
Theoretical studies of heteroatom-doping in TiO2 to enhance the electron injection in dye-sensitized solar cells
Abstract
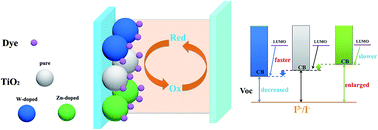
Density functional calculations have been explored to analyze the structural, electronic and charge transfer properties of a doped TiO2 substrate and catechol–TiO2 interfaces for dye-sensitized solar cells. The results demonstrate that the dopant W6+ moves the CB (conduction band) edge downward and introduces 5d-unoccupied orbitals located in the CB bottom of the TiO2. On the other hand, the dopant Zn2+ shifts the CB edge upward with an insertion of 3d-occupied orbitals into the VB (valence band). In catechol–TiO2 systems, W6+ enlarges the energy difference between the LUMO of catechol and LUMO of TiO2, which enlarges the driving force for electron injection in turn and results in an increased short circuit current (Jsc). Our modeling injection dynamics and quantitative Bader analysis of the interfacial charge transfer have revealed that the catechol–TiO2 doped with W6+ provides faster and more electron injection for dye sensitized solar cells (DSSCs). While the Zn2+ doped system exhibits lower electron efficiency due to the minimized energy difference between the catechol LUMO and TiO2 CB.
 Please wait while we load your content...
Please wait while we load your content...

