
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the electro-mechanical performance of polypyrrole through 3- and 3,4-methyl substituted copolymers†
D.
Melling
*ab,
S. A.
Wilson
a and
E. W. H.
Jager
b
aInstitute for Medical Science and Technology, University of Dundee, Wilson House, 1 Wurzburg Loan, Dundee MediPark, Dundee, DD21FD, UK. E-mail: d.melling@dundee.ac.uk
bDepartment of Physics, Chemistry and Biology (IFM), Linköping University, Linköping, 581 83, Sweden
First published on 29th September 2015
Abstract
Conducting polymers such as polypyrrole are biocompatible materials used in bioelectronic applications and microactuators for mechanobiology and soft microrobotics. The materials are commonly electrochemically synthesised from an electrolyte solution comprising pyrrole monomers and a salt, which is incorporated as the counter ion. This electrosynthesis results in polypyrrole forming a three-dimensional network with extensive cross-linking in both the alpha and beta positions, which impacts the electro-mechanical performance. In this study we adopt a ‘blocking strategy’ to restrict and control cross-linking and chain branching through beta substitution of the monomer to investigate the effect of crosslinking on the electroactive properties. Methyl groups where used as blocking groups to minimise the impact on the pyrrole ring system. Pyrrole, 3- and 3,4-methyl substituted pyrrole monomers were electro-polymerised both as homo-polymers and as a series of co-polymer films. The electroactive performance of the films was characterised by measuring their electrochemical responses and their reversible and non-reversible film thickness changes. This showed that altering the degree of crosslinking through this blocking strategy had a large impact on the reversible and irreversible volume change. These results elaborate the importance of the polymer structure in the actuator performance, an aspect that has hitherto received little attention.
1. Introduction
Conjugated Polymers (CP) are attractive biocompatible materials for bioelectronic devices since they show both electronic and ionic conductivity. Their range of conductivity can be from tens to several thousands of S cm−1 depending upon the monomer (pyrrole, thiophene and aniline) and dopant.1–5 This makes them an interesting candidate for interfacing electronics with biological tissue. CPs have been used for controlled drug delivery,6,7 biosensors,8,9 neural electrodes,10,11 wettability switches,12,13 and controlling cell adhesion, proliferation and differentiation.14–18 They are also commonly used as the electroactive material driving polymer (micro-)actuators.19–25 Such CP actuators have been used in applications ranging from medical devices, including a steerable guide wire and a PTCA balloon seal,23 to microfabricated chips for mechanobiology that provide mechanical stimulation to single cells.26Following the early demonstrations,19,20 researchers have sought to improve CP actuator performance, particularly with regard to increased strain and increased strain rate. A full range of optimisation studies utilizing easily accessible synthesis parameters have been performed for polypyrrole (PPy) films as follows: polymerisation potential,27 current density,28 monomer and dopant concentrations, the type of dopant,29–32 the type of solvent,30 the temperature,33 and pH.34 However, these optimisations have led to only a modest improvement, but they did make clear that the volume change that occurs during redox switching of PPy is dependent upon the mass transport (ions and solvent) that occurs between the polymer and electrolyte. The observed response is dependent on the total mass flow and the characteristic response of the polymer network to the influx of ions and solvent. Progress has been made into how the ‘state of compaction’ of the polymer network changes during electromechanical stimulation35,36 yielding structural information.37 To-date there have been few studies on PPy films with purposely adapted network structures. This represents an alternative strategy to achieve the sought-after performance gains.
Electropolymerised polypyrroles are known to be highly crosslinked polymers.38 Both simulations and XPS studies estimate branching and crosslinking in PPy to be 33% and 22% respectively.39,40 Unfortunately, the use of XPS has not become widely accepted as a method for determining crosslinking and there is currently no established technique for determining crosslinking in these intractable polymers. In the case of crosslinked polymers, the molecular weight parameter loses its meaning, and it is the density of crosslinking that is strongly correlated to physical properties such as solvent-swellability. For non-crosslinked polymer films molecular weight information is of greater significance, but can be difficult to obtain. Molecular weight determination for the homopolymer, poly(3,4-dimethylpyrrole), indicates that it consists of chains of approximately 1000 pyrrole units.41 Electrochemically polymerised PPy films are typically highly disordered (amorphous) and show only diffuse X-ray scattering, including those formed using DBS dopant.42,43
N- and beta-substituted polypyrroles have been synthesised with the strategic aim of increasing their functionality,44–48 enhancing solubility and processability,49–52 increasing adhesion to substrates53,54 and producing novel composite polymers.55 By extension it should be possible to alter the degree of crosslinking of PPy by chemical substitution of the monomer to achieve a leap in actuation performance, however this approach, has received little attention to-date. Tominaga et al. increased the levels of crosslinking in PPy with the aim of decreasing electrochemical creep.56 They did this by supplementing the crosslinking that occurs naturally as a result of beta-substitution during electropolymerisation with additional N-alkyl crosslinks. This did indeed result in a decrease in electrochemical creep but also resulted in a decrease in actuation strain.
Here, a synthetic approach for decreasing the degree of crosslinking in PPy by introducing beta-substituted pyrrole monomers resulting in pyrrole-copolymers is presented. The effect of decreasing crosslinking on the actuation performance of PPy is investigated using a relatively new approach based on laser scanning micrometry (LSM) which allows non-contact out-of-plan strain measurements to be made in an unloaded condition.57 CP actuation is characterised by macroscopic volume change and hence strain measurements are critical to any experimental evaluation.58–64 LSM allows strain to be characterised without isotonic or isometric loading. This is particularly important when considering thin films, as these effects are strongly influenced by the substrate interaction. Hence the uncertainty associated with bending beam measurements is avoided when using the non-contact LSM technique.58,59,63,64
In this study 3- and 3,4-substituted pyrrole monomers were used (Fig. 1). Beta substitution of the pyrrole ring blocks substitution that naturally leads to branching and crosslinking during electropolymerisation. Increased beta substitution therefore reduces the opportunities for branching and crosslinking during polymerisation. The methyl group was selected as a suitable blocking group due to its small size and minimal effect on the on the electronic properties of the pyrrole ring system65–67 and delocalisation between adjacent monomer units along the polymer chain as occurs in the oxidised form of the polymer.68 The methyl group, unlike long alkyl chains, has a minimal effect upon the conductivity of polypyrrole.67 Both pyrrole and 3-methyl-1H-pyrrole monomers are available commercially; however, 3,4-dimethyl-1H-pyrrole monomer is not and was synthesised for the purposes of this study.
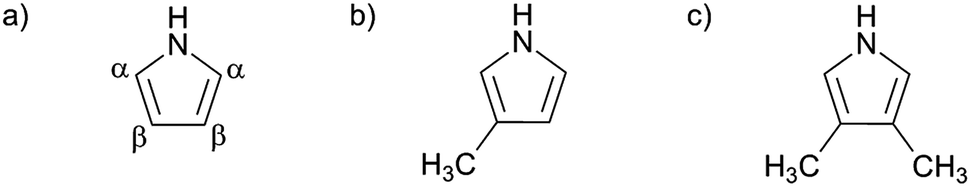 | ||
| Fig. 1 Monomers employed: (a) pyrrole (showing alpha and beta positions) (b) 3-methyl-1H-pyrrole and (c) 3,4-dimethyl-1H-pyrrole. | ||
Hence, in addition to unmodified pyrrole monomer (Py), we have used the mono-substituted 3-methyl-1H-pyrrole (3MPy) and di-substituted 3,4-dimethyl-1H-pyrrole (3,4DMPy) monomers both synthesised into homopolymers and copolymers with Py. Compared to the homopolymer PPy, lower levels of branching and crosslinking are anticipated for homopolymers formed from 3MPy which has just one beta position available for substitution. For 3,4DMPy, which has both beta positions substituted, no branching or crosslinking is possible; this monomer can only form linear, non-branched and non-crosslinked, homopolymers. In addition to the homopolymers, copolymers were electropolymerised using solutions containing mixed monomers at different ratios.4,50 By combining different volume fractions of substituted and non-substituted pyrrole monomers, different levels of crosslinking will occur.
2. Experimental section
2.1 Materials and methods
Pyrrole (Aldrich, 98%), 3-methyl-1H-pyrrole (TCI Europe, >98%) and 3,4-dimethyl-1H-pyrrole were purified by vacuum distillation prior to use. Sodium dodecylbenzenesulfonate (NaDBS, TCI Europe, hard type, mixture) and sodium dodecylbenzenesulfonate (Aldrich, technical grade) was used as received. Ultrapure water was obtained from Millipore Milli-Q equipment. Gold wire (Goodfellows UK, 99.95%, temper: as drawn) of diameter 0.5 mm was used as received.The 3,4-dimethyl-1H-pyrrole was not commercially available and was synthesised using the published procedure developed by Ichimura et al.69 using the following chemicals obtained from Sigma-Aldrich and used as received: ethylcarbamate (urethane, ≥99%), pyridine (anhydrous, 99.8%), toluene (anhydrous, 99.8%), thionyl chloride (ReagentPlus®, ≥99%), 2,3-dimethyl-1,3-butadiene (98%), methanol (anhydrous, 99.8%) and potassium hydroxide (Reagent grade, 90%, flakes).69
All electrochemical studies were performed using an Autolab electrochemical workstation (Metrohm µAUTOLAB III potentiostat–galvanostat) controlled using a personal computer with GPES electrochemical software.
A 0.5 mm diameter gold wire was used to construct a working electrode with an exposed (non-insulated) length of 10 mm (surface area 15.7 mm2) on which polypyrrole films were electrogenerated. Platinised (2.5 µm) titanium anode mesh (Ti-shop, 1 mm thick) of length 100 mm and width 25 mm was used as a counter electrode. The reference electrode was a BASi Ag/AgCl (3 M, NaCl gel saturated with AgCl) electrode with the tip being stored between uses in 3 M NaCl (aq.). All potentials reported in this work were referenced to this electrode. All the experiments were performed at a room temperature (25 ± 2 °C).
Porous gold working electrodes where prepared by electroplating a 5 µm layer of porous gold on the surface of the AuWE to increase the adhesion of the films.70 The surface was first cleaned by heating in ‘Piranha’ solution (H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2:H2O = 1:2:4) at 70 °C for 5 minutes followed by thorough rinsing with deionised water. An aqueous mixture of two types of commercial gold electroplating salt (from Enthone) was employed in the ratio: 10 ml Neutronex™ 309A:300 ml Neutronex™ 309B:190 ml Milli-Q water. A ‘seeding’ period of 120 s at −0.7 V was undertaken followed by a ‘growth’ period of 390 s at −0.9 V to deposit a 5 µm layer of porous gold.
:H2O2:H2O = 1:2:4) at 70 °C for 5 minutes followed by thorough rinsing with deionised water. An aqueous mixture of two types of commercial gold electroplating salt (from Enthone) was employed in the ratio: 10 ml Neutronex™ 309A:300 ml Neutronex™ 309B:190 ml Milli-Q water. A ‘seeding’ period of 120 s at −0.7 V was undertaken followed by a ‘growth’ period of 390 s at −0.9 V to deposit a 5 µm layer of porous gold.
2.2 Synthetic procedures
All of the films were subjected to a conditioning period of 200 s at a potential of 0 V prior to being actuated to assess the level of baseline noise and display the initial film thickness. Films were then actuated using a square wave potential (0 V (200 s), −1 V (200 s)) until they reached a stable state (fully irreversibly expanded state). We have described in detail the use of LSM for determining the actuation performance of polypyrrole films elsewhere.57
:NH4OH:H2O2 (5:1:1) at 80 °C (RCA-SC1 cleaning) for 10–20 minutes depending on how resistant the film was to removal.
3. Results and discussion
3.1 Synthesis of 3,4-dimethyl-1H-pyrrole
The synthesis of pyrrole monomers with substituents exclusively in the beta-positions is not easy to achieve. Electrophilic substitution of the pyrrole ring occurs preferentially in the more reactive alpha (2 and 5) positions.48 When beta-substitution does occur it is usually accompanied by substitution in the alpha positions. This results in the formation of mixed products, typically positional isomers, which can be difficult to separate. One strategy for achieving exclusive substitution of the beta-positions is to use protecting groups which can prevent substitution of the pyrrole nitrogen and the alpha positions.71 However, even with the use of a protecting group, substitution of the beta positions can lead to a mixture of 3- and 3,4-substituted products, which require separation and lowers the yield of the di-substituted product.For these reasons we used the synthesis developed by Ichimura et al.69 This elegant synthesis has just two steps (Fig. 2) and is relatively easy to undertake. It does not involve the selective substitution of the pyrrole ring. Instead the pyrrole ring is formed by ring-closure with the methyl groups already in appropriate positions within one of the starting materials (2,3-dimethylbuta-1,3-diene). The first step in the reaction is a ‘Diels–Alder’ reaction between 2,3-dimethylbuta-1,3-diene and ethyl carbamate, resulting in the formation of a non-aromatic 6-membered cyclic intermediate. In the second (final) step the intermediate undergoes a reduction in ring size (6 to 5 member ring) and aromatisation to give 3,4-dimethyl-1H-pyrrole. The strength of the alkali plays a key role in the formation of the pyrrole with higher yields being obtained with stronger concentrations of alkali; the use of methanolic alkali also giving higher yields than aqueous alkali.
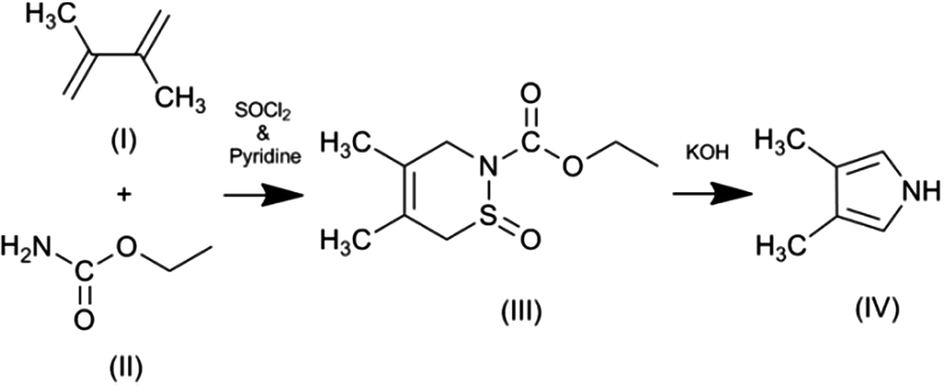 | ||
| Fig. 2 Synthesis of 3,4-dimethyl-1H-pyrrole. (I) 2,3-dimethyl-1,3-butadiene, (II) ethylcarbamate (urethane), (III) 2-ethoxycarbonyl-3,6-dihydro-4,5-dimethyl-1,2-thiazine-1-oxide (IV) 3,4-dimethyl-1H-pyrrole. | ||
We successfully synthesised the 3,4-dimethyl-1H-pyrrole monomer starting from ethyl carbamate. Both the structure and purity of our product was confirmed by spectroscopic techniques and elemental analysis (Fig. S3–S7†). In summary: 1H NMR (400 MHz, CDCl3, δ): 7.75 (s, br, 1H, NH), 6.53 (d, J = 2.52 Hz, 2H, Cα), 2.05 (s, 6H; CH3); 13C NMR (400 MHz, CDCl3, δ): 118.13 (Cβ), 115.49 (Cα), 9.94 (CH3); IR (KBr): ν = 3420.61 (S) (broad, pyrrollic NH (free) stretch), 3250 (m) (shoulder, pyrrollic NH (H-bonded) stretch), 2922.04 (w) (–CH3), 2863.05 (w) (–CH3), 1677.09 (m) (Py ring C–C), 1448.95 (w) (–CH3 δ), 1384.32 (w) (–CH3 γ), 1107.54 (w), 1069.06 (w), 564.57 (w) (broad). EIMS m/z (%): 95 (65) [M+], 94 (100) [M+ − H], 80 (27), 67 (16), 53 (11), 41 (17), 39 (25) 28 (15); anal. calcd for C6H9N: C 75.74, H 9.53, N 14.71; found: C 75.73, H 9.51, N 14.83.
3.2 Homopolymer films
Potentiodynamic synthesis of 3,4DMPPy(DBS) and 3MPPy(DBS) at the surface of a gold working-electrode (AuWE) was undertaken using cyclic voltammetry (CV) to determine suitable conditions for polymerisation and actuation. Fig. 3 shows that both 3,4DMPy and 3MPy monomers start to polymerise at potentials in the region of 0.3–0.4 V vs. Ag/AgCl and have oxidation and reduction peaks around −0.1 V and −0.6 V respectively, similar to PPy(DBS).72 An electrical potential of 0.55 V was selected as suitable potential to potentiostatically polymerise all subsequent films and potentials of 0 V and −1 V employed to fully actuate the films.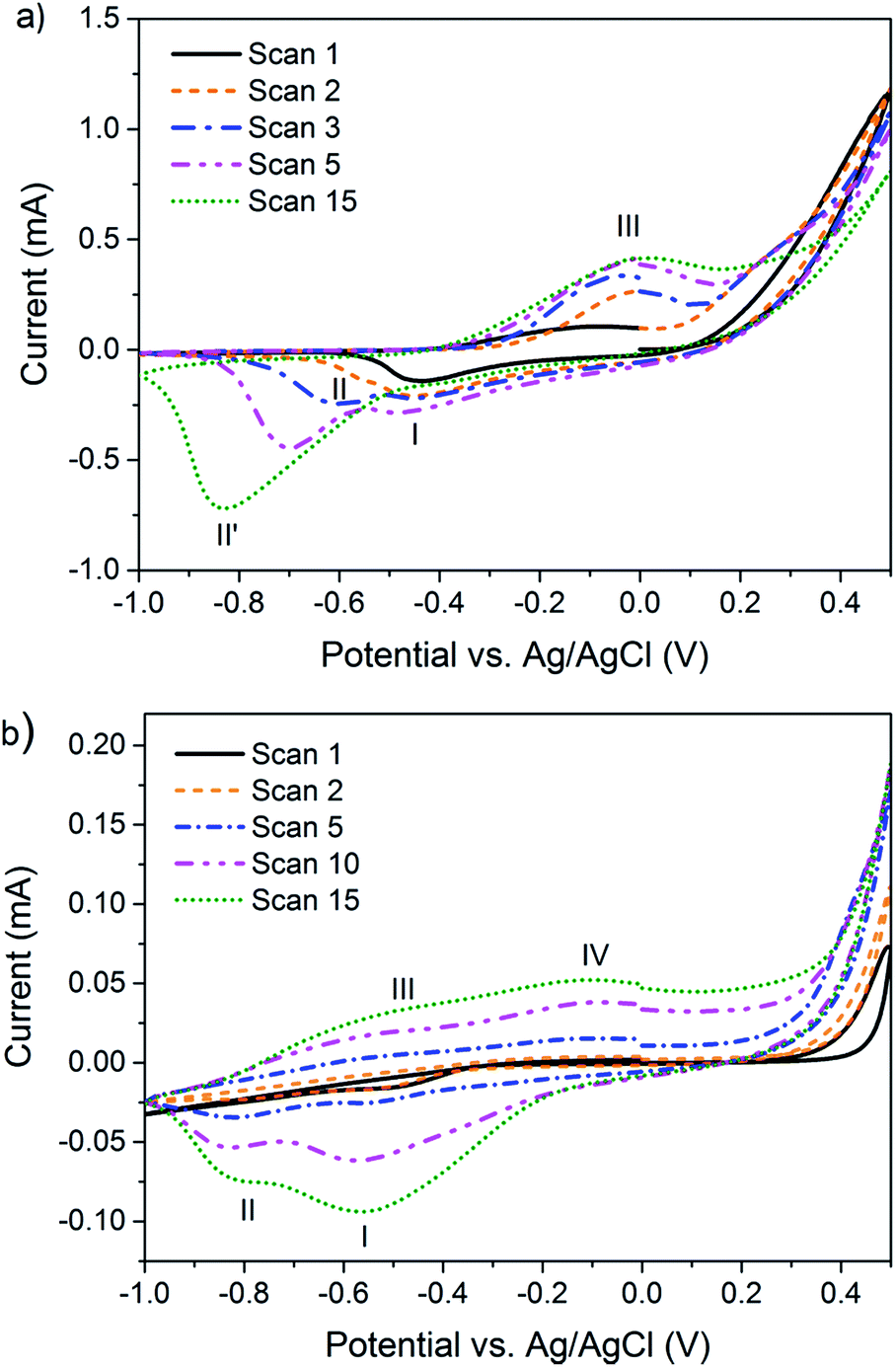 | ||
| Fig. 3 Voltammograms for (a) 3,4DMPPy(DBS) and (b) 3MPPy(DBS) potentiodynamically synthesised at a gold working electrode employing 0.1 M solutions of aqueous monomer and NaDBS electrolyte at a scan rate of 0.05 V s−1. | ||
Both voltammograms show the evolution of the oxidation and reduction peaks with cycle number. The voltammograms for 3,4DMPy show a single peak (I) during the first reduction scan which increases slightly and then disappears in subsequent scans. Another peak (II) appears during the second reduction scan which increases in area and shifts (maxima) from −0.6 V to −0.80 V in later scans. A single oxidation peak (III) is displayed for 3,4DMPy during the oxidation scan which increases in area with increasing scan number.
The voltammograms for 3MPy also display a single reduction peak (I) during the first scan with a second peak (II) appearing during the second scan which is present in all subsequent scans. Both reduction peaks increase in area with scan number, with the increase in peak (I) being the greatest. The oxidation peak is very broad and appears to be comprised of two broad overlapping peaks (III and IV).
The potentiodynamic synthesis of PPy(DBS) films carried out in aqueous solutions at slow scan rates is known to produce a double cathodic peak, and is evidence of the co-existence of two kinds of polymer structure.72 The first structure (peak I) is formed during the polymerisation process. This polymeric structure is modified to a more stable structure (peak II) at a lower potential, later on in the reduction process.
The potentiodynamic polymerisation of both 3,4DMPPy(DBS) and 3MPPy(DBS) produced black films on the surface of the wire AuWE similar to PPy films. Given that crosslinking is not possible for 3,4DMPPy(DBS) polymer, such solid films must be the result of non-bonding interactions such as pi-stacking, intercalculation with the DBS dopant, or the entanglement of long ‘linear’ (unbranched/non-crosslinked) chains.73–75 When cyclic voltammetry was stopped (open circuit) the solid black 3,4DMPPy(DBS) film formed a transparent phase at its surface over a period of approximately 30 minutes when kept in contact with aqueous electrolyte. Both phases are shown in Fig. 4a. One explanation might be that given enough time the non-crosslinked 3,4DMPPy chains are free to move apart and allow water to enter the network as a result of osmotic and hydrophilic forces and swell the polymer.76 This would explain why it forms at the surface in contact with the aqueous electrolyte. Further characterisation of this apparent gel-like material is needed.
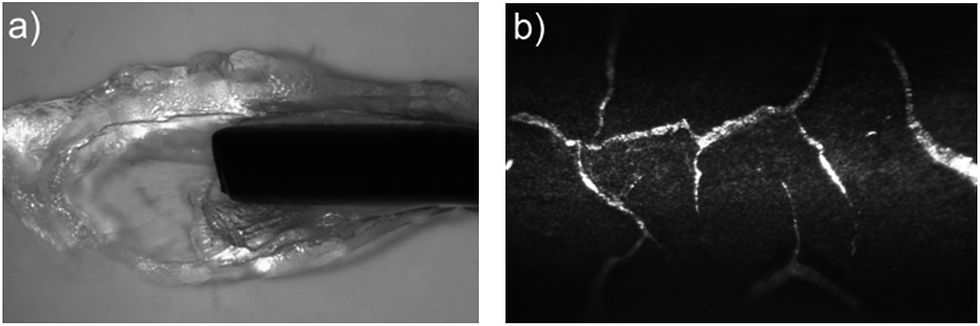 | ||
| Fig. 4 (a) The two phases formed during (black solid phase) and after (transparent phase) CV of 0.01 M 3,4DMPy and 0.1 M NaDBS (aq.) at a scan rate of 0.05 V s−1. (b) A highly fractured homopolymer film of 3MPPy(DBS) formed during actuation revealing the gold substrate. | ||
The 3MPPy(DBS) films and all copolymer films (described in the next section) did not form this transparent phase during either potentiodynamic or potentiostatic synthesis. Unlike 3,4DMPPy(DBS), 3MPPy(DBS) and the copolymers still have unsubstituted beta-positions in which crosslinking can occur thus preventing formation of this highly swollen phase.
To characterise the actuation performance of the homopolymers films, 5 µm thick 3MPPy(DBS) and 3,4DMPPy(DBS) films were electrosynthesised onto the Au wire WE using a constant potential of 0.55 V vs. Ag/AgCl in an aqueous 0.1 M monomer and 0.1 M NaDBS solution. Electrosynthesis was undertaken in situ within the measuring region of the LSM. This allowed film growth to be monitoring directly in real time ensuring that all of the films were of the same thickness. The films were actuated using a square wave potential (0 V for 200 s, −1 V for 200 s) and all failed by brittle fracture and delamination within a few cycles and most typically during the first cycle. Microscopic examination of the actuated films revealed a highly fractured surface as shown in Fig. 4b. These homopolymer films had lower mechanical strength than the PPy(DBS) films of the same thickness which could be actuated for many cycles without any fracture. The decrease in mechanical strength of these films is consistent with lower levels of crosslinking.68 Furthermore the potentiostatic synthesis of PPy films results in oxidised films which are more compact and containing less water and straighter and stiffer chain segments than the reduced polymer.77 The low levels of crosslinking present in the 3MPPy(DBS) films and absence of crosslinking in the 3,4DMPPy(DBS) films might be more prone to being compacted, containing straighter and stiffer chain segments than PPy(DBS) at a polymerisation potential of 0.55 V which would explain their brittle behaviour.68,78
Interestingly the transparent phase observed for potentiodynamically synthesised 3,4DMPPy(DBS) films during cyclic voltammetry was not observed for potentiostatically synthesised 3,4DMPPy(DBS) films. The as-polymerised (non-actuated) 3,4-DMPPy films were more resistant to removal from the AuWE than PPy films when heated with RCA-SC1 wash, suggesting a denser, more chemical resistant structure for 3,4-DMPPy films compared to PPy films. Once the 3,4-DMPPy films had been actuated this increased resistance to chemical cleaning was no longer evident suggesting the structure had changed as a result of the actuation process.
3.3 Copolymer films
Copolymer films were electropolymerised using polymerisation solutions comprised of mixed monomers.4,79,80 The 5 µm copolymer films of poly(3MPy-co-Py)(DBS) and poly(3,4DMPy-co-Py)(DBS) were electrosynthesised using mixed monomer solutions containing different volume fractions of 0.1 M Py and 3MPy or 3,4DMPy respectively at a constant potential of 0.55 V vs. Ag/AgCl. We were able to synthesise copolymer films using methyl pyrrole ratios ranging from 0.03 to 0.50 resulting in smooth, adherent films (we will indicate the fraction by writing the number in front of the name, e.g. 0.04 poly(3MPy-co-Py)(DBS) means a pyrrole copolymer synthesised from 0.04 fraction of 3MPy and 0.96 Py). Given that the rate of polymerisation of 3MPy and 3,4DMPy was greater that of Py, the amount of substituted monomer incorporated into the copolymer films will be higher than that indicated by the fraction of substituted monomer in the polymerisation solution. This can be seen by the fact that already small additions of beta-substituted monomers result in large effects in the resulting PPy films.Thereafter, the copolymer films were actuated using a square wave potential of 0 V (200 s) and −1 V (200 s) and their actuation performance assessed using LSM. The actuation behaviour of these films could be divided into two regimes depending on the fraction of the methyl substituted pyrrole monomer in the synthesis electrolyte. Poly(3MPy-co-Py)(DBS) copolymer films electrosynthesised using solutions with a 3DMPy fraction > 0.04, and poly(3,4DMPy-co-Py)(DBS) copolymer films containing a 3,4DMPy fraction > 0.028, initially underwent deformation before failing by delamination after a small number of cycles. These films typically displayed large changes in reversible and irreversible expansion as shown in Fig. 5. The irreversible expansion at times showed instabilities (‘kinks’) which were unpredictable in their nature.
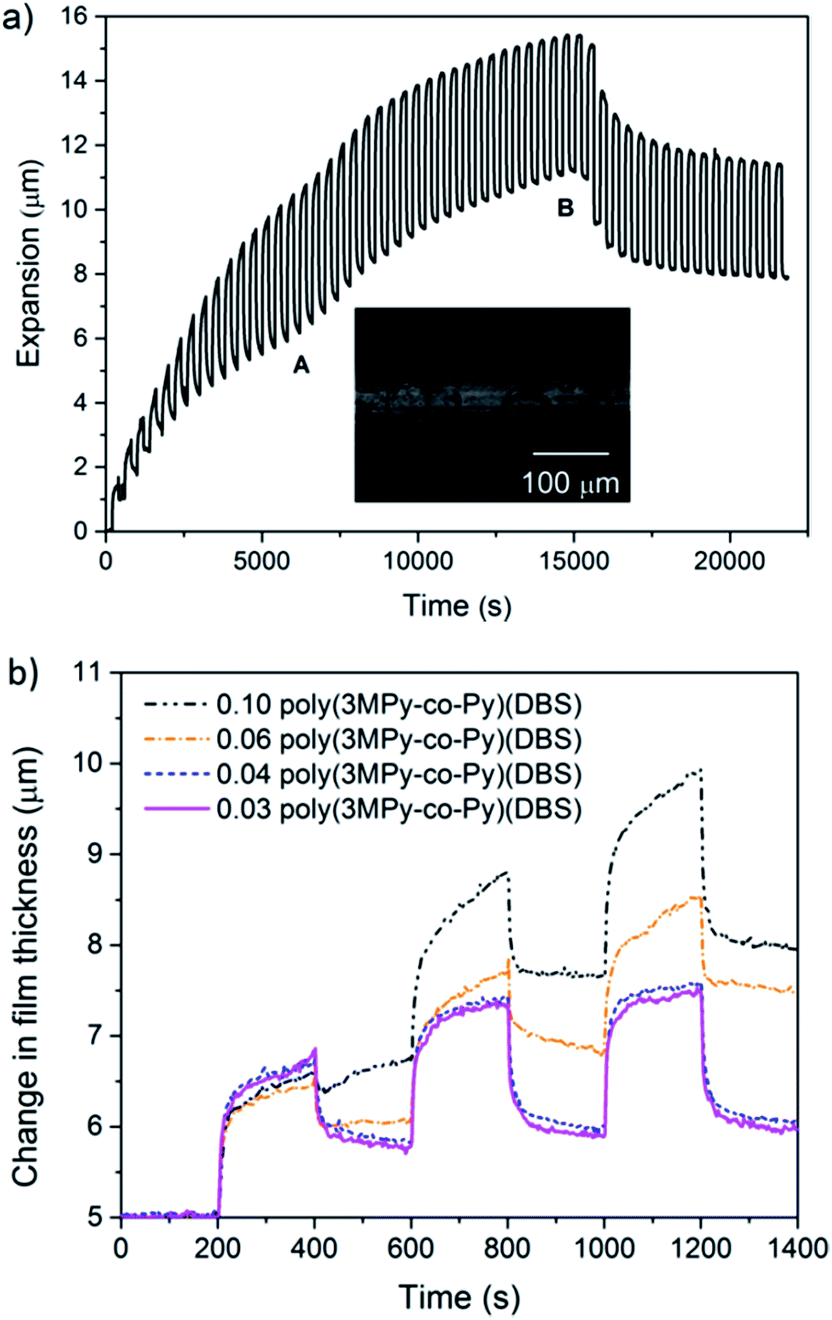 | ||
| Fig. 5 (a) A typical actuation profile for a 0.06 poly(3MPy-co-Py)(DBS) copolymer film. The profile displays very large increases in reversible and irreversible expansions. In region A deformation, like that shown in the inset, occurs. At point B delamination occurs. (b) An overlay of the first three cycles of 0.03 to 0.10 poly(3MPy-co-Py)(DBS) copolymer films showing the existence of a ‘threshold’ fraction above which plastic deformation occurs. | ||
Microscopic examination of these actuated copolymer films showed evidence of deformation, typically manifested as rings or ridges in the polymer films as shown in the inset in Fig. 5a. This behaviour would explain the ‘kinks’ displayed by the profile.
It was found that both the poly(3MPy-co-Py)(DBS) and poly(3,4DMPy-co-Py)(DBS) films polymerised on porous gold working electrodes (pAuWE) using solutions with a 3MPy fraction ≤ 0.04 and 3,4DMPy ≤ 0.028 respectively, could be actuated long-term, producing stable reversible volume change (Fig. 6).
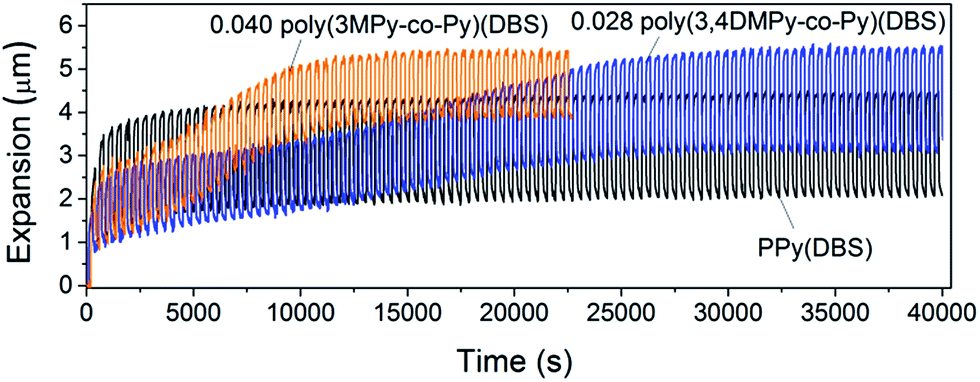 | ||
| Fig. 6 Overlays showing a typical actuation sequences for 5 µm films of PPy(DBS), 0.040 poly(3MPy-co-Py)(DBS) and 0.028 poly(3,4DMPy-co-Py)(DBS). | ||
The existence of a ‘threshold fraction’ separating the two regimes is illustrated in Fig. 5b, for poly(3MPy-co-Py)(DBS). The difference (increase) in irreversible expansion on going from 0.03 to 0.04 poly(3MPy-co-Py)(DBS) copolymers is relatively small and gradual during the first three cycles. In contrast above 0.04 the irreversible expansion increases rapidly and is very large, increasing with increasing fraction of 3MPy used in the synthesis solutions. Similar behaviour was seen for poly(3,4DMPy-co-Py)(DBS).
Fig. 7a shows the variation in maximum reversible expansion with time (cycle number). During the first 25 cycles (<10000 s) the largest reversible expansion for all three materials occurred in the first reduction scan, which then decreased significantly by the second or third cycle.81 The reversible expansion of PPy(DBS) increases steadily with time (cycle number) until it eventually stabilised after approximately 100 cycles (40000 s). The 0.028 poly(3,4DMPy-co-Py)(DBS) showed similar behaviour, undergoing relatively large increase in reversible expansion with increasing cycle number, which stabilised after approximately 100 cycles. This might be explained by the increased free volume being generated by conformational chain movements and swelling of the polymer network with time.82 Furthermore the conductivity of the polymer network might be increased as the copolymer chains are stretched.83,84 In contrast 0.040 poly(3MPy-co-Py)(DBS) showed a slight decrease in reversible expansion with cycle number by about 17000 s. Any increase in conductivity by stretching the polymer network might be off-set by a concomitant decrease in conductivity caused by increasing separation of neighbouring chain as a result of a more highly swollen network.
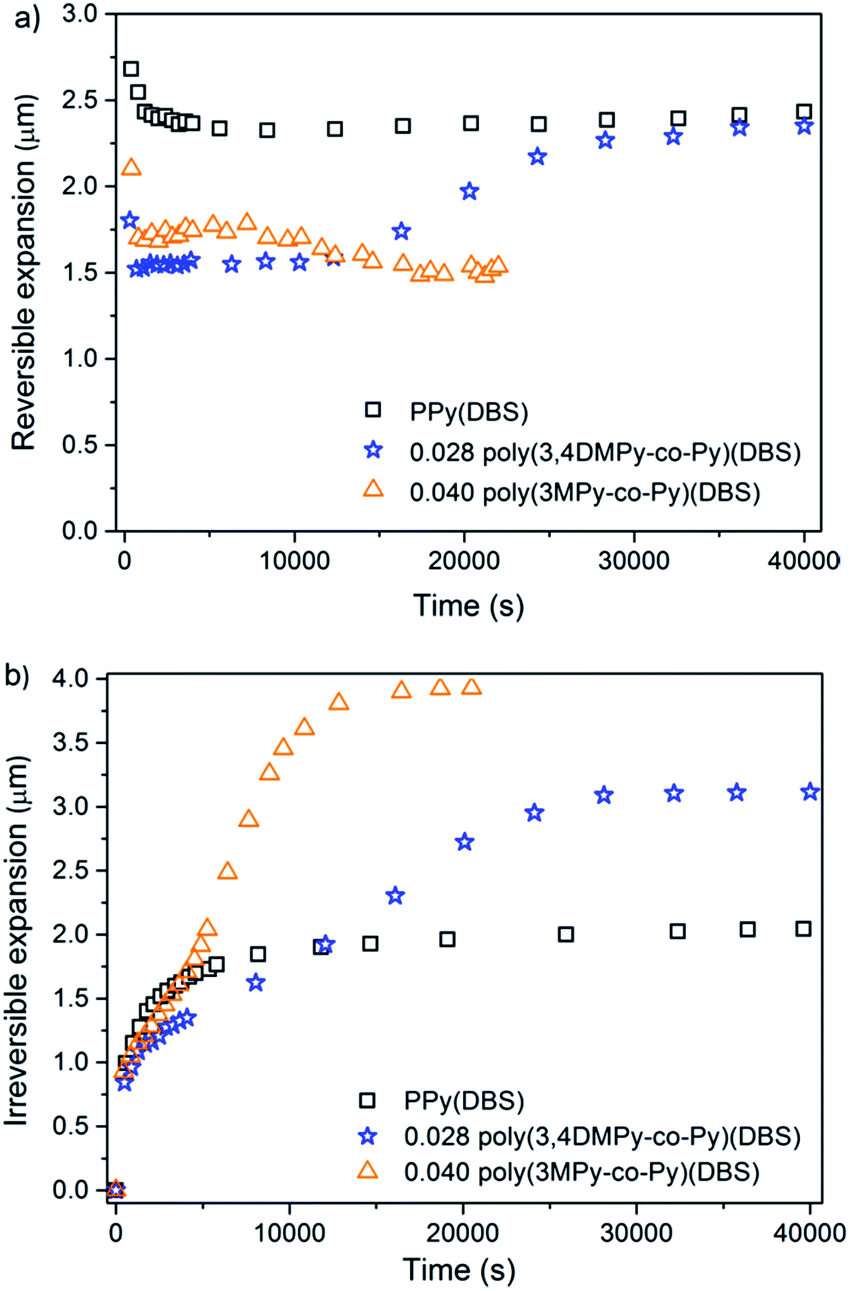 | ||
| Fig. 7 (a) The reversible expansion of PPy(DBS), 0.040 poly(3MPy-co-Py)(DBS) and 0.028 poly(3,4DMPy-co-Py)(DBS) with time. (b) The irreversible expansion of PPy(DBS), 0.040 poly(3MPy-co-Py)(DBS) and 0.028 poly(3,4DMPy-co-Py)(DBS) with time. | ||
Fig. 7b shows the variation in irreversible expansion for the three materials with time. All of the copolymers undergo greater irreversible expansion compared to the PPy(DBS), with the greatest irreversible expansion displayed by 0.040 poly(3MPy-co-Py)(DBS). Interestingly, although the maximum irreversible expansion of the copolymers are ultimately greater than that of the PPy films in the stable state, the irreversible expansion of the PPy film is greater than the copolymer films early on in the actuation sequence. The maximum irreversible expansion of the 0.040 poly(3MPy-co-Py)(DBS) films take approximately ten cycles to become greater than that of the PPy(DBS). This delay is even greater for the 0.028 poly(3,4DMPy-co-Py)(DBS) films which take approximately 35 cycles to ‘overtake' the irreversible expansion of the PPy(DBS) films.
Cation transport, which is dominating in these PPy(DBS) materials,21,22 is strongly affected by the state of the polymer matrix.81 The more compact the polymer, the slower the ion transport.77 Films containing more linear polymer chains are able to pack more closely and be subject to greater hydrogen bonding and pi-stacking between chains than a highly branched and crosslinked network. Such films would be more densely packed and display lower ion transport rates compared to films with a more open matrix structure.77 Electron diffraction patterns have been obtained for polypyrroles due to relatively small crystalline regions embedded in an amorphous matrix.85,86 The lamella structure proposed for PPy(DBS) films may be capable of even greater order.73 This might explain the slower increase of the irreversible expansion observed for the copolymers displayed in Fig. 7b. These might contain a matrix with more and/or larger crystalline regions. These regions would take longer for ions and solvent to penetrate and overcome intramolecular forces holding them more closely together.
Both the 0.028 poly(3,4-DMPy-co-Py)(DBS) and 0.040 poly(3MPy-co-Py)(DBS) films display larger total maximum expansions (reversible + irreversible) than PPy(DBS) in the stable state. This can readily be seen by comparing the relative positions of the top of the actuation curves in Fig. 6. The total maximum expansions for these copolymer samples are very similar.
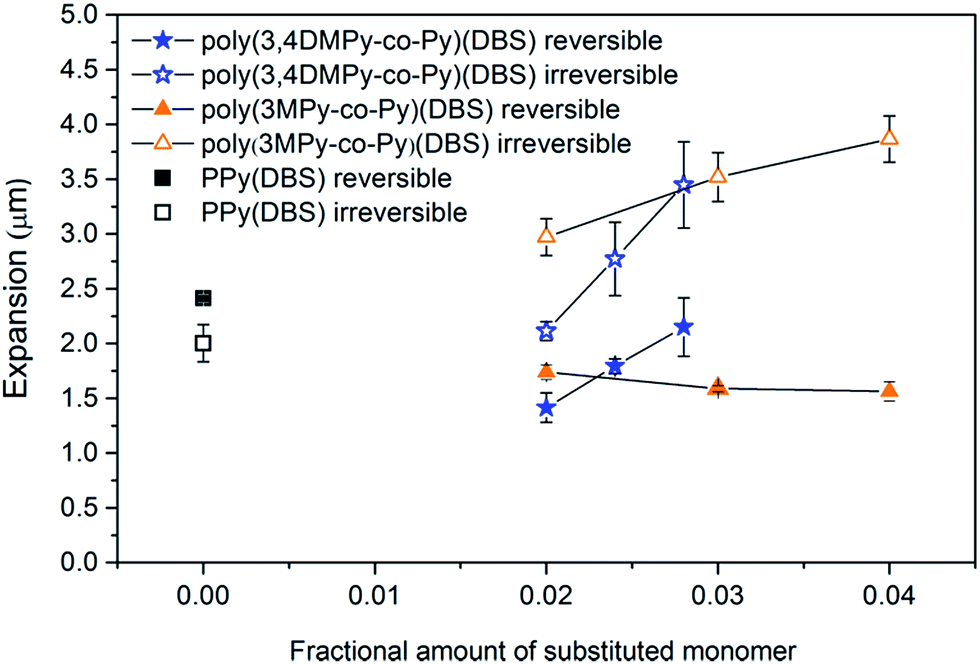 | ||
| Fig. 8 The variation in maximum reversible and irreversible expansion in the stable state with increasing substituted monomer content (n = 3). Fraction 0.00 means PPy(DBS) homopolymer. Error bar = 1SD. | ||
The variation in the mean maximum expansion rate in the stable state of the copolymer films during the reduction and oxidation scans with substituted monomer content is displayed in Fig. 9. The variation in expansion rate can be seen to mirror the changes in reversible expansion for the copolymer films. Although the expansion rate of the poly(3,4DMPy-co-Py)(DBS) films is less than the PPy(DBS) films, the speed increases with increasing 3,4DMPy content. In contrast, the expansion rate of the poly(3MPy-co-Py)(DBS) films decreases with increasing 3MPy content. Comparing the expansion rate during the reduction scan with the expansion rate (more correctly the contraction rate) during the oxidation scan, there is little difference observed for both the PPy and poly(3,4DMPy-co-Py)(DBS) films. However, in general, the expansion rate during the reduction scan is greater than during oxidation scan for the poly(3MPy-co-Py)(DBS) films.
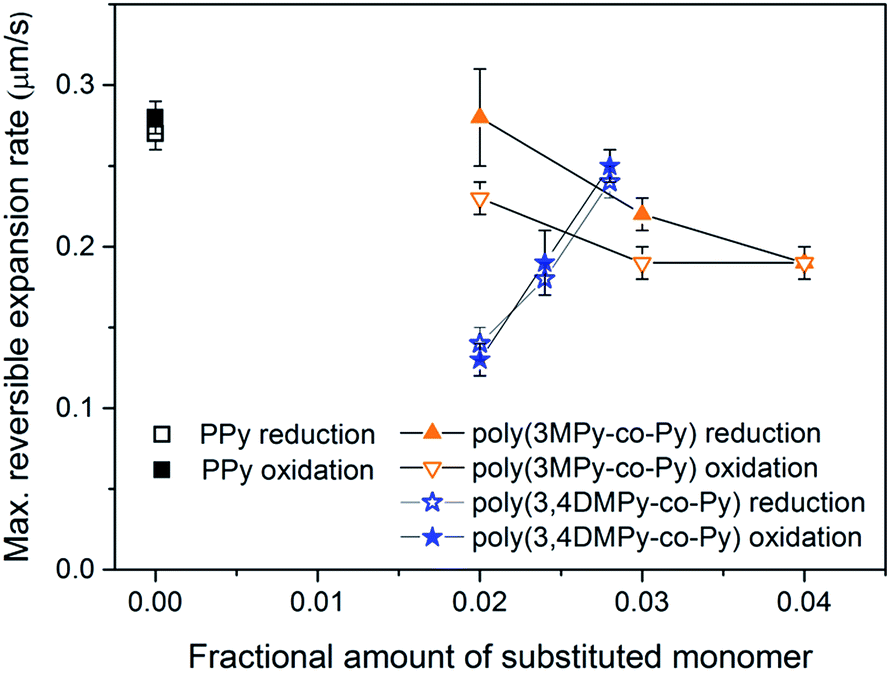 | ||
| Fig. 9 Maximum reversible expansion rates in the stable state for poly(3MPy-co-Py)(DBS)(DBS), poly(3,4-DMPy-co-Py)(DBS) and PPy(DBS) during the reduction and oxidation scans. Note: actuation data was recorded using a data point interval of 0.05 s during the stable state. Error bar = 1SD. | ||
The variation in the mean reduction charge in the stable state, recorded during the first 20 seconds of the cycle, with substituted monomer content is shown in Fig. 10. The bulk of the redox charge flow occurs within this time. Comparing the reversible expansion (Fig. 8) with the consumed charge (Fig. 10) there is indeed less reversible expansion and less charge for both types of copolymer compared to polypyrrole. However, the changes in reversible expansion and charge do not correlate (e.g. for 0.04 poly(3MPy-co-Py)(DBS) −7.8% decrease in charge results in a −35.2% decrease in reversible expansion relative to PPy(DBS)) as should be expected since these are faradaic machines.87 On the other hand the reversible expansion of poly(3,4DMPy-co-Py)(DBS) increases while the charge decreases with increasing 3,4DMPy fraction. Whereas for poly(3MPy-co-Py)(DBS) the reversible expansion decreases whilst the charge is more or less stable with increasing 3MPy fraction. In addition, the reversible expansion of poly(3,4DMPy-co-Py)(DBS) undergoes a 54% increase with increasing cycle number (see Fig. 6, 1.52 µm at cycle 2 to 2.34 µm at steady state), and yet there is essentially no change in charge (Fig. S8†).
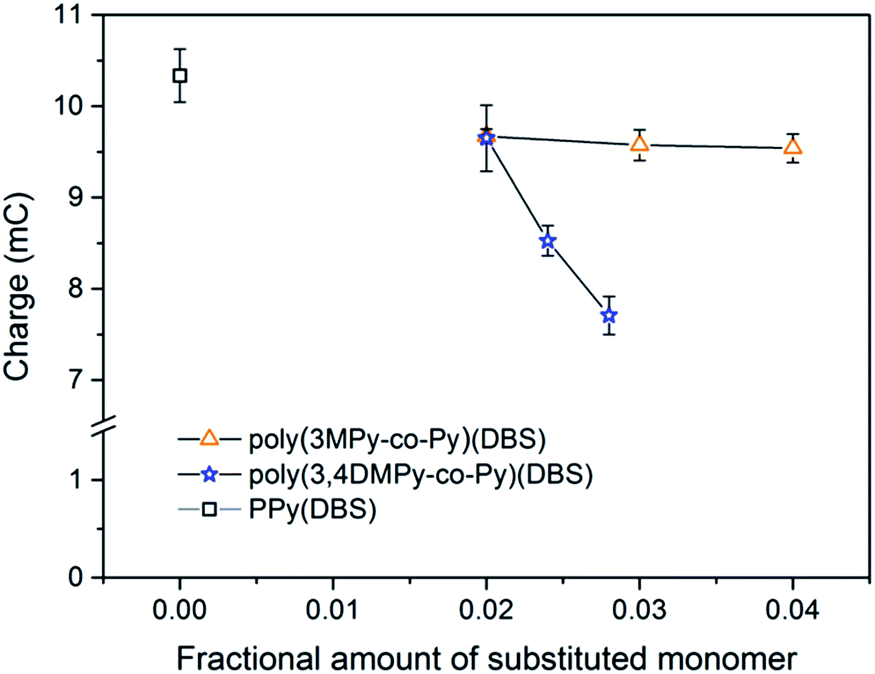 | ||
| Fig. 10 The variation of reduction charge in the stable state for poly(3MPy-co-Py)(DBS)(DBS), poly(3,4DMPy-co-Py)(DBS) and PPy(DBS). The charge was recorded during the first 20 s of the reduction scan, Error bar = 1SD. | ||
The reduced reversible expansion of the copolymers compared to polypyrrole that accompanies the introduction of substituted pyrrole monomers can only be explained to minor extent due to a reduced faradaic charge exchange (due to the electronic and steric effects66 of the methyl group) as could be expected. However, from this data is it clear that there is also a large contribution due to changes in the structure of the polymer network.
:100 respectively. When actuated these films displayed very large irreversible expansions which increased with cycle number. They displayed signs of deformation and delamination after a few tens of cycles. The reversible expansion of the films increased then decreased with increasing cycle number as they reached maximal expansion prior to delamination. In this sense, these copolymer films displayed similar behaviour to the poly(3MPy-co-Py)(DBS) and poly(3,4DMPy-co-Py)(DBS) above their threshold levels and therefore were not studied further.
4. Conclusions
We have probed the relationship between network structure and the actuation performance of polypyrrole films by synthesizing beta-substituted polypyrroles (homopolymers and copolymers) and comparing their actuation with polypyrrole. Beta-substitution of the polymer prevents (‘blocks’) crosslinking and branching that occurs at the beta-positions. Increasing the amount of beta-substitution in the polymer network results a decrease in crosslinking and branching.We have shown that a decrease in crosslinking (increased blocking) results in an increase in irreversible expansion. This is attributed to an increase in solvent swelling and electrochemical creep61,88 accompanying decreasing levels of crosslinking; behaviour which is well documented for other types of polymers and gels.89
All of the substituted homopolymer films were of low mechanical strength and underwent brittle fracture and delamination within a few cycles when actuated. Prior to failure they underwent very large irreversible expansion. In these materials the low levels of crosslinking impact upon the strength of the materials. Crosslinking is necessary to maintain the strength of an actuating film.90
The reversible expansion of the copolymer films is less than polypyrrole in the range investigated (below the threshold levels). The reversible expansion of the poly(3,4DMPy-co-Py)(DBS) increases with increasing 3,4DMPy content, whereas poly(3MPy-co-Py)(DBS) undergoes a small decreases with increasing 3DMPy content. The increase in reversible expansion of poly(3,4DMPy-co-Py)(DBS) with increasing 3,4DMPy is relatively large; for 0.28 poly(3,4DMPy-co-Py)(DBS) it is very close to that of PPy(DBS). This behaviour cannot be explained by electronic effects alone. We did not see a correlation between the change in the redox charge and the change in reversible expansion as should be expected for these faradaic machines.87 Moreover, methyl groups are expected to have a relatively small effect on the electronic properties of the pyrrole ring system65,66 and delocalisation between adjacent monomer units along the polymer chain as occurs in the oxidised form of the polymer68 and hence minimal effect on conductivity.67 Therefore there appears to be a ‘structural’ or ‘polymer network’ effect.
Apart from their use as actuating materials, numerous applications exist for these new materials. They might be capable of being used to control the release of relatively large, biologically relevant, molecules. At present only small molecules can be released from within the matrix of conventional polypyrrole films as large molecules become trapped within the highly crosslinked matrix. The forms of polypyrrole developed in this work which have low levels of crosslinking and become highly swollen might allow larger molecules to be release and higher loadings of the films.
Another potential application for these materials is in the mechanostimulation of cells.26 Here the materials would be capable of applying a larger total strain (reversible + irreversible) to the cells whilst maintaining the cells in a state of continuous strain (irreversible) between strokes.
At present there is no established technique for directly determining the crosslinking density of conducting polymers. However, based on the work presented here, we propose that changes in crosslinking can be indirectly monitored by observing changes in irreversible expansion; an increase in irreversible expansion reveals a decrease in crosslinking and vice versa. This view is in agreement with the work of Tominaga et al. who demonstrated that an increase in N-crosslinking results in a decrease in the irreversible expansion of PPy films.56 Indeed, the ‘solvent swellability’ of polymers is commonly used to monitor changes in crosslinking in important commercial polymers such as crosslinked polyethylene e.g. American Standard Test Method: ASTM D2765. We are currently investigating methods for directly monitoring changes in crosslinking.
Abbreviations
| Py | Pyrrole |
| DBS | Dodecylbenzene sulphonate |
| 3MPy | 3-Methyl-1H-pyrrole |
| 3,4DMPy | 3,4-Dimethyl-1H-pyrrole |
| PPy | Polypyrrole |
| 3MPPy | 3-Methylpolypyrrole |
| 3,4DMPPy | 3,4-Dimethylpolypyrrole |
| poly(3MPy-co-Py) | 3-Methylpolypyrrole-polypyrrole copolymer |
| poly(3,4DMPy-co-Py) | 3,4-Dimethylpolypyrrole-polypyrrole copolymer |
| DEPT | Distortionless enhancement by polarisation transfer. |
Acknowledgements
The authors wish to express their sincere gratitude to Prof. Magnus Berggren Linköping University (LiU-ITN), Prof. Anthony Turner Linköping University (LiU-IFM) and Dr Alex Skordos (Cranfield University) for their support. Funding has been supplied by EPSRC grant: EPP/504880/1, EU-FP7-Erasmus, European Science Foundation COST Action MP1003 ESNAM (European Scientific Network for Artificial Muscles) and COST-STSM-MP1003-11581, Swedish Foundation for Strategic Research (SSF), and Linköping University.References
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS PubMed.
- D. Kumar and R. C. Sharma, Eur. Polym. J., 1998, 34, 1053–1060 CrossRef CAS.
- G. Tourillon and F. Garnier, J. Phys. Chem., 1983, 87, 2289–2292 CrossRef CAS.
- K. K. Kanazawa, A. F. Diaz, M. T. Krounbi and G. B. Street, Synth. Met., 1981, 4, 119–130 CrossRef CAS.
- G. B. Street, T. C. Clarke, R. H. Geiss, V. Y. Lee, A. Nazzal, P. Pfluger and J. C. Scott, J. Phys., Colloq., 1983, 44(C3), 599 CrossRef.
- L. L. Miller, B. Zinger and Q. X. Zhou, J. Am. Chem. Soc., 1987, 109, 2267–2272 CrossRef CAS.
- M. Pyo and J. R. Reynolds, Chem. Mater., 1996, 8, 128–133 CrossRef CAS.
- A. Kros, S. W. F. M. van Hövell, N. A. J. M. Sommerdijk and R. J. M. Nolte, Adv. Mater., 2001, 13, 1555–1557 CrossRef CAS.
- W. J. Sung and Y. H. Bae, Biosens. Bioelectron., 2003, 18, 1231–1239 CrossRef CAS.
- X. Cui and D. C. Martin, Sens. Actuators, A, 2003, 103, 384–394 CrossRef CAS.
- T. M. Higgins, S. E. Moulton, K. J. Gilmore, G. G. Wallace and M. In Het Panhuis, Soft Matter, 2011, 7, 4690–4695 RSC.
- J. Isaksson, C. Tengstedt, M. Fahlman, N. Robinson and M. Berggren, Adv. Mater., 2004, 16, 316–320 CrossRef CAS PubMed.
- J. Causley, S. Stitzel, S. Brady, D. Diamond and G. Wallace, Synth. Met., 2005, 151, 60–64 CrossRef CAS PubMed.
- J. Y. Wong, R. Langer and D. E. Ingber, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3201–3204 CrossRef CAS.
- C. E. Schmidt, V. R. Shastri, J. P. Vacanti and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8948–8953 CrossRef CAS.
- K. Svennersten, M. H. Bolin, E. W. H. Jager, M. Berggren and A. Richter-Dahlfors, Biomaterials, 2009, 30, 6257–6264 CrossRef CAS PubMed.
- A. Herland, K. M. Persson, V. Lundin, M. Fahlman, M. Berggren, E. W. H. Jager and A. I. Teixeira, Angew. Chem., Int. Ed., 2011, 50, 12529–12533 CrossRef CAS PubMed.
- A. Kotwal and C. E. Schmidt, Biomaterials, 2001, 22, 1055–1064 CrossRef CAS.
- Q. Pei and O. Inganäs, Adv. Mater., 1992, 4, 277–278 CrossRef CAS PubMed.
- T. F. Otero, E. Angulo, J. Rodríguez and C. Santamaría, J. Electroanal. Chem., 1992, 341, 369–375 CrossRef CAS.
- E. W. H. Jager, E. Smela and O. Inganas, Science, 2000, 290, 1540–1545 CrossRef CAS.
- E. Smela, Adv. Mater., 2003, 15, 481–494 CrossRef CAS PubMed.
- S. A. Wilson, R. P. J. Jourdain, Q. Zhang, R. A. Dorey, C. R. Bowen, M. Willander, Q. U. Wahab, M. Willander, S. M. Al-hilli, O. Nur, E. Quandt, C. Johansson, E. Pagounis, M. Kohl, J. Matovic, B. Samel, W. van der Wijngaart, E. W. H. Jager, D. Carlsson, Z. Djinovic, M. Wegener, C. Moldovan, E. Abad, M. Wendlandt, C. Rusu and K. Persson, Mater. Sci. Eng., R, 2007, 56, 1–129 CrossRef PubMed.
- G. Alici, V. Devaud, P. Renaud and G. Spinks, J. Micromech. Microeng., 2009, 19(2), 025017 CrossRef.
- A. Khaldi, C. Plesse, C. Soyer, E. Cattan, F. Vidal, C. Legrand and D. Teyssié, Appl. Phys. Lett., 2011, 98(16), 164101 CrossRef PubMed.
- K. Svennersten, M. Berggren, A. Richter-Dahlfors and E. W. H. Jager, Lab Chip, 2011, 11, 3287–3293 RSC.
- M. Pyo, C. C. Bohn, E. Smela, J. R. Reynolds and A. B. Brennan, Chem. Mater., 2003, 15, 916–922 CrossRef CAS.
- S. Maw, E. Smela, K. Yoshida, P. Sommer-Larsen and R. B. Stein, Sens. Actuators, A, 2001, 89, 175–184 CrossRef CAS.
- S. Maw, E. Smela, K. Yoshida and R. B. Stein, Synth. Met., 2005, 155, 18–26 CrossRef CAS PubMed.
- S. Hara, T. Zama, W. Takashima and K. Kaneto, Polym. J., 2004, 36, 151–161 CrossRef CAS.
- S. Hara, T. Zama, W. Takashima and K. Kaneto, Polym. J., 2004, 36, 933–936 CrossRef CAS.
- L. Bay, N. Mogensen, S. Skaarup, P. Sommer-Larsen, M. Jørgensen and K. West, Macromolecules, 2002, 35, 9345–9351 CrossRef CAS.
- C. O. Yoon, H. K. Sung, J. H. Kim, E. Barsoukov, J. H. Kim and H. Lee, Synth. Met., 1999, 99, 201–212 CrossRef CAS.
- S. Shimoda and E. Smela, Electrochim. Acta, 1998, 44, 219–238 CrossRef CAS.
- T. F. Otero, H. Grande and J. Rodríguez, J. Electroanal. Chem., 1995, 394, 211–216 CrossRef.
- T. F. Otero, H. Grande and J. Rodrîguez, Electrochim. Acta, 1996, 41, 1863–1869 CrossRef CAS.
- T. F. Otero and J. G. Martinez, Chem. Mater., 2012, 24, 4093–4099 CrossRef CAS.
- J. M. Ribó, A. Dicko, M. A. Vallès, J. Claret, P. Dallemer, N. Ferrer-Anglada, R. Bonnett and D. Bloor, Polymer, 1993, 34, 1047–1053 CrossRef.
- E. Yurtsever, O. Esentürk, H. Önder Pamuk and M. Yurtsever, Synth. Met., 1999, 98, 229–236 CrossRef.
- J. Joo, J. K. Lee, J. S. Baeck, K. H. Kim, E. J. Oh and J. Epstein, Synth. Met., 2001, 117, 45–51 CrossRef CAS.
- A. Nazzal and G. B. Street, J. Chem. Soc., Chem. Commun., 1984, 2, 83–84 RSC.
- K. K. Kanazawa, A. F. Diaz, W. D. Gill, P. M. Grant, G. B. Street, G. P. Gardini and J. F. Kwak, Synth. Met., 1979, 1, 329–336 CrossRef.
- C. Tohumcu, R. Taş and M. Can, Ionics, 2014, 20, 1687–1692 CrossRef CAS.
- A. Deronzier and J. C. Moutet, Acc. Chem. Res., 1989, 22, 249–255 CrossRef CAS.
- F. Garnier, H. Korri-Youssoufi, P. Srivastava, B. Mandrand and T. Delair, Synth. Met., 1999, 100, 89–94 CrossRef CAS.
- J. C. Moutet, E. Saint-Aman, F. Tran-Van, P. Angibeaud and J. P. Utille, Adv. Mater., 1992, 4, 511–513 CrossRef CAS PubMed.
- H. Hasegawa, M. Kijima and H. Shirakawa, Synth. Met., 1997, 84, 177–178 CrossRef CAS.
- N. J. L. Guernion and W. Hayes, Curr. Org. Chem., 2004, 8, 637–651 CrossRef CAS.
- H. Masuda, S. Tanaka and K. Kaeriyama, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1831–1840 CrossRef CAS PubMed.
- S. A. Ashraf, F. Chen, C. O. Too and G. G. Wallace, Polymer, 1996, 37, 2811–2819 CrossRef CAS.
- E. E. Havinga, W. Ten Hoeve, E. W. Meijer and H. Wynberg, Chem. Mater., 1989, 1, 650–659 CrossRef CAS.
- P. G. Pickup, J. Electroanal. Chem., 1987, 225, 273–280 CrossRef CAS.
- F. Lallemand, D. Auguste, C. Amato, L. Hevesi, J. Delhalle and Z. Mekhalif, Electrochim. Acta, 2007, 52, 4334–4341 CrossRef CAS PubMed.
- E. Smela, Langmuir, 1998, 14, 2996–3002 CrossRef CAS.
- R. Cagnolati, M. Lucchesi, P. A. Rolla, V. Castelvetro, F. Ciardelli and A. Colligiani, Synth. Met., 1992, 46, 127–131 CrossRef CAS.
- K. Tominaga, K. Hamai, B. Gupta, Y. Kudoh, W. Takashima, R. Prakash and K. Kaneto, Phys. Procedia, 2011, 14, 143–146 CrossRef CAS PubMed.
- D. Melling, S. Wilson and E. W. H. Jager, Smart Mater. Struct., 2013, 22, 104021, DOI:10.1088/0964-1726/22/10/104021.
- Q. Pei and O. Inganäs, J. Phys. Chem., 1992, 96, 10507–10514 CrossRef CAS.
- Q. Pei and O. Inganäs, J. Phys. Chem., 1993, 97, 6034–6041 CrossRef CAS.
- P. Chiarelli, A. Della Santa, D. de Rossi and A. Mazzoldi, J. Intell. Mater. Syst. Struct., 1995, 6, 32–37 CrossRef CAS PubMed.
- J. D. Madden, D. Rinderknecht, P. A. Anquetil and I. W. Hunter, Sens. Actuators, A, 2007, 133, 210–217 CrossRef CAS PubMed.
- E. Smela and N. Gadegaard, J. Phys. Chem. B, 2001, 105, 9395–9405 CrossRef CAS.
- E. Smela, M. Kallenbach and J. Holdenried, J. Microelectromech. Syst., 1999, 8, 373–383 CrossRef CAS.
- M. Benslimane and P. Gravesen, Proc. SPIE-Int. Soc. Opt. Eng., 1999, 3669, 87–97 CrossRef CAS PubMed.
- B. G. Street, C. T. Clarke, H. R. Geiss, Y. V. Lee, A. Nazzal, P. Pfluger and C. J. Scott, J. Phys., Colloq., 1983, 44, C3-599–C593-606 CrossRef.
- J. L. Brédas, G. B. Street, B. Thémans and J. M. André, J. Chem. Phys., 1985, 83, 1323–1329 CrossRef PubMed.
- R. C. Foitzik, A. Kaynak, J. Beckmann and F. M. Pfeffer, Synth. Met., 2005, 155, 185–190 CrossRef CAS PubMed.
- K. Yakushi, L. J. Lauchlan, G. B. Street and J. L. Brédas, J. Chem. Phys., 1984, 81, 4133–4137 CrossRef CAS PubMed.
- K. Ichimura, S. Ichikawa and K. Imamura, Bull. Chem. Soc. Jpn., 1976, 49, 1157–1158 CrossRef CAS.
- Y. Liu, Q. Gan, S. Baig and E. Smela, J. Phys. Chem. C, 2007, 111, 11329–11338 CAS.
- B. L. Bray, P. H. Mathies, R. Naef, D. R. Solas, T. T. Tidwell, D. R. Artis and J. M. Muchowski, J. Org. Chem., 1990, 55, 6317–6328 CrossRef CAS.
- F. J. G. Frutos, T. F. Otero and A. J. F. Romero, Electrochim. Acta, 2007, 52, 3621–3629 CrossRef CAS PubMed.
- W. Wernet, M. Monkenbusch and G. Wegner, Mol. Cryst. Liq. Cryst., 1985, 118, 193–197 CrossRef CAS PubMed.
- Y. Dai and E. Blaisten-Barojas, J. Chem. Phys., 2010, 133(3), 034905 CrossRef PubMed.
- A. Nazzal and G. B. Street, J. Chem. Soc., Chem. Commun., 1984, 83–84 RSC.
- L. Bay, T. Jacobsen, S. Skaarup and K. West, J. Phys. Chem. B, 2001, 105, 8492–8497 CrossRef CAS.
- X. Wang and E. Smela, J. Phys. Chem. C, 2009, 113, 369–381 CAS.
- T. F. Otero, J. Mater. Chem. B, 2013, 1, 3754–3767 RSC.
- M. J. González-Tejera, M. V. García, E. Sánchez de la Blanca, M. I. Redondo, M. A. Raso and I. Carrillo, Thin Solid Films, 2007, 515, 6805–6811 CrossRef PubMed.
- Q. Pei and O. Inganäs, Synth. Met., 1992, 46, 353–357 CrossRef CAS.
- X. Wang and E. Smela, J. Phys. Chem. C, 2009, 113, 359–368 CAS.
- T. F. Otero, H. Grande and J. Rodríguez, J. Phys. Chem. B, 1997, 101, 8525–8533 CrossRef CAS.
- R. Pytel, E. Thomas and I. Hunter, Chem. Mater., 2006, 18, 861–863 CrossRef CAS.
- R. Z. Pytel, E. L. Thomas, Y. Chen and I. W. Hunter, Polymer, 2008, 49, 1338–1349 CrossRef CAS PubMed.
- R. H. Geiss, G. B. Street, W. Volksen and J. Economy, IBM J. Res. Dev., 1983, 27, 321–329 CrossRef CAS.
- J. C. Scott, J. L. Bredas, K. Yakushi, P. Pfluger and G. B. Street, Synth. Met., 1984, 9, 165–172 CrossRef CAS.
- L. Valero, J. Arias-Pardilla, J. Cauich-Rodríguez, M. A. Smit and T. F. Otero, Electrochim. Acta, 2011, 56, 3721–3726 CrossRef CAS PubMed.
- K. Kaneto, T. Shinonome, K. Tominaga and W. Takashima, Jpn. J. Appl. Phys., 2011, 50(9), 091601 CrossRef.
- N. G. Bel'nikevich, T. V. Budtova, S. A. Vesnebolotskaya and G. K. Elyashevich, Russ. J. Appl. Chem., 2008, 81, 1818–1820 CrossRef.
- K. M. Persson, R. Karlsson, K. Svennersten, S. Löffler, E. W. H. Jager, A. Richter-Dahlfors, P. Konradsson and M. Berggren, Adv. Mater., 2011, 23, 4403–4408 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 principle of LSM actuation measurement, Fig. S2 metrics used to characterise actuation performance, Fig. S3–S7 confirmation of the structure and purity of 3,4-dimethyl-1H-pyrrole, Fig. S8 comparison of charge consumed for the prepared films during cycle 10 and the stable state. See DOI: 10.1039/c5ra15587h |
| This journal is © The Royal Society of Chemistry 2015 |