
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Termini capping of metal-poly-His peptide complexes induces the formation of α-helix†
Eyal
Simonovsky
ab,
Henryk
Kozlowski
*c and
Yifat
Miller
*ab
aDepartment of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel
bIlse Katz Institute for Nanoscale Science and Technology, Beer-Sheva 84105, Israel. E-mail: ymiller@bgu.ac.il
cFaculty of Chemistry, University of Wroclaw, 50-383 Wroclaw, Poland. E-mail: henryk.kozlowski@chem.uni.wroc.pl
First published on 1st December 2015
Abstract
An ensemble of structures of metal-hexa-histidine-tag capped and uncapped peptides has been studied using molecular dynamics simulations. The metal-binding peptides are polymorphic for both capped and uncapped peptides. The capping of the peptide termini promotes α-helical conformations that are induced by the metal binding sites.
His-tags are specific sequences containing His repeats, such as six to nine subsequent histidyl residues. They are used for purification of recombinant proteins by immobilized-metal affinity chromatography (IMAC).1 The typical (His)6-tag is linked to the C- or N-terminus of the protein (which is meant to be purified), and it serves as a molecular ‘anchor’ that binds to a metal ion (copper or nickel), immobilized by chelation with nitrilotriacetic acid (NTA) bound to a solid support. Such polyhistidyl tags can be also found in nature.2 More than 2000 histidine-rich proteins (HRPs) have been found in microorganisms including 60% and 82% of archaeal and bacterial species, respectively. Proteins containing histidine-rich domains play a critical role in metal regulation and homeostasis.3,4 Further functions of histidine-rich proteins in eukaryotes have been reported,5,6e.g. antimicrobial activities in the oral cavity (e.g., histatins from saliva),7 heme polymerization (e.g., HRP-2 from Plasmodium falciparum),8 or heparin binding (e.g., serum glycoprotein HRG found in humans).9
Chemically synthesised peptides have free amino and carboxy termini, being electrically charged in general. In order to remove this charge, peptide ends are often modified by N-terminal acetylation and/or C-terminal amidation, i.e., capping of the ends of peptides. We have recently studied the coordination of Cu2+ with capped (His)6-tag peptide using experimental and computational tools.10 The structure of the metal-binding His-tag is mainly determined by the metal binding sites. One can assume that the termini capping of the metal-binding His-tag will not affect its conformational structure; however, we suggest that the capping of the peptide termini significantly shifts the energy landscape of the metal-peptide complexes, affects its conformational structure and actually allows the stabilization of different conformations.
The effect of termini capping groups on the structure of metal-binding peptides was not the subject of many studies; however the effect of termini capping on the structure of non-metal-binding peptides is well documented in the literature. Several works reported that termini capping affected the self-assembly of peptides.11–13 In the specific aspect of the capping of peptides with α-helical structures, N-acetylation was found to increase the helix propensities of peptides.14,15 Furthermore, helical propensity was induced in a polypeptide chain by linking specially designed capped N-terminal groups.15 In addition, the stabilization and destabilization of helices affected by various N- or C-termini groups in capped peptides had been extensively studied by several groups.15–17 It had been also suggested that terminal polar and charged groups of capped peptides promote formation of α-helices.18–23 These studies were focused on free soluble monomeric peptides. However, in metal-binding peptides, the peptide structure is “locked” via the metal-binding site. To the best of our knowledge, the effect of termini capping on metal-binding poly-His peptides, and specifically, on complexes in which metal ions induce formation of helices has not been investigated so far at the atomic resolution.
In this study we show for the first time that capping of both N- and C-termini of a metal-binding poly-His peptide [Cu2+-(His)6] promotes α-helical conformations and that these helical conformations are induced by specific metal binding sites. The Kabsch & Sander method24 for secondary structural analysis has showed that for some of the metal-binding sites, the capped peptides were arranged in helical conformations while the uncapped peptides did not show helical propensities. Energy calculations and population analysis showed that the α-helical conformations are more stable and preferred than the non-helical conformations. Interestingly, spontaneous additional interactions to the constructed Cu2+-(His)6 peptides of imidazoles and/or free carboxyl groups of the C-termini of the uncapped peptides with the Cu2+ are produced during the molecular dynamics (MD) simulations. These additional interactions contribute to the stabilization of the metal-induced α-helical conformations for the capped peptides.
A total of twelve Cu2+-(His)6 peptide models had been derived from the six different binding sites: six models for the capped peptide (A models) and six models for the uncapped peptide (B models) (Fig. 1a). The capped peptide models were constructed using the widely used N-terminus acetylation and C-terminus amidation. The Cu2+ binds to different sets of two imidazoles of the peptide, representing the different binding sites (Fig. 1b) proposed by our previous study.10 The constructed initial models are detailed in the ESI.†
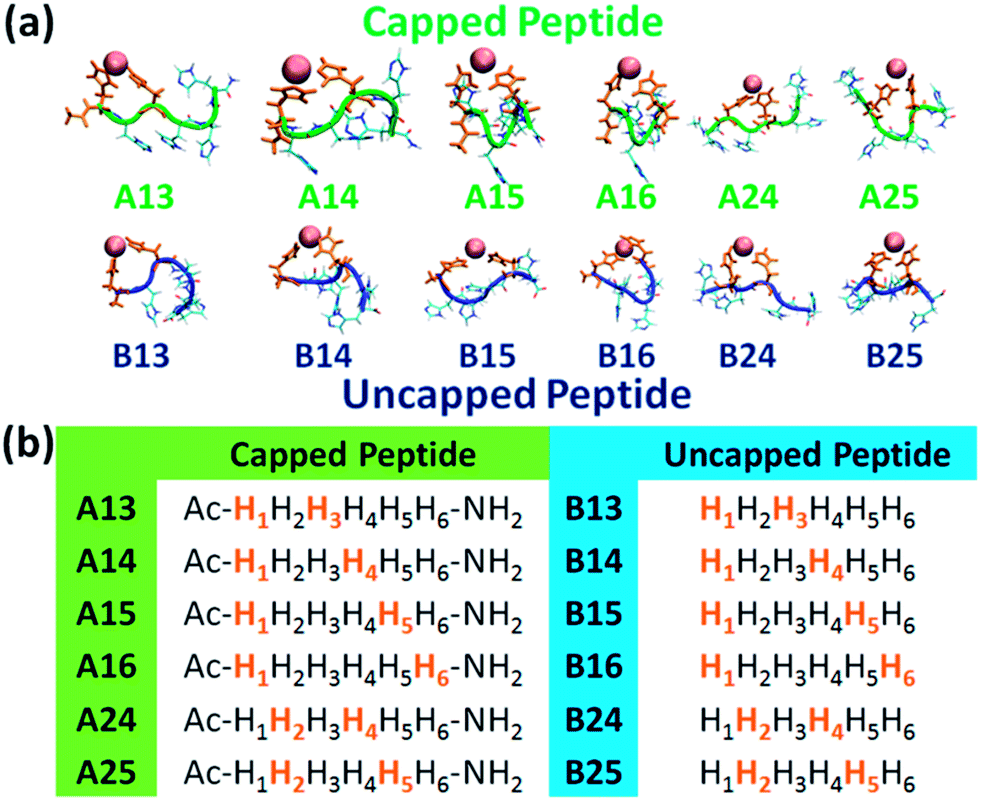 | ||
| Fig. 1 (a) The simulated twelve Cu2+-(His)6 capped peptide and uncapped peptide models. (b) The sequence of the capped and uncapped peptides. His residues that are colored in orange bind to Cu2+. | ||
In order to study the relative conformational energies and the structures of the different possible metal binding peptide models, each model was explicitly solvated and minimized at physiological pH. All-atom explicit MD simulations were then performed in the NPT ensemble at 310 K for 100 ns, using the NAMD program25 with the all-atom CHARMM27 (ref. 26 and 27) force-field with CMAP correction.28 The preference of each model for a specific arrangement has been obtained by comparing the relative conformational energies of all constructed models, using the Generalized Born Method with Molecular Volume (GBMV).29,30 The relative conformational energies can be compared only for models that have the same sequence and the same number of atoms; thus, the relative conformational energies had been computed separately for the six capped peptide simulated models (A13, A14, A15, A16, A24, and A25) and for the six uncapped peptide simulated models (B13, B14, B15, B16, B24, and B25). By applying Monte Carlo (MC) simulations, the relative probability of model n was evaluated as Pn = Nn/Ntotal, where Pn is the population of model n, Nn is the total number of conformations visited for model n, and Ntotal is the total steps. The advantages of using Monte Carlo (MC) simulations to estimate conformer probability lie in their good numerical stability and the control that they allow of transition probabilities among several conformers, as we extensively applied in similar systems.31–33 Finally, the secondary structure of each simulated peptide had been determined by using the Kabsch & Sander method,24 which is named the “Define Secondary Structure of Proteins” (DSSP) method. For each snapshot along the 100 ns (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 snapshots) the His residues of the peptides, excluding the termini residues (His1 and His6), were analysed using the DSSP method that provides structural properties of “α-helix”, “β-strand” or “not α-helix or β-strand” for each residue along the peptide sequence.
000 snapshots) the His residues of the peptides, excluding the termini residues (His1 and His6), were analysed using the DSSP method that provides structural properties of “α-helix”, “β-strand” or “not α-helix or β-strand” for each residue along the peptide sequence.
A total of 3000 conformations of the six capped peptide models (500 conformations for each model) and a total of 3000 conformations of the six capped peptide models (500 conformations for each model) were used to construct the energy landscape of the six capped peptide models and six uncapped peptide models (Table S1, ESI†). In addition, we evaluated the populations of the models using MC simulations (Fig. 2). The estimated energies and populations of the different models illustrate polymorphic states, i.e. similar populations. Yet, among the capped peptide models, models A15 and A16 have the highest population (above 20%). Interestingly, these models have strong propensities of α-helical structure (Fig. 1). Among the uncapped peptide models there is no single model with higher population, but several models with similar populations (∼20%): B13, B24, and B25 and with no α-helical structures (Fig. 1). We suggest that the negative charge in the C-terminal of the uncapped peptide promote conformational change and thus leads to a rugged energy landscape, i.e. the negative charge in the C-terminal of the uncapped peptide leads to polymorphism. We further suggest that the capping of the peptide leads to population shift and to the conformational change of metal binding peptides. Interestingly, the most significant population shifts appear in model A16/B16. While model B16 does not show α-helical structures, model A16 shows α-helical structures (Fig. 1). The differences in the populations' values for this model (A16 vs. B16) show the highest differences compare to the other models (Fig. 2). The conformational change from a “non-helical” structure to a “helical” structure significantly changed the populations.
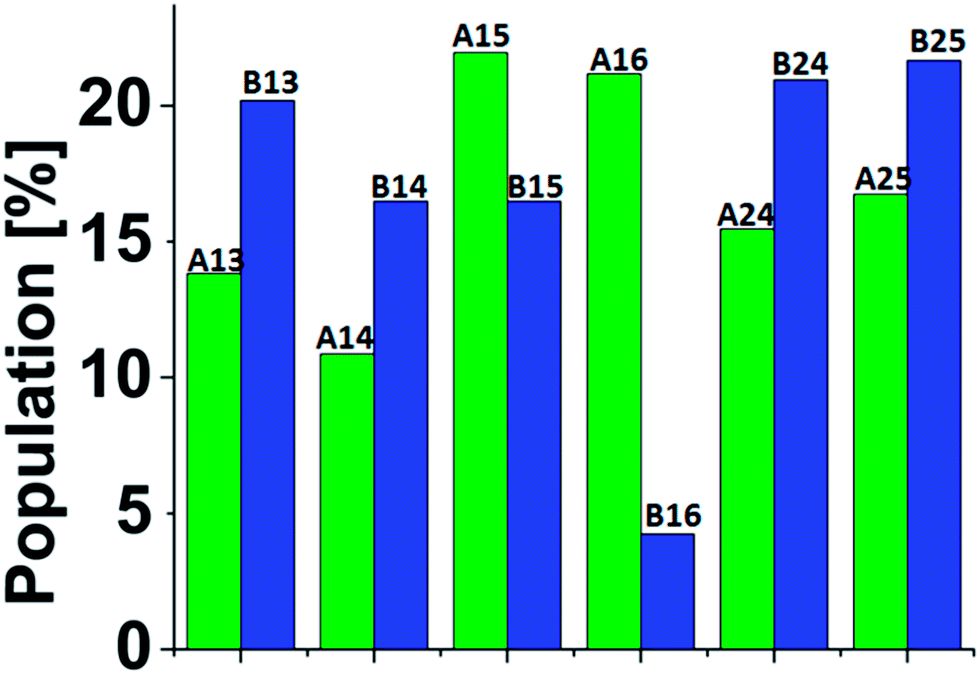 | ||
| Fig. 2 Populations of the simulated six Cu2+-(His)6 capped (green) and six uncapped (blue) peptide models. The populations had been estimated via Monte Carlo simulations (see ESI and text†). | ||
Interestingly, the conformational changes of A16/B16 models from a “non-helical” structure to a “helical” structure are also confirmed by the DSSP secondary structure analysis (Fig. 3). As seen in Fig. 2 the difference in the population between the capped and the uncapped peptide models A15 and B15, respectively, is ∼10%. In these models the Cu2+ binds to i and i+4 His residues (i.e. H1 and H5), and thus the Cu2+ fixes the peptide backbone to form α-helical conformation in the capped peptide A15 – above 60% (Fig. 3). Previous experimental studies by Ghadiri et al.34,35 Ruan et al.36 and Siedlecka et al.37 illustrated designed capped metal-binding peptides. In some of these studies, the metal binds to residues i and i+4 along the sequence.34–36 This binding site, i and i+4 along the peptide sequence, yields stable α-helical conformations, which are induced by metal binding nucleation.34–36 In the absence of metals, these peptides had not been arranged in α-helical conformations. Herein, we demonstrate a first computational study that validates the experimental observation at the atomic resolution and more specifically for metal-poly-His peptides. These experimental observations34–36 and our computational study suggest that the widely used metal-binding capped peptides in general, and specifically, metal-binding capped His-tag peptides, may induce α-helical conformations and thus serve as helix nucleation initiators.
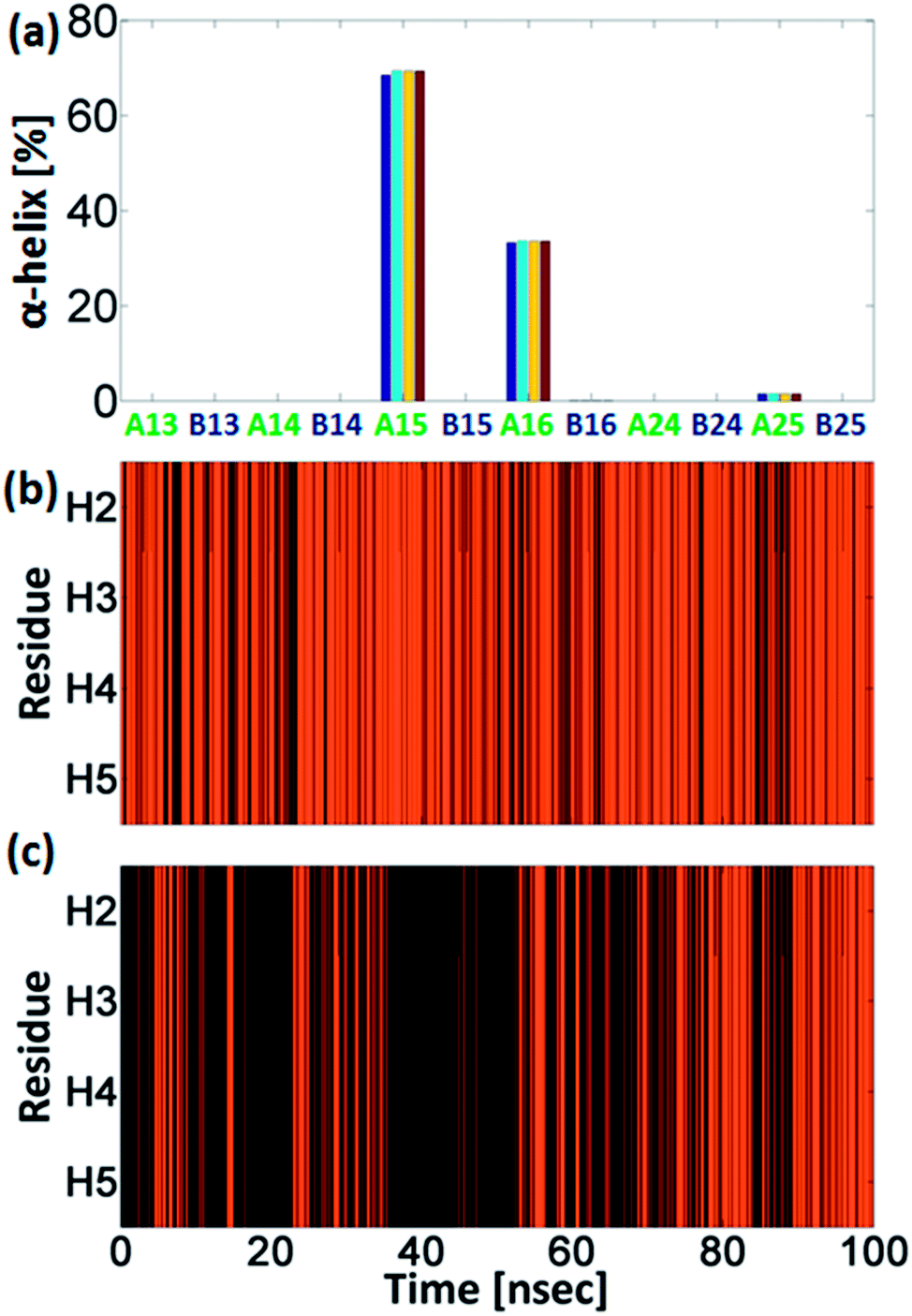 | ||
| Fig. 3 (a) The percentage of α-helical properties of residues H2 (blue), H3 (light blue), H4 (yellow) and H5 (red) for the capped and the uncapped peptide models via DSSP analysis. Model A15 shows the highest percentage of α-helix properties; explicit DSSP analysis along the time of the simulations of residues H2–H5 for models A15 (b) and A16 (c). Orange lines represent α-helix and black lines represent neither α-helix nor β-strand. | ||
Interestingly, the uncapped peptide B15 does not present an α-helical conformation, although the Cu2+ binds to i and i+4 residues. Following the MD simulations, one can see that a spontaneous interaction between the Cu2+ and His4 had been observed and thus “disturbs” the formation of the i and i+4 binding site and consequently does not allow the peptide to be organized in an α-helical conformation. The α-helical conformations have been also found in two other capped peptide models: A16 and A25 with smaller probability (Fig. 3). During the MD simulations of the capped peptide A16 model, spontaneous interactions between the Cu2+ and His5 had been observed (Fig. 4). These interactions “mimic” the binding site i and i+4, which is illustrated in model A15 and thus one can see properties of α-helical conformation – ∼30% (Fig. 3). The capped peptide model A25 illustrates a relatively small percentage of α-helical conformation – less than 5% (Fig. 3), in this case, it does not “mimic” the i and i+4 binding site, by forming spontaneous interactions (Fig. 1 and S1, ESI†). The other capped peptide models A13, A14 and A24 that do not “mimic” the binding site i and i+4, do not demonstrate properties of α-helical conformations. Finally, all the uncapped peptides, including B15, B16 and B25, did not show properties of α-helical conformations (Fig. 3). In summary, spontaneous interactions that “mimic” the i and i+4 binding site in the capped peptide contribute to the formation of helical structure, but spontaneous interactions in the uncapped counterparts peptides “disturb” the formation of helical structure. We do not refer to the formation of helical structure due to residue–residue interactions or residues–solvent interactions.
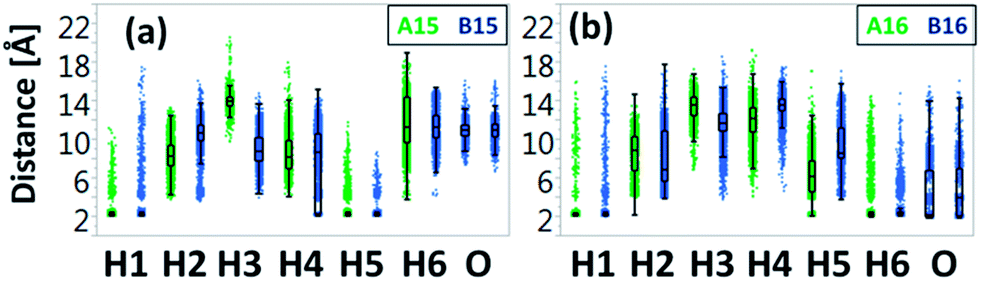 | ||
| Fig. 4 The Cu2+-Nε (His) atom and Cu2+-O (C-terminal carboxyl group) atom distance distribution for each His residue obtained from MD simulations for models A15 and B15 (a) and for models A16 and B16 (b). The vertical lines within each box represent the median distance values. | ||
One of the interesting results that had been obtained from our simulations is the formation of spontaneous interactions between the Cu2+ and the negative charged oxygen atoms of the carboxylic groups in the C-terminal of the three uncapped peptide models B14, B16 and B25 (Fig. 3 and S1, ESI†). These spontaneous interactions' events were not permanent in the simulations, but one cannot neglect that they occur. We cannot explain why these events occur only in these three uncapped peptides and not in the other three uncapped peptides. Yet, all the six uncapped Cu2+-(His)6 peptides did not illustrate α-helical conformations. We suggest that the negative charge in the C-termini of these uncapped peptides interrupts the expected helix dipole. Consequently, the negative charge in the C-termini yields repulsive interactions between the C-terminal carboxylate and the neighbouring amide bond. Previous studies showed that positive charge in the C-termini of capped peptides yields stable α-helical conformations,23 while negative charge destabilizes α-helical conformations.16
It is of an interest to examine the effect of only singly-capped (C- or N-terminal) peptide on the formation of α-helix in the Cu2+-His6. Among all the six studied models we chose the A15 to examine the singly-capped peptide. The justification for this choice is due to two reasons. First, this model is the most populated model that fit the rule of i and i+4 (Fig. 4). Second, it illustrates the highest percentage of helical structure (Fig. 3a). Fig. 5a and b demonstrate the simulated models A15, B15, C15 (singly-capped N-terminal) and D15 (singly-capped C-terminal) models. One can see that C15 has less than 5% of helical properties and D15 shows ∼0.3% of helical properties (Fig. 5c). These relatively small probabilities are negligible and thus lead to the conclusion that singly-capped peptide prevents the formation of α-helical structure in Cu2+-His6. Yet, it was previously suggested that N-acetylation was found to increase the helix propensities of peptides,14,15 therefore the simulated model C15 has very small properties of helical structure (less than 5%). D15 has a smaller percentage of helical properties, due to the lack of N-acetylation.
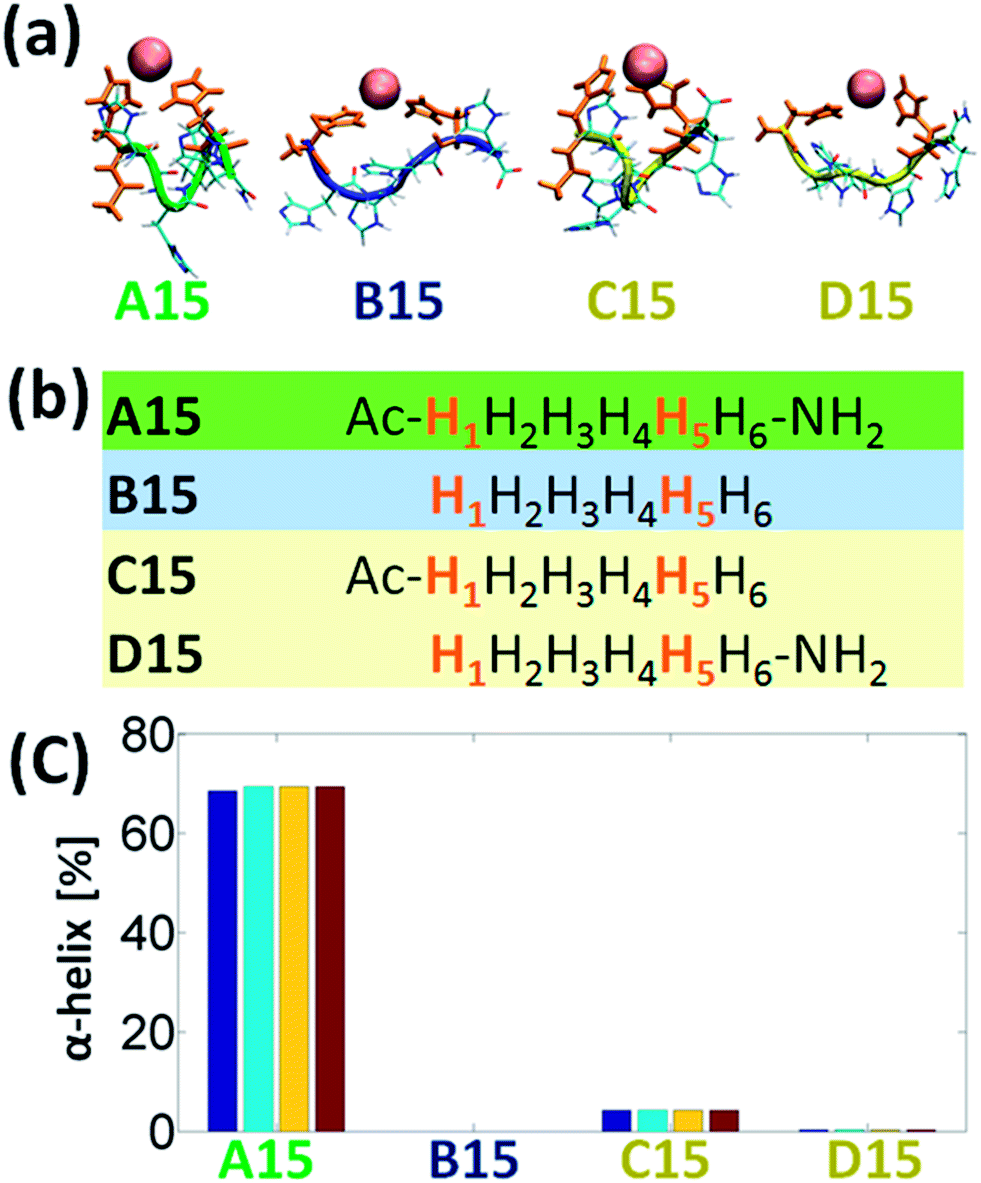 | ||
| Fig. 5 (a) The simulated Cu2+-(His)6 peptides: A15-capped peptide, B15-uncapped peptide, C15-singly capped N-terminal peptide and D15-singly capped C-terminal peptide. (b) The sequence of peptides A15, B15, C15 and D15. (c) The percentage of α-helical properties of residues H2 (blue), H3 (light blue), H4 (yellow) and H5 (red) for the capped and the uncapped peptide models via DSSP analysis. | ||
Finally, one can see from Fig. S1,† that although D15 exhibits the rule of i and i+4, it does not show helical properties, due to the uncapped N-terminal. However, C15 which also exhibits this rule and shows very small properties of helical structure, interact with the free carboxylic groups (Fig. S1†), similarly as obtained for B14, B16, and B25, and thus does not show helical structure.
Conclusions
Our study leads to several conclusions: first, capping of the termini of metal-binding poly-His peptides play important role in conformational change inducing the formation of α-helical conformation. Second, capping of the termini of metal-binding peptides yields to the formation of polymorphic states. Third, singly-capped (C- or N-terminal) peptide prevents the formation of α-helical structure in the Cu2+-His6. Fourth, our study suggests that capping and uncapping of the termini of peptides/proteins (with absence and with presence of metal ions), may differently affect the structural properties of the peptides/proteins. Therefore, both capped and uncapped peptides/proteins should be investigated in aim to compare the differences in the structural characterization.Acknowledgements
This research was supported by Grant No. 2011128 from the United States-Israel Binational Science Foundation (BSF). All simulations were performed using the high-performance computational facilities of the Miller lab in the BGU HPC computational center. The support of the BGU HPC computational center staff is greatly appreciated.Notes and references
- D. S. Waugh, Trends Biotechnol., 2005, 23, 316–320 CrossRef CAS PubMed.
- M. Rowinska-Zyrek, D. Witkowska, S. Potocki, M. Remelli and H. Kozlowski, New J. Chem., 2013, 37, 58–70 RSC.
- L. Martino, Y. Z. He, K. L. D. Hands-Taylor, E. R. Valentine, G. Kelly, C. Giancola and M. R. Conte, FEBS J., 2009, 276, 4529–4544 CrossRef CAS PubMed.
- D. Witkowska, M. Rowinska-Zyrek, G. Valensin and H. Kozlowski, Coord. Chem. Rev., 2012, 256, 133–148 CrossRef CAS.
- G. Schmitt-Ulms, S. Ehsani, J. C. Watts, D. Westaway and H. Wille, PLoS One, 2009, 4, e7208, DOI:10.1371/journal.pone.0007208.
- P. Stanczak, D. Valensin, E. Porciatti, E. Jankowska, Z. Grzonka, E. Molteni, E. Gaggelli, G. Valensin and H. Kozlowski, Biochemistry, 2006, 45, 12227–12239 CrossRef CAS PubMed.
- M. J. Oudhoff, J. G. M. Bolscher, K. Nazmi, H. Kalay, W. van't Hof, A. V. N. Amerongen and E. C. I. Veerman, FEBS J., 2008, 22, 3805–3812 CAS.
- A. Trampuz, M. Jereb, I. Muzlovic and R. M. Prabhu, Critical Care, 2003, 7, 315–323 CrossRef PubMed.
- M. K. Burch, M. N. Blackburn and W. T. Morgan, Biochemistry, 1987, 26, 7477–7482 CrossRef CAS PubMed.
- J. Watly, E. Simonovsky, R. Wieczorek, N. Barbosa, Y. Miller and H. Kozlowski, Inorg. Chem., 2014, 53, 6675–6683 CrossRef CAS PubMed.
- M. Andreasen, K. K. Skeby, S. Zhang, E. H. Nielsen, L. H. Klausen, H. Frahm, G. Christiansen, T. Skrydstrup, M. Dong, B. Schiott and D. Otzen, Biochemistry, 2014, 53, 6968–6980 CrossRef CAS PubMed.
- K. Tao, J. Wang, P. Zhou, C. Wang, H. Xu, X. Zhao and J. R. Lu, Langmuir, 2011, 27, 2723–2730 CrossRef CAS PubMed.
- V. Castelletto, I. W. Hamley, C. Cenker, U. Olsson, J. Adamcik, R. Mezzenga, J. F. Miravet, B. Escuder and F. Rodriguez-Llansola, J. Phys. Chem. B, 2011, 115, 2107–2116 CrossRef CAS PubMed.
- A. Chakrabartty, A. J. Doig and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11332–11336 CrossRef CAS.
- W. Maison, E. Arce, P. Renold, R. J. Kennedy and D. S. Kemp, J. Am. Chem. Soc., 2001, 123, 10245–10254 CrossRef CAS PubMed.
- M. Kuemin, S. Schweizer, C. Ochsenfeld and H. Wennemers, J. Am. Chem. Soc., 2009, 131, 15474–15482 CrossRef CAS PubMed.
- J. P. Schneider and W. F. DeGrado, J. Am. Chem. Soc., 1998, 120, 2764–2767 CrossRef CAS.
- L. G. Presta and G. D. Rose, Science, 1988, 240, 1632–1641 CAS.
- B. Forood, E. J. Feliciano and K. P. Nambiar, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 838–842 CrossRef CAS.
- B. Forood, H. K. Reddy and K. P. Nambiar, J. Am. Chem. Soc., 1994, 116, 6935–6936 CrossRef CAS.
- B. Forood, E. J. Feliciano and K. P. Nambiar, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 838–842 CrossRef CAS.
- J. S. Richardson and D. C. Richardson, Science, 1988, 240, 1648–1652 CAS.
- D. E. Blagdon and M. Goodman, Biopolymers, 1975, 14, 241–245 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- L. Kale, R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan and K. Schulten, J. Comput. Phys., 1999, 151, 283–312 CrossRef CAS.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- P. L. Freddolino, S. Park, B. Roux and K. Schulten, Biophys. J., 2009, 96, 3772–3780 CrossRef CAS PubMed.
- M. S. Lee, M. Feig, F. R. Salsbury and C. L. Brooks, J. Comput. Chem., 2003, 24, 1348–1356 CrossRef CAS PubMed.
- M. S. Lee, F. R. Salsbury and C. L. Brooks, J. Chem. Phys., 2002, 116, 10606–10614 CrossRef CAS.
- V. Wineman-Fisher, Y. Atsmon-Raz and Y. Miller, Biomacromolecules, 2015, 16, 156–165 CrossRef CAS PubMed.
- F. Pontecchiani, E. Simonovsky, R. Wieczorek, N. Barbosa, M. Rowinska-Zyrek, S. Potocki, M. Remelli, Y. Miller and H. Kozlowski, Dalton Trans., 2014, 43, 16680–16689 RSC.
- Y. Raz, J. Adler, A. Vogel, H. A. Scheidt, T. Haupl, B. Abel, D. Huster and Y. Miller, Phys. Chem. Chem. Phys., 2014, 16, 7710–7717 RSC.
- M. R. Ghadiri and A. K. Fernholz, J. Am. Chem. Soc., 1990, 112, 9633–9635 CrossRef CAS.
- M. R. Ghadiri and C. Choi, J. Am. Chem. Soc., 1990, 112, 1630–1632 CrossRef CAS.
- F. Q. Ruan, Y. Q. Chen and P. B. Hopkins, J. Am. Chem. Soc., 1990, 112, 9403–9404 CrossRef CAS.
- M. Siedlecka, G. Goch, A. Ejchart, H. Sticht and A. Bierzynski, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 903–908 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Material and methods and analysis details. See DOI: 10.1039/c5ra15385a |
| This journal is © The Royal Society of Chemistry 2015 |