission light of UV-LED chips. Furthermore, two routes are discovered to realize the white emission in a single LGW host. When codoping Tm3+, Tb3+, and Eu3+ ions into a LGW host, a white emission is realized by simply adjusting the doping concentration of Eu3+ ions, in which the energy transfer from Tb3+ to Eu3+ ions was found to play an important role. On the other hand, a white emission can also be achieved by utilizing the principle of energy transfer and the appropriate tuning of Tm3+ and Dy3+ contents in LGW:Tm3+, Dy3+. Moreover, we investigate the energy transfer mechanism between Tm3+ and Dy3+ ions in LGW:Tm3+, Dy3+, which results in tunable emission colors from such phosphor materials. These results indicate that the as-prepared products may be potentially applied as candidates of single-phase white-light-emitting phosphors for application in WLEDs.
2. Experimental
2.1 Materials and synthesis
A series of rare-earth-doped LiGd(WO4)2 phosphors were synthesized through the solid-state reaction. Li2CO3 (A.R.), WO3 (A.R.), Gd2O3 (99.99%), Tm2O3 (99.99%), Tb4O7 (99.99%), Dy2O3 (99.99%) and Eu2O3 (99.99%) were used as the starting materials. Stoichiometric amounts of the raw materials were first thoroughly mixed by grinding them in an agate mortar with an appropriate amount of ethanol and then dried at 80 °C for 0.5 h. After regrinding for 10 min, the powder mixtures were loaded into a crucible and transferred to a box furnace to calcine at 800 °C for 6 h in air with a heating rate of 3 °C min−1. Finally, the prepared samples were cooled to room temperature and then ground once more for further measurements.
2.2 Characterization
The X-ray diffraction (XRD) patterns of the samples were recorded on a Rigaku D/max 2400 X-ray diffractometer using Cu Kα radiation (λ = 0.15405 nm). Fourier transform infrared (FTIR) spectra were performed on a JASCO FT/IR-460 plus spectrometer using a KBr disk technique. Photoluminescence (PL) emission and photoluminescence excitation (PLE) spectra as well as quantum efficiency were carried out on a Hitachi F-7000 spectrophotometer equipped with a 150 W xenon lamp as the excitation source. The luminescence decay curves were measured using an FLS920 spectrofluorometer (Edinburgh Instruments). All the measurements were performed at room temperature.
3. Results and discussion
3.1 Phase identification and structure
The representative XRD patterns of LGW and Tm3+, Tb3+, Dy3+, and Eu3+ ion doped LGW samples as well as the standard data of LiGd(WO4)2 (JCPDS no. 04-008-0355) are presented in Fig. 1. It is obvious that the diffraction peaks of all these samples are well assigned to the pure tetragonal phase of LiGd(WO4)2 [space group: I41/a (88)] according to the JCPDS file 04-008-0355, and no traces of impurity phases are observed, indicating that the Tm3+, Tb3+, Dy3+ and Eu3+ ions can be completely dissolved into this host and their introductions do not cause any significant changes to the crystal structure. LiGd(WO4)2 crystals belong to a scheelite-type tetragonal structure, which have the cell parameters of a = b = 5.20 Å, c = 11.17 Å, V = 302.03 Å3 and Z = 2.28 Fig. 2 shows the unit cell structure of LiGd(WO4)2 and the coordination environments of the Gd3+ (Li+) and W6+ sites. It can be seen that the central W6+ is coordinated by four oxygen atoms at a tetrahedral site and the Li+ and Gd3+ cations are randomly distributed over the same site positions and are coordinated by eight oxygen atoms to form a distorted polyhedron with symmetry S4 (without an inversion center).29–31 Based on the similar effective ionic radii and charge balance, the Tm3+, Tb3+, Dy3+ and Eu3+ ions are expected to occupy Gd3+ sites in LiGd(WO4)2.
 |
| | Fig. 1 XRD patterns of representative LGW host (a), LGW:5% Tm3+ (b), LGW:5% Tb3+ (c), LGW:5% Dy3+ (d), LGW:5% Eu3+ (e), LGW:2% Tm3+, 8% Dy3+ (f), and LGW:2% Tm3+, 4% Tb3+, 7% Eu3+ (g) samples as well as the standard data of LiGd(WO4)2 (JCPDS no. 04-008-0355). | |
 |
| | Fig. 2 The unit cell structure of LGW and the coordination environment of cations in the host lattice. | |
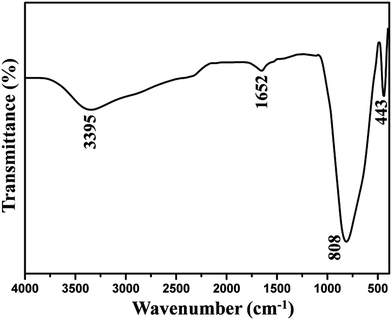
To determine the chemical constituents of the sample, FT-IR spectroscopy was performed on the pure LGW phosphor, as shown in Fig. 3. A broad absorption band at 3395 cm−1 is ascribed to the stretching vibration of O–H, which belongs to physically absorbed water on the surface of the product from air. And a weak absorption band at 1652 cm−1 is attributed to the bending vibration of O–H. The set of bands below 1000 cm−1 are characteristic vibrations of W–O bands (from the WO42− group). A strong absorption band at 808 cm−1 and a weak band at 443 cm−1 are caused by the stretching vibration of W–O and bending vibration of W–O, respectively.32
 |
| | Fig. 3 FT-IR spectra of pure LGW. | |
3.2 Photoluminescence properties
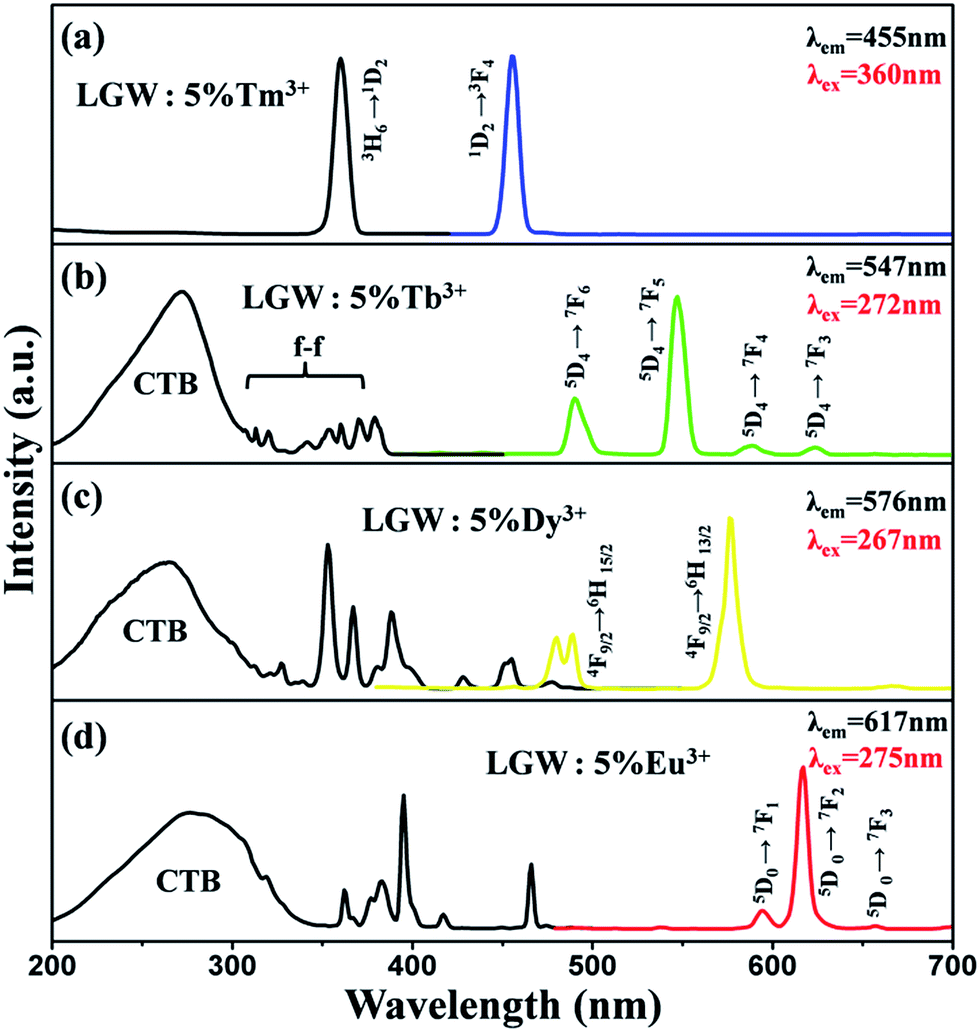
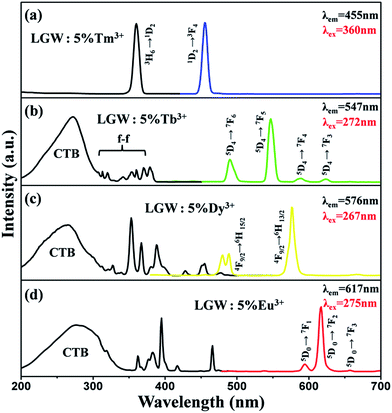
Fig. 4 illustrates the PLE and PL spectra of LGW:5% Tm3+, LGW:5% Tb3+, LGW:5% Dy3+ and LGW:5% Eu3+ samples, respectively. The excitation spectrum of LGW:5% Tm3+ shown in Fig. 4a contains a sharp absorption peak at 360 nm attributed to the 3H6 → 1D2 transition of Tm3+,33 which matches well with the UV-LED chips. Upon excitation at 360 nm, the LGW:5% Tm3+ phosphor shows an intense blue emission attributed to the 1D2 → 3F4 transition (455 nm),34 indicating it could be an outstanding candidate as a blue phosphor for UV-LED chips. Fig. 4b shows the PL excitation and emission spectra of LGW:5% Tb3+ phosphors. The excitation spectrum was obtained by monitoring the emission of Tb3+ (547 nm, 5D4 → 7F5). It is observed that the excitation spectrum can be divided into two parts: one is a broad band with a maximum at 272 nm, which is attributed to the charge-transfer absorption from the 2p orbitals of oxygen to the 5d orbitals of tungsten within the WO42− groups and the 4f8 → 4f75d transition of Tb3+;35 the other one is composed of a series of narrow bands from 300 to 400 nm, which correspond to the characteristic f–f transitions of Tb3+ within its 4f8 configuration.25 Under 272 nm excitation, the obtained emission spectrum of LGW:5% Tb3+ consists of f–f transition lines within a 4f8 electron configuration of Tb3+, i.e., 5D4 → 7F6 (490 nm) in the blue region and 5D4 → 7F5 (547 nm) in the green region, as well as 5D4 → 7F4 (590 nm) and 5D4 → 7F3 (623 nm) in the red region.36 The strongest one is located at 547 nm corresponding to the 5D4 → 7F5 transition of Tb3+. As for LGW:5% Dy3+ phosphors, the excitation spectra monitoring the emission at 576 nm corresponding to the 4F9/2 → 6H13/2 transition of Dy3+ is shown in Fig. 4c. It can be seen that the excitation spectrum of LGW:5% Dy3+ consists of a strong broad band from 200 to 320 nm with a maximum at 267 nm due to the WO42− groups as discussed above. In the longer wavelength region from 310 to 500 nm, some sharp lines due to the f–f transitions of Dy3+ are observed, which are ascribed to the transitions from the 6H15/2 ground state to the different excited states of Dy3+, i.e., 327 nm (6P3/2), 353 nm (6P7/2), 367 nm (6P5/2), 388 nm (4I13/2), 428 nm (4G11/2), 455 nm (4I15/2), and 477 nm (4F9/2), respectively.37 Upon the excitation at 267 nm, LGW:5% Dy3+ phosphors obtain blue and yellow luminescence, corresponding to the 4F9/2 → 6H15/2 (478 and 488 nm) and the 4F9/2 → 6H13/2 (576 nm) transitions of Dy3+ ions, respectively. The 4F9/2 → 6H15/2 transition is the magnetic dipole transition, which hardly varies with the crystal field strength or coordination environment around Dy3+ ions. However, the forced electric dipole transition 4F9/2 → 6H13/2 transition (ΔJ = 2) is hypersensitive to the chemical environment around Dy3+ ions in the host lattice.38 When Dy3+ ions locate at low-symmetry sites without an inversion center, the yellow emission 4F9/2 → 6H13/2 transition is often prominent in the emission spectra. From the results in Fig. 4c (right), the intensity of the 4F9/2 → 6H13/2 transition is much stronger than that of the 4F9/2 → 6H15/2 transition, indicating that Dy3+ ions are located in an asymmetric cation environment, which is consistent with a symmetric S4 without an inversion center. At last, Fig. 4d shows the PL excitation and emission spectra of LGW:5% Eu3+ phosphors. When monitoring the red emission of Eu3+ (617 nm, 5D0 → 7F2), the PLE spectrum of LGW:5% Eu3+ reveals a broad band ranging from 200 to 350 nm ascribed to the CTB of the WO42− groups and the O2−–Eu3+ charge transfer transition from an oxygen 2p state excited to an Eu3+ 4f state.26 In addition, we can easily distinguish the characteristic lines located at 362, 383, 395, 417, and 466 nm corresponding to the transitions of Eu3+ ions from the ground level 7F0 to the 5D4, 5L7, 5L6, 5D3, and 5D2 excited levels, respectively.39 The PL spectrum of the LGW:5% Eu3+ phosphor is obtained by exciting at 275 nm. It can be found that the strongest emission peak of LGW:5% Eu3+ is located at 617 nm, which originates from the 5D0 → 7F2 transition of Eu3+. Two other weak peaks at 594 and 657 nm are attributed to the 5D0 → 7F1 and 5D0 → 7F3 transitions of Eu3+, respectively. It is well-known that Eu3+ is an excellent structural probe for investigating the local environment in a host lattice. The electric dipole transition 5D0 → 7F2 is a hypersensitive one, and the emission intensity is strongly influenced by the local environment surrounding Eu3+ ions. When Eu3+ ions are located at a low symmetry site, the 5D0 → 7F2 transition often dominates in the emission spectrum. Nevertheless, the magnetic dipole transition (5D0 → 7F1) is independent to the local crystal field environment.40 In the present case, the emission intensity of electric transition is much stronger than that of magnetic dipole transition, indicating that the Eu3+ ions occupy the low symmetry sites, which is in accordance with the emission spectrum of LGW:5% Dy3+. As illustrated in Fig. 4a–d, there are some excitation peaks from 350 to 420 nm in the PLE spectrum of Tm3+, Tb3+, Dy3+ and Eu3+ doped LGW samples, which indicate that those phosphors can match well with the dominant emission band of a UV-LED chip. In addition, the presence of the excitation peak for WO42− groups in the excitation spectrum of Tb3+/Dy3+/Eu3+ implies that there is an energy transfer from WO42− groups of the host to Tb3+/Dy3+/Eu3+ ions in phosphors.41 However, we do not observe a charge transfer band for WO42− groups in the PLE spectrum of LGW:Tm3+, certifying the inexistence of the energy transfer from WO42− to Tm3+.
 |
| | Fig. 4 PL and PLE spectra of (a) LGW:5% Tm3+, (b) LGW:5% Tb3+, (c) LGW:5% Dy3+ and (d) LGW:5% Eu3+ samples. | |
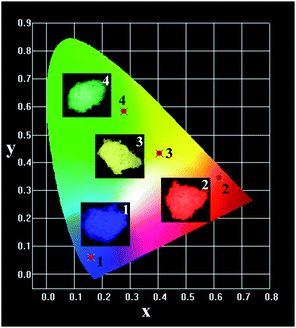
A better understanding of the actual emission color of the phosphor is very important for application in lighting and display devices. Fig. 5 shows the Commission Internationale de L’Eclairage (CIE) chromaticity coordinates and the corresponding digital luminescence photographs of LGW:5% Tm3+, LGW:5% Tb3+, LGW:5% Dy3+ and LGW:5% Eu3+ phosphors. The CIE chromaticity coordinates of LGW:Tm3+/Tb3+/Dy3+/Eu3+ phosphors can vary from (0.160, 0.061) for LGW:5% Tm3+ to (0.277, 0.583) for LGW:5% Tb3+, (0.406, 0.431) for LGW:5% Dy3+, and (0.619, 0.345) for LGW:5% Eu3+, which correspond to the color changes from blue to green, yellow, and red, respectively.
 |
| | Fig. 5 CIE color coordinate diagram of LGW doped with 5% (1) Tm3+, (2) Eu3+, (3) Dy3+, and (4) Tb3+. The insets are their corresponding digital photographs taken under 365 nm excitation. | |
The aim of this study is to realize the white light emission in a single-phase host for UV-excited WLEDs. For this purpose, an appropriate combined amount of the luminescent ions Tm3+, Tb3+, and Eu3+ as emitters of blue, green, and red light codoped into a LGW host was selected for white light generation. Therefore, a series of LGW:2% Tm3+, 4% Tb3+, x% Eu3+ (x = 0, 1, 3, 5, 7) samples have been prepared. The PL spectra of the as-prepared samples under excitation at 360 nm and the corresponding CIE chromaticity diagram are presented in Fig. 6. Under excitation at 360 nm, the emission color varies from light green (which is represented by point 1) to rose red (point 5), and in particular the white light is realized (point 3) by simply controlling the doping concentration of Eu3+ ions (Fig. 6b). Therefore, a novel single-phase white-emitting phosphor is successfully obtained by simultaneously blending the blue, green, and red emissions of Tm3+, Tb3+, and Eu3+ ions in a LGW host under UV light excitation.
 |
| | Fig. 6 (a) PL emission spectra of LGW:2% Tm3+, 4% Tb3+, x% Eu3+ samples with different Eu3+ concentrations (0 ≤ x ≤ 7). (b) CIE coordinate diagram of LGW:2% Tm3+, 4% Tb3+, x% Eu3+ with different Eu3+ concentrations: (1) x = 0; (2) x = 1; (3) x = 3; (4) x = 5; and (5) x = 7. The insets are their corresponding digital photographs taken under 365 nm excitation. | |
Detailed information of LGW:2% Tm3+, 4% Tb3+, x% Eu3+ (x = 0, 1, 3, 5, 7) phosphors for the CIE chromaticity coordinates and CCTs are listed in Table 1. Moreover, it is worth noting that although the concentration of Tb3+ was fixed at 4%, as the Eu3+-doping concentration increased, the emission intensity of Eu3+ was enhanced clearly whereas the emission intensity of Tb3+ decreased steadily (Fig. 6a), indicating that there is an energy transfer process from Tb3+ to Eu3+ ions. As reported, the luminescence intensities of various rare earth ions can be enhanced or quenched by an energy transfer from other codoping rare earth ions.42 The energy transfer between Tb3+ and Eu3+ is a well-known phenomenon since the 5D4 → 7F6,5,4,3 emissions of Tb3+ are effectively overlapped with the 7F0,1 → 5D0,1,2 absorptions of Eu3+.43 So far, the energy transfer phenomenon from Tb3+ to Eu3+ has been extensively investigated in many inorganic hosts, such as molybdates,36 fluorides,44 germanates,45 tungstates,46 oxyhalides,47 niobates,48 and so on. In order to provide further evidence to validate the energy transfer from Tb3+ to Eu3+ ions in the LGW host lattice, the fluorescence decay curves of Tb3+ in LGW:2% Tm3+, 4% Tb3+, x% Eu3+ (0 ≤ x ≤ 7) samples excited at 272 nm, monitoring the emission of Tb3+ ions at 547 nm, were measured and shown in Fig. 7. The decay curves fit well with a single exponential formula as follows:49
| | |
I = I0exp(−t/τ)
| (1) |
where
I and
I0 are the luminescence intensities at time
t and 0, and
τ is the luminescence lifetime. For the LGW:2% Tm
3+, 4% Tb
3+,
x% Eu
3+ (0 ≤
x ≤ 7) samples, the lifetime of Tb
3+ decreases with increasing Eu
3+ concentration, which are 5.26, 4.55, 4.31, 4.14 and 3.87 ms for
x = 0, 1, 3, 5 and 7, respectively. This is powerful proof for the energy transfer from Tb
3+ to Eu
3+ ions.
Table 1 Comparison of the CIE chromaticity coordinates and CCT (K) for LGW:2% Tm3+, 4% Tb3+, x% Eu3+ phosphors excited under 360 nm UV radiation
| Sample no. |
Sample composition |
CIE coordinates (x, y) |
CCT (K) |
| 1 |
x = 0 |
(0.279, 0.418) |
7344 |
| 2 |
x = 1 |
(0.321, 0.356) |
5993 |
| 3 |
x = 3 |
(0.343, 0.327) |
4980 |
| 4 |
x = 5 |
(0.377, 0.296) |
3159 |
| 5 |
x = 7 |
(0.435, 0.280) |
1961 |
 |
| | Fig. 7 The decay curves for the luminescence of Tb3+ ions in LGW:2% Tm3+, 4% Tb3+, x% Eu3+ samples with changing Eu3+ concentrations (excited at 272 nm, monitored at 547 nm). | |
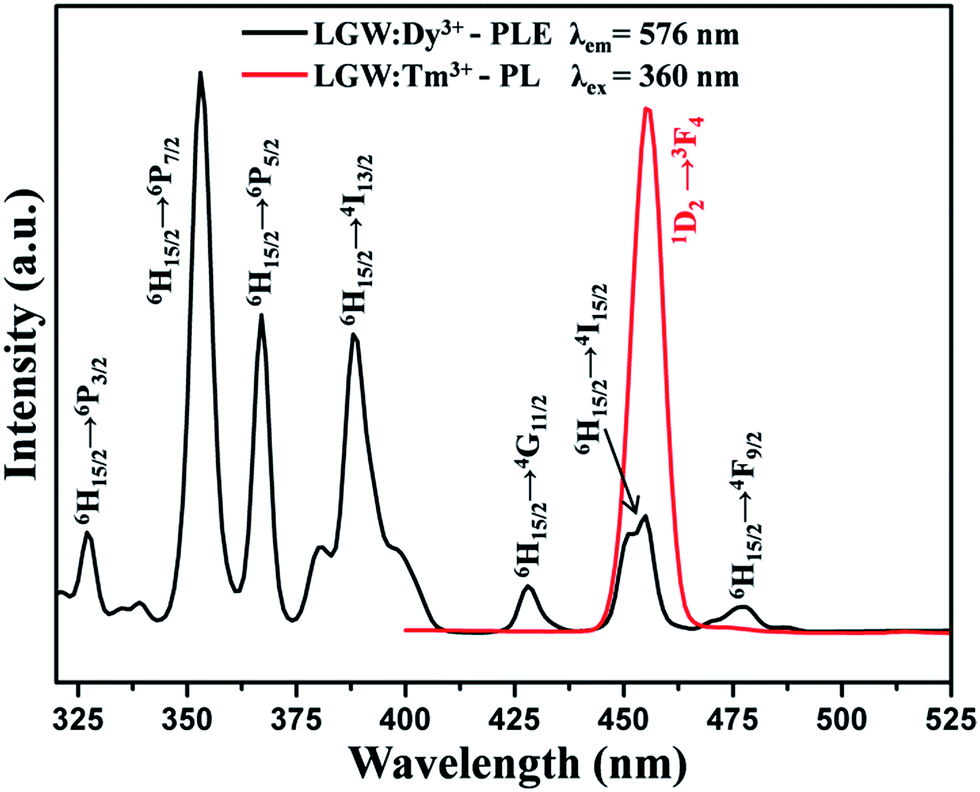
Interestingly, white light emission can also be realized by combining the blue and yellow emissions of Tm3+ and Dy3+ ions in a single host under UV light excitation. As shown in Fig. 8, the comparison of the PL spectrum of LGW:Tm3+ and the PLE spectrum of LGW:Dy3+ reveals a significant spectral overlap between the emission of Tm3+ and the excitation of Dy3+. Therefore, an effective resonance-type energy transfer from Tm3+ to Dy3+ in a LGW host is expected. In order to further investigate the energy transfer process from Tm3+ to Dy3+ and to study the impact of doping concentration on the luminescence properties of phosphors, a series of LGW:2% Tm3+, x% Dy3+ (x = 0, 2, 4, 6, 8, 10) samples were prepared. The PL spectra of LGW:2% Tm3+, x% Dy3+ under 360 nm excitation are illustrated in Fig. 9. The doping concentration of Tm3+ was fixed at 2%, while the Dy3+ content changes from 0% to 10%. We can observe that as the amount of Dy3+ content increases, the emission intensity of Tm3+ gradually decreases, whereas the emission intensity of Dy3+ first increases to an optimum concentration of 6%, and then begins to decrease because of the concentration quenching effect, which confirms the existence of energy transfer from Tm3+ to Dy3+ ions. Meanwhile, those single-phase LGW:2% Tm3+, x% Dy3+ phosphors exhibit abundant color-tunable emissions through an effective energy transfer process and controlling the doping concentration of Dy3+. The most interesting part of this approach is its ability to realize white light emission from a particular sample of LGW:2% Tm3+, 6% Dy3+. This has been confirmed by direct exposure to ultraviolet (UV) light as well as from the CIE coordinate calculation (discussed later). In addition, the fluorescence decay lifetime measurements for Tm3+ emission (λex = 360 nm, λem = 455 nm) with increasing Dy3+ concentration in LGW:2% Tm3+, x% Dy3+ were conducted to further confirm the occurrence of energy transfer from Tm3+ to Dy3+. Fig. 10 shows the decay curves of Tm3+ emission in LGW:2% Tm3+, x% Dy3+ samples. The decay curves can be fitted successfully by a second-order exponential function as given by the following equation:50
| | |
I = A1exp(−t/τ1) + A2exp(−t/τ2)
| (2) |
where
I represents the luminescence intensity;
A1 and
A2 are constants;
t is the time;
τ1 and
τ2 are rapid and slow decay times for exponential components, respectively. As a result, the average decay times (
τ*) can be determined using the following equation:
| | |
τ* = (A1τ12 + A2τ22)/(A1τ1 + A2τ2)
| (3) |
 |
| | Fig. 8 Spectral overlap between the PL spectrum of LGW:Tm3+ (red line) and PLE spectrum of LGW:Dy3+ (black line). | |
 |
| | Fig. 9 PL emission spectra of LGW:2% Tm3+, x% Dy3+ samples with different Dy3+ concentrations (x = 0, 2, 4, 6, 8, and 10). | |
 |
| | Fig. 10 Photoluminescence decay curves of Tm3+ in LGW:2% Tm3+, x% Dy3+ with changing Dy3+ concentrations. | |
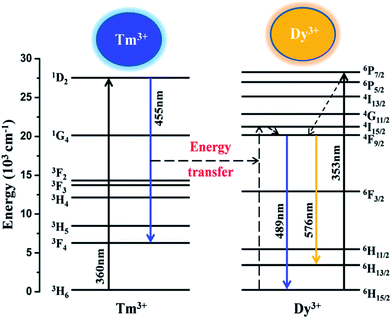
On the basis of eqn (3), the average lifetimes of Tm3+ are determined to be 9.56, 7.82, 6.63, 5.31 and 4.54 μs for Dy3+ concentrations of 0%, 4%, 6%, 8%, and 10%, respectively. The decrease in the lifetimes of Tm3+ with increasing Dy3+ concentration strongly demonstrates the existence of energy transfer from Tm3+ to Dy3+ ions. Fig. 11 illustrates a schematic diagram of the energy transfer process from Tm3+ to Dy3+ in a LGW:Tm3+, Dy3+ phosphor. As shown in Fig. 11, the energy gap between 1D2 and 3F4 of Tm3+ matches well with that between 6H15/2 and 4I15/2 of Dy3+, which makes the energy transfer from Tm3+ to Dy3+ ions efficient. It illustrates the existence of energy transfer from Tm3+ to Dy3+ when they are codoped into the LGW host and provides a necessary condition for synthesizing the single-phase color-tunable phosphors.
 |
| | Fig. 11 A simple model expressing the energy transfer from Tm3+ to Dy3+ in LGW:Tm3+, Dy3. | |
3.3 Energy transfer mechanism
The energy transfer efficiency (ηT) from Tm3+ to Dy3+ in LGW:2% Tm3+, x% Dy3+ systems can be calculated using the following equation:51where ηT is the energy transfer efficiency, and IS and IS0 are the luminescence intensities of Tm3+ ions in the presence and absence of Dy3+, respectively. As shown in Fig. 12, the intensity of Tm3+ emission and the energy transfer efficiency from Tm3+ to Dy3+ in a LGW:2% Tm3+, x% Dy3+ system are plotted as a function of Dy3+ concentration. One can find that with the increase of Dy3+ concentration, the intensity of Tm3+ emission decreases gradually, while the energy transfer efficiency steadily increases to reach a maximum at 84% when using UV irradiation of 360 nm as the excitation wavelength, indicating that the energy transfer from Tm3+ to Dy3+ becomes more and more efficient.
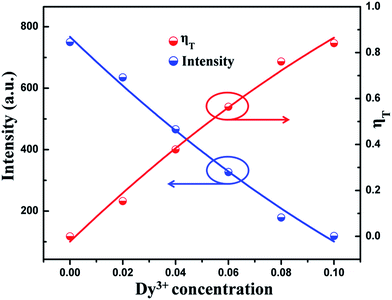 |
| | Fig. 12 Dependence of the emission intensity of Tm3+ and the energy transfer efficiency (ηT) on Dy3+ concentration in LGW:2% Tm3+, x% Dy3+ (x = 0, 2, 4, 6, 8, 10) phosphors. | |
In order to determine the energy transfer mechanism from Tm3+ to Dy3+ ions, it is necessary to know the critical distance (Rc) between the sensitizer (Tm3+) and activator (Dy3+). With the increase of Dy3+ content, the distance between Tm3+ and Dy3+ ions becomes shorter and shorter, thus the probability of energy migration increases. When the distance becomes small enough, concentration quenching occurs and energy migration is hindered. Therefore, the critical distance (Rc) for energy transfer from Tm3+ to Dy3+ ions can be calculated using the concentration quenching method. According to Blasse,52,53 the critical distance between Tm3+ and Dy3+ ions can be expressed by
| |
 | (5) |
where
V refers to the volume of the unit cell,
N is the number of available sites for the dopant in the unit cell, and
Xc is the total content of Tm
3+ and Dy
3+, at which the luminescence intensity of Tm
3+ is half that of the sample in the absence of Dy
3+ ions. For the LGW host,
V = 302.03 Å
3,
N = 2, and
Xc = 0.07. Therefore, the
Rc value is calculated to be about 16.03 Å. Generally, non-radiative energy transfer from a sensitizer to an activator usually takes place through an exchange interaction or electric multipolar interactions.
54 The value of
Rc calculated above implies a small possibility of an exchange interaction because the exchange interaction is predominant only for about 5 Å.
55 As a consequence, we can conclude the energy transfer mechanisms from Tm
3+ to Dy
3+ ions are dominated by electric multipolar interactions. According to Dexter’s energy transfer formula of multipolar interaction and Reisfeld’s approximation, the following relationship can be used:
56| |
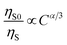 | (6) |
where
ηS0 and
ηS represent the luminescence quantum efficiencies of Tm
3+ ions in the absence and presence of Dy
3+ ions, respectively.
C is the total doping concentration of Tm
3+ and Dy
3+ ions. The value of
α = 6, 8, and 10 corresponds to dipole–dipole, dipole–quadrupole, and quadrupole–quadrupole interactions, respectively. The values of
ηS0/
ηS also can be approximately calculated by the ratio of related luminescence intensities as
57| |
 | (7) |
where
IS0 and
IS refer to the luminescence intensity of Tm
3+ ions without and with Dy
3+ ions, respectively. The relationship between
IS0/
IS and
Cα/3 based on the above equation is shown in
Fig. 13. It can be clearly seen that when
α = 8, the linear fitting result is the best. Therefore, the energy transfer from Tm
3+ to Dy
3+ ions takes place through the dipole–quadrupole interaction mechanism.
 |
| | Fig. 13 Dependence of IS0/IS of Tm3+ emission on Tm3+ and Dy3+ ion concentration: (a) C6/3, (b) C8/3, and (c) C10/3. | |
Fig. 14 shows the CIE chromaticity coordinates and the representative digital luminescence photographs of single-phase emission-tunable phosphors LGW:2% Tm3+, x% Dy3+. The CIE chromaticity coordinates for the samples were determined on the basis of their corresponding PL spectrum, and were calculated as (0.258, 0.207), (0.294, 0.257), (0.335, 0.321), (0.349, 0.348) and (0.368, 0.368) for LGW:2% Tm3+, x% Dy3+ with x = 2, 4, 6, 8 and 10. For WLED applications, the quantum efficiency (QE) is an important parameter for phosphors. Therefore, the white-light-emitting phosphors, LGW:2% Tm3+, 4% Tb3+, 3% Eu3+ and LGW:2% Tm3+, 6% Dy3+, were measured. The QE of LGW:2% Tm3+, 4% Tb3+, 3% Eu3+ is 9%, while that of LGW:2% Tm3+, 6% Dy3+ is 28%. It is believed that the quantum efficiency of these white-light-emitting phosphors could be further improved by optimizing the synthetic conditions.
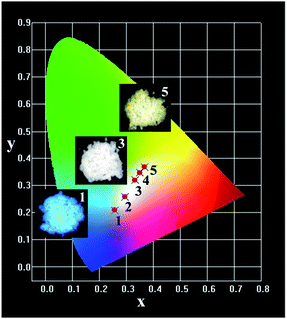 |
| | Fig. 14 CIE color coordinate diagram of LGW:2% Tm3+, x% Dy3+ with different Dy3+ concentrations: (1) x = 2; (2) x = 4; (3) x = 6; (4) x = 8; and (5) x = 10. The insets are their corresponding digital photographs taken under 365 nm excitation. | |
4. Conclusion
In summary, a series of LiGd(WO4)2:Re3+ (Re = Tm, Tb, Dy, Eu) phosphors have been prepared by the conventional solid-state reaction method. Under UV light excitation, the LGW:Tm3+, LGW:Tb3+, LGW:Dy3+ and LGW:Eu3+ phosphors show the characteristic emissions of Tm3+ (blue), Tb3+ (green), Dy3+ (yellow) and Eu3+ (red). More interestingly, two routes are disclosed to realize the white emission via the energy transfer process between different rare earth ions in a single LGW host. By codoping Tm3+, Tb3+, Eu3+ into the LGW host, white light emission was obtained under 360 nm excitation which is accessible with UV-LED chips. In addition, the LGW:2% Tm3+, x% Dy3+ phosphors show tunable color from light blue to light yellow, and in particular the white light emission is also realized based on the energy transfer from Tm3+ to Dy3+ ions under UV excitation. In detail, the energy transfer mechanism from Tm3+ to Dy3+ ions has been demonstrated to be through dipole–quadrupole interaction. These results indicate that the present materials could potentially serve as single-phase white-light-emitting phosphors for application in UV-excited WLEDs.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21476045) and the Foundation of key laboratory for micro/nano technology and system of Liaoning province (20140401).
Notes and references
- T. R. Kuykendall, A. M. Schwartzberg and S. Aloni, Adv. Mater., 2015, 27, 5805 CrossRef CAS PubMed.
- Z. Zhang, D. Liu, D. Li, K. Huang, Y. Zhang, Z. Shi, R. Xie, M.-Y. Han, Y. Wang and W. Yang, Chem. Mater., 2015, 27, 1405 CrossRef CAS.
- A. Layek, P. C. Stanish, V. Chirmanov and P. V. Radovanovic, Chem. Mater., 2015, 27, 1021 CrossRef CAS.
- P. Pust, V. Weiler, C. Hecht, A. Tucks, A. S. Wochnik, A. K. Henss, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891 CrossRef CAS PubMed.
- J. Chen, Y. Liu, L. Mei, H. Liu, M. Fang and Z. Huang, Sci. Rep., 2015, 5, 9673 CrossRef CAS PubMed.
- M. M. Shang, C. X. Li and J. Lin, Chem. Soc. Rev., 2014, 43, 1372 RSC.
- S. Nakamura, Angew. Chem., Int. Ed., 2015, 54, 7770 CrossRef CAS PubMed.
- A. Marchuk and W. Schnick, Angew. Chem., Int. Ed., 2015, 54, 2383 CrossRef CAS PubMed.
- X. Li, J. D. Budai, F. Liu, J. Y. Howe, J. Zhang, X.-J. Wang, Z. Gu, C. Sun, R. S. Meltzer and Z. Pan, Light: Sci. Appl., 2013, 2, e50 CrossRef.
- K. Li, M. Shang, Y. Zhang, J. Fan, H. Lian and J. Lin, J. Mater. Chem. C, 2015, 3, 7096 RSC.
- M. H. Fang, H. D. Nguyen, C. C. Lin and R. S. Liu, J. Mater. Chem. C, 2015, 3, 7277 RSC.
- W. Lu, Y. Jia, Q. Zhao, W. Lv and H. You, Chem. Commun., 2014, 50, 2635 RSC.
- J. Ding, Q. Wu, Y. Li, Q. Long, C. Wang and Y. Wang, Dalton Trans., 2015, 44, 9630 RSC.
- G. Li, Y. Zhang, D. Geng, M. Shang, C. Peng, Z. Cheng and J. Lin, ACS Appl. Mater. Interfaces, 2012, 4, 296 CAS.
- H. Liu, Y. Luo, Z. Mao, L. Liao and Z. Xia, J. Mater. Chem. C, 2014, 2, 1619 RSC.
- W. R. Liu, C. H. Huang, C. W. Yeh, J. C. Tsai, Y. C. Chiu, Y. T. Yeh and R. S. Liu, Inorg. Chem., 2012, 51, 9636 CrossRef CAS PubMed.
- W. Tang and F. Zhang, Eur. J. Inorg. Chem., 2014, 3387 Search PubMed.
- P. Li, Z. Wang, Z. Yang and Q. Guo, J. Mater. Chem. C, 2014, 2, 7823 RSC.
- X. Zhang and M. Gong, Dalton Trans., 2014, 43, 2465 RSC.
- W. Lv, Y. Jia, Q. Zhao, M. Jiao, B. Shao, W. Lü and H. You, RSC Adv., 2014, 4, 7588 RSC.
- Y. Jia, R. Pang, H. Li, W. Sun, J. Fu, L. Jiang, S. Zhang, Q. Su, C. Li and R. S. Liu, Dalton Trans., 2015, 44, 11399 RSC.
- J. Zhou and Z. Xia, J. Mater. Chem. C, 2015, 3, 7552 RSC.
- Z. Ci, Q. Sun, M. Sun, X. Jiang, S. Qin and Y. Wang, J. Mater. Chem. C, 2014, 2, 5850 RSC.
- A. M. Kaczmarek and R. van Deun, Chem. Soc. Rev., 2013, 42, 8835 RSC.
- X. Liu, W. Hou, X. Yang and J. Liang, CrystEngComm, 2014, 16, 1268 RSC.
- Y. Liu, G. Liu, J. Wang, X. Dong and W. Yu, Inorg. Chem., 2014, 53, 11457 CrossRef CAS PubMed.
- Z. Wang, J. Zhong, H. Jiang, J. Wang and H. Liang, Cryst. Growth Des., 2014, 14, 3767 CAS.
- B. Rekik, M. Derbal, O. Benamara and K. Lebbou, J. Cryst. Growth, 2014, 405, 11 CrossRef CAS.
- X. Huang, Z. Lin, L. Zhang, J. Chen and G. Wang, Cryst. Growth Des., 2006, 6, 2271 CAS.
- J. M. Postema, W. T. Fu and D. J. W. Ijdo, J. Solid State Chem., 2011, 184, 2004 CrossRef CAS.
- L. Li, Y. Liu, R. Li, Z. Leng and S. Gan, RSC Adv., 2015, 5, 7049 RSC.
- A. J. Peter and I. B. Shameem Banu, J. Mater. Sci.: Mater. Electron., 2014, 25, 2771 CrossRef CAS.
- G. Li, C. Li, C. Zhang, Z. Cheng, Z. Quan, C. Peng and J. Lin, J. Mater. Chem., 2009, 19, 8936 RSC.
- L. Wu, Y. Zhang, M. Y. Gui, P. Z. Lu, L. X. Zhao, S. Tian, Y. F. Kong and J. J. Xu, J. Mater. Chem., 2012, 22, 6463 RSC.
- H. Qian, J. Zhang and L. Yin, RSC Adv., 2013, 3, 9029 RSC.
- L. Hou, S. Cui, Z. Fu, Z. Wu, X. Fu and J. H. Jeong, Dalton Trans., 2014, 43, 5382 RSC.
- L. Jing, X. Liu, Y. Li and Y. Wang, J. Lumin., 2015, 162, 185 CrossRef CAS.
- Y. Zhang, W. Gong, J. Yu, H. Pang, Q. Song and G. Ning, RSC Adv., 2015, 5, 62527 RSC.
- T. Ishigaki, A. Torisaka, K. Nomizu, P. Madhusudan, K. Uematsu, K. Toda and M. Sato, Dalton Trans., 2013, 42, 4781 RSC.
- Y. Luo, Z. Xia, B. Lei and Y. Liu, RSC Adv., 2013, 3, 22206 RSC.
- Y. Su, L. Li and G. Li, J. Mater. Chem., 2009, 19, 2316 RSC.
- X. Li, Y. Zhang, D. Geng, J. Lian, G. Zhang, Z. Hou and J. Lin, J. Mater. Chem. C, 2014, 2, 9924 RSC.
- E. Nakazawa and S. Shionoya, J. Chem. Phys., 1967, 47, 3211 CrossRef CAS.
- C. Lorbeer and A. V. Mudring, J. Phys. Chem. C, 2013, 117, 12229 CAS.
- J. Zhou and Z. Xia, J. Mater. Chem. C, 2014, 2, 6978 RSC.
- Z. Hou, Z. Cheng, G. Li, W. Wang, C. Peng, C. Li, P. Ma, D. Yang, X. Kang and J. Lin, Nanoscale, 2011, 3, 1568 RSC.
- G. Li, Z. Hou, C. Peng, W. Wang, Z. Cheng, C. Li, H. Lian and J. Lin, Adv. Funct. Mater., 2010, 20, 3446 CrossRef CAS.
- K. Li, Y. Zhang, X. Li, M. Shang, H. Lian and J. Lin, Phys. Chem. Chem. Phys., 2015, 17, 4283 RSC.
- S. P. Lee, C. H. Huang and T. M. Chen, J. Mater. Chem. C, 2014, 2, 8925 RSC.
- C. H. Huang and T. M. Chen, J. Phys. Chem. C, 2011, 115, 2349 CAS.
- W. Wu and Z. Xia, RSC Adv., 2013, 3, 6051 RSC.
- S. Das, C. Y. Yang, H. C. Lin and C. H. Lu, RSC Adv., 2014, 4, 64956 RSC.
- G. Blasse, Philips Res. Rep., 1969, 24, 131 CAS.
- R. Reisfeld and L. Boehm, J. Solid State Chem., 1972, 4, 417 CrossRef CAS.
- W. J. Yang, L. Y. Luo, T. M. Chen and N. S. Wang, Chem. Mater., 2005, 17, 3883 CrossRef CAS.
- D. L. Dexter and J. A. Schulman, J. Chem. Phys., 1954, 22, 1063 CrossRef CAS.
- J. Sun, Z. Lian, G. Shen and D. Shen, RSC Adv., 2013, 3, 18395 RSC.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.