
New insights into synthesis and oligomerization of ε-lactams derived from the terpenoid ketone (−)-menthone†
Abstract
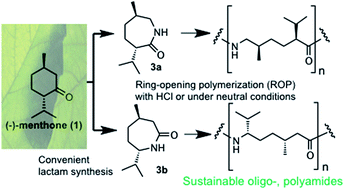
Two regioisomeric lactams, which are derived from terpenoid ketone (−)-menthone in two steps, are oligomerized in an easy acid-induced procedure to obtain oligoamides that contain alkyl groups and stereocenters; for this, a nucleophilic oligomerization via a non-ionic propagating site (neutral conditions) and a polycondensation are also possible. Furthermore, a regioselective synthesis is demonstrated where (−)-menthone is transformed into one of these lactams in a one-step procedure via Beckmann rearrangement without isolation of any oxime intermediate using the reagent hydroxylamine-O-sulfonic acid (HOSA). These concise oligoamide syntheses smooth the way to novel sustainable long-chain polyamides with highly interesting structures.
 Please wait while we load your content...
Please wait while we load your content...

