
Bromo–nitro substitution on a tertiary α carbon—a previously uncharacterized facet of the Kornblum substitution†
Matthew J. Leonard‡
*,
Peter G. McKay and
Anthony R. Lingham
RMIT University, 1 Bowen street, Melbourne 3001, Australia. E-mail: Matthew.Leonard742@gmail.com
First published on 3rd September 2015
Abstract
Sodium nitrite in dimethylformamide substitutes nitro for bromine alpha to an amide carbonyl in high yield at a tertiary site. Hammett plots show a strongly positive ρ value (+0.67), indicating a negatively-charged transition state, in contrast to the typical SN1/SN2 mechanism domain for Kornblum substitutions.
Introduction
The Kornblum substitution is the replacement of a halogen on an organic compound by a nitro group by using sodium nitrite (NaNO2) as a nitrite source and DMF as a solvent. The reaction proceeds in high yields at room temperature and does not require anhydrous conditions. It was discovered and widely characterized by Nathan Kornblum (Fig. 1) at Purdue University, Indiana in the 1950s.1–3 | ||
| Fig. 1 Nathan Kornblum. | ||
In 1991 Noboru Ono published a textbook that summarized the key methods for the preparation of nitro compounds4 followed by an updated version in 2001 (ref. 5) (while writing this book, Ono collaborated with Kornblum, who was late in his career and died shortly after). Ono named the substitution “the Kornblum reaction” in both the 1991 and 2001 versions.4,5 However, in 2002 the term “Kornblum reaction” was used by Mamedov et al. to refer to the Kornblum oxidation,6 which is a different reaction that was also elucidated by Kornblum. Yet a third reaction has also been named after Kornblum, namely the Kornblum–DeLaMare rearrangement.7 We propose that the X–NO2 substitution that was characterized by Kornblum be described as the ‘Kornblum substitution’. We here discuss and further characterize the Kornblum substitution.
The Kornblum substitution was summarized by Ono and others as occurring on primary and secondary halogeno compounds, but not tertiary where a HX elimination product is consistently observed4,5,8,9 (Fig. 2). Kornblum's original observations2 support this view.
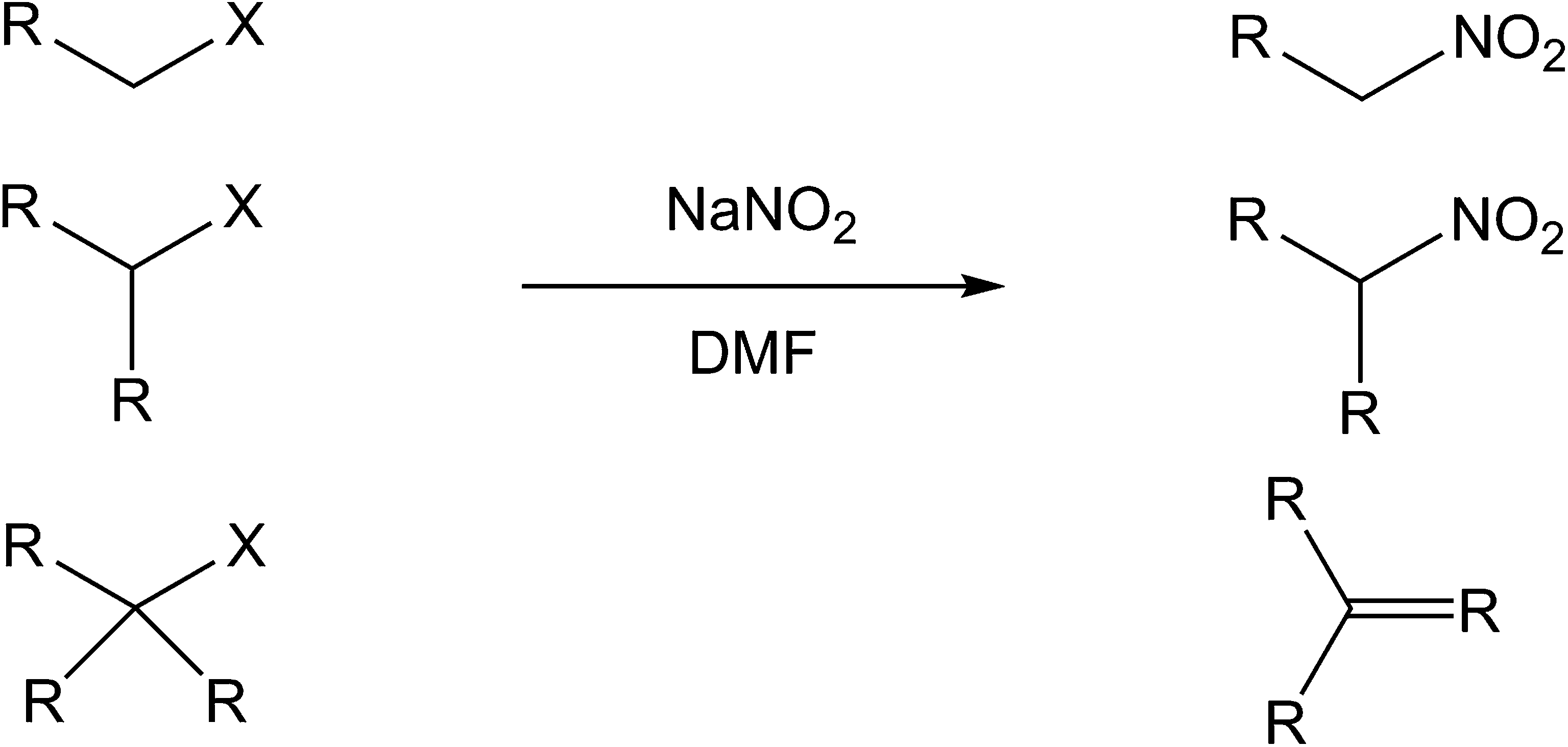 | ||
| Fig. 2 The Kornblum substitution. | ||
However, we have found that the Kornblum substitution does proceed on a tertiary centre that is alpha to an anilide carbonyl group. We have hence used the Kornblum substitution to prepare an α-nitroisobutyranilide (2) in order to perform an alternative synthesis of the hydantoin anti-baldness compound RU58841, a process that we published in 2014.10 The reaction was simple and performed in high yield with low cost materials (Fig. 3).
 | ||
| Fig. 3 An α-nitroisobutyranilide from an α-bromoisobutyranilide. | ||
We had discovered that it was possible to do a Kornblum substitution on the α-bromoisobutyranilide (1) when we observed that the product of 1 treated with NaNO2 in DMF appeared as an M-89 signal on a GC-MS. We found that an aryl isocyanate (Ar–NCO) was forming with the loss of 2-nitropropane under the high temperature conditions of the GC-MS injector port, but that at room temperature the α-bromoisobutyranilide (1) readily formed the α-nitroisobutyranilide (2), which could be crystallized by the addition of water.
We note that among Kornblum's original writings on the topic, in one paper Kornblum described the substitution as occurring on primary and secondary carbons alpha to a carbonyl.11 He subsequently placed a patent on this process for the preparation of α-nitroesters from α-haloesters which included an example of a tertiary nitro compound – the preparation of ethyl α-nitroisobutyrate (4) from ethyl α-bromoisobutyrate (3)12 (Fig. 4).
 | ||
| Fig. 4 Preparation of ethyl α-nitroisobutyrate from Kornblum's 1957 patent.12 | ||
The example in this patent of a tertiary halo–nitro substitution alpha to a carbonyl seems to have remained unnoticed; in 1971 Sayo et al. used a longer four-step synthesis to achieve a library of α-nitroisobutyranilides,13 which could have been done in two steps if they had used the pathway shown in Fig. 3.
The Kornblum substitution was subsequently performed on a tertiary alpha carbon twice more by other workers, neither of whom commented on the novelty of the substitution's occurring at a tertiary halo carbon. In 1957 Kissinger and Ungnade14 stated that they followed Kornblum's method from his 1956 paper2 to prepare ethyl α-nitroisobutyrate (4) from ethyl α-bromoisobutyrate (3); in 1977 Gelbard and Colonna15 carried out the Kornblum substitution on tertiary halo ethyl esters in order to characterize the effectiveness of a new type of nitrite resin. These three reports have gone generally unnoticed by the organic synthesis community; later publications in the 1990s and 2000s still regarded the Kornblum substitution as not proceeding on a tertiary centre.16–18 Kornblum spoke entirely in terms of SN2 and SN1 mechanisms, and the general belief is that a bromo–nitro substitution will not proceed at such sites due to steric hindrance on the tertiary carbon, which impedes an SN2 pathway.19 It does encourage SN1, but the nitrite ion is an ambident nucleophile,20 and SN1 substitution by nitrite is known usually to involve nucleophilic attack by the harder oxygen atom (acting under charge control21), giving an alkyl nitrite product (R–O–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).8 SN2, on the other hand, sees nucleophilic attack occur from the softer nitrogen atom (under orbital control) to furnish an alkyl nitro compound (R–NO2).8 The concept that an SN1 or an SN2 process will control the product of an attack by an ambident nucleophile has been called “Kornblum's rule”.20
O).8 SN2, on the other hand, sees nucleophilic attack occur from the softer nitrogen atom (under orbital control) to furnish an alkyl nitro compound (R–NO2).8 The concept that an SN1 or an SN2 process will control the product of an attack by an ambident nucleophile has been called “Kornblum's rule”.20
A 1997 paper by Glushkov and co-workers22 shows evidence that Kornblum's rule does not apply to tertiary halo carbons with an alpha carbonyl. The authors expected that Kornblum's rule would see thiocyanate ions (SCN−) attacking the carbocation from a tertiary halo compound in an SN1 manner to form an isothiocyanate (R–NCS), but instead from their substrates they observed thiocyanate products (Rʹ–SCN) (Fig. 5), which are the expected result of an SN2 substitution (attack by the more polarizable atom on the ambident nucleophile).
 | ||
| Fig. 5 Chloro–thiocyanate substitution as observed by Glushkov et al.22 | ||
Glushkov et al. postulated that the destabilizing effect of an alpha carbonyl prevented the formation of a carbocation and caused their substitution to occur by an SN2 process.22 Their language, like many others', suggests that they consider SN1 and SN2 to be the only two options.
It has been frequently noted that nucleophilic substitution reactions alpha to a carbonyl show atypical properties.23,24 They are, in particular, unusually fast,25–31 though how much so depends on the nucleophile and other circumstances.32–40 Various mechanistic reasons have been proposed for this. Some authors argue that addition takes place initially at the carbonyl, followed by either a 1,2-shift of the nucleophile to the alpha position,41–47 or, alternatively, formation of an epoxide that reacts with further nucleophile at the alpha position.40,48–55 Other authors reject this and contend that the reacting nucleophile makes an ordinary SN2-like attack at the carbon bearing the leaving group, but is assisted by interaction with carbonyl π* antibonding orbitals that temporarily accept electron density (often described as conjugation with the p orbitals or the π system, or as an enolate-like transition state),34,37,38,56–65 or alternatively by purely electrostatic effects.23,66–69 More recently a halfway position between these two extremes has been urged: that the attacking nucleophile bridges the carbonyl and the alpha carbon (and, by the principle of microscopic reversibility, the leaving group must also bridge both positions).70 This mechanism has been supported by recent computational studies,71–73 some of which suggest that there is a bifurcation in the potential energy surface after the transition state, a situation in which conventional transition state theory breaks down and molecular dynamics may become important.74–77 It has also been suggested that substitution is by SN1 reaction, and that this is accelerated by neighbouring group participation by the carbonyl, creating a 2H-oxirenium cation,78 or by prior enolisation on the other side, creating an allylic system;79 other suggestions include via a carbene produced from an enolate,67 and through nucleophilic attack at the halogen.32,41,67 Evidence for each mechanism, and against other mechanisms, has been found by different workers in different reactions, and many writers give evidence that different mechanisms dominate in different circumstances.23,32,55,73
In most papers Kornblum generally described the substitution as SN2, but in one paper he described it as more SN2 than SN1 in nature, but with properties of both.80 As the substitution's proceeding on a tertiary centre is at odds with Kornblum's stated SN2 mechanism, we suspected that a different mechanism was operative. We have therefore prepared a library of α-nitroisobutyranilides to show prima facie trends of the rate of Br–NO2 substitution, and to prepare Hammett plots from the rate data to indicate any charge in the transition state that would hint at the mechanism.
Results and discussion
In order to prepare a library of α-bromoisobutyranilides, anilines of varied substitution were selected with both greater and lesser electron withdrawing capacity than R = phenyl and also aniline itself (Table 1). The library of α-bromoisobutyranilides were then each exposed to a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of sodium nitrite to reactant compound using DMF as solvent at ambient temperature. The rate of conversion was monitored by taking hourly aliquots for GC-MS analysis. It was observed that the more electron-withdrawn the compound, the faster the bromo–nitro substitution took place.
:1 molar ratio of sodium nitrite to reactant compound using DMF as solvent at ambient temperature. The rate of conversion was monitored by taking hourly aliquots for GC-MS analysis. It was observed that the more electron-withdrawn the compound, the faster the bromo–nitro substitution took place.
| Bromo compound | Nitro compound | R-Substituents | |
|---|---|---|---|
 |
1 | 2 | p-Cyano-m-trifluoromethyl |
| 5 | 6 | H | |
| 7 | 8 | p-Methyl | |
| 9 | 10 | o-Carboethoxy | |
| 11 | 12 | o-Nitro | |
| 13 | 14 | m-Nitro | |
| 15 | 16 | p-Nitro | |
| 17 | 18 | o-Bromo | |
| 19 | 20 | o-Chloro | |
| 21 | 22 | m-Chloro | |
| 23 | 24 | p-Chloro | |
| 25 | 26 | o-Methoxy | |
| 27 | 28 | m-Methoxy | |
| 29 | 30 | p-Methoxy | |
| 31 | 32 | Benzyl in place of phenyl | |
| 33 | 34 | n-Butyl in place of phenyl | |
| 35 | 36 | 2,4-Dinitro |
As well as α-bromoisobutyranilides, which have an aryl group beyond the amide nitrogen, two compounds with an alkyl group in place of the aryl, n-butyl and benzyl, were also prepared, and found to undergo bromo–nitro substitution but at a much reduced rate compared with the aryl compounds. An additional α-bromoisobutyranilide with 2,4-dinitro substitution (35) was not characterized due to extreme difficulty of isolation but its rate of Br–NO2 substitution to give 36 could be easily monitored. It is therefore included in the graph to show the additional increase in rate when the compound's R group had the electron withdrawing capacity of two nitro groups and, as expected, it proceeds much faster than the mono-nitros and the CN/CF3 substituted compounds (1).
Some general trends in the bromo–nitro substitution were immediately apparent before any calculations were applied to the data. The first principle that overrides all others is that the reaction goes faster when the R group is more electron withdrawing, no matter how the R group is configured. Changes such as switching between ortho, meta and para substituted groups have a comparatively small effect on rate.
An overview of the rate data is shown in Fig. 6. The % substrate is plotted logarithmically: most compounds showed close to pseudo-first order behaviour. The sodium nitrite was present in ten-fold excess; it has limited solubility in DMF and excess solid was present. The solution was rapidly stirred, keeping the nitrite concentration constantly near saturation. Approximate straight lines of best fit are shown.
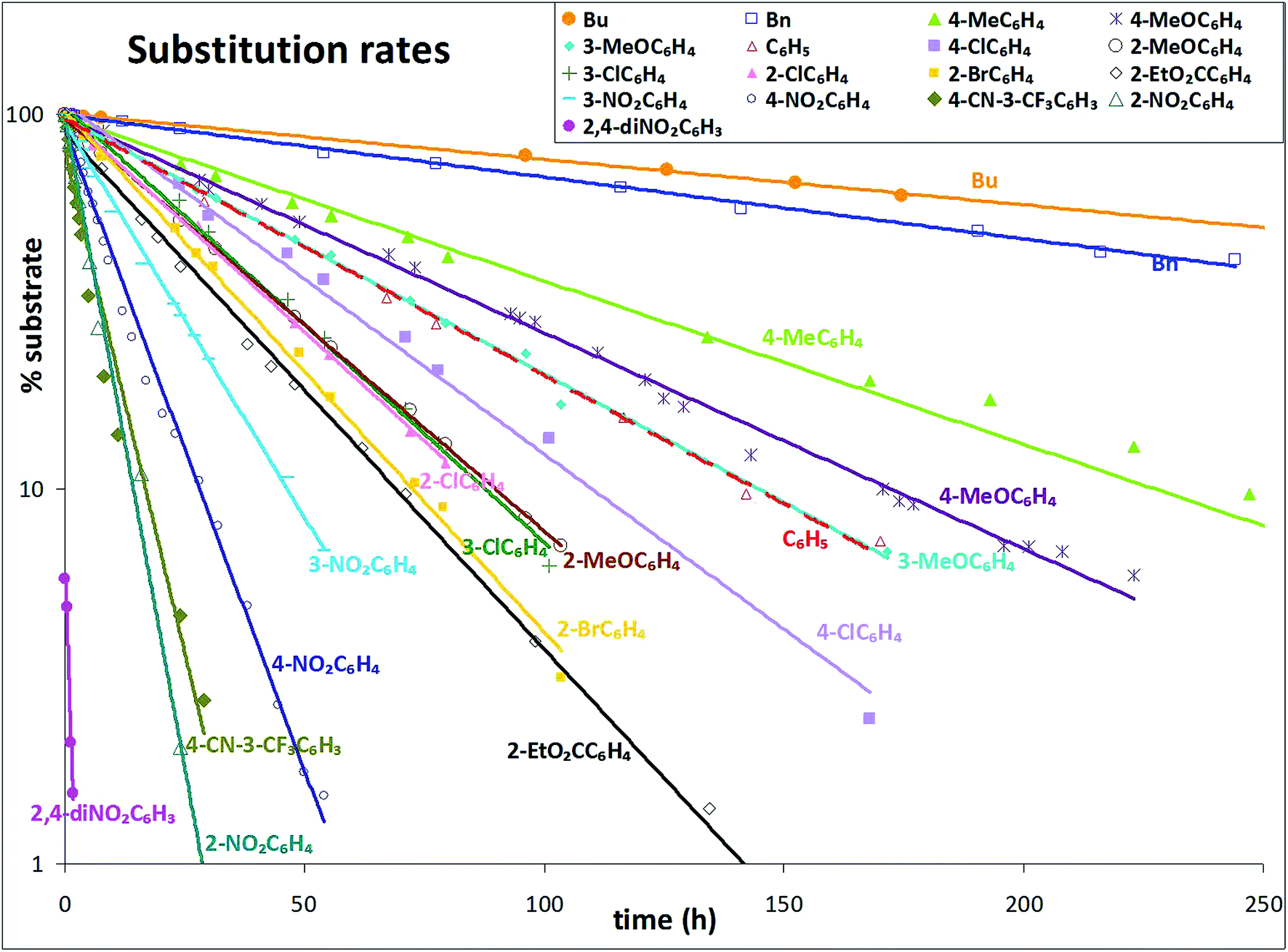 | ||
| Fig. 6 Substitution rate for tertiary α-bromoisobutyranilides. | ||
It was observed that reactions of ortho substituted α-bromoisobutyranilides proceeded faster than the equivalent meta or para isomers for the three substituents, methoxy, chloro and nitro, for which we had data. It may be that a substituent on the aryl ring closer to the site of halo–nitro substitution facilitates the substitution through some form of steric acceleration. This is shown in Fig. 7–9.
 | ||
| Fig. 7 Substitution rate for methoxy substituted α-bromoisobutyranilides. | ||
 | ||
| Fig. 8 Substitution rate for chloro substituted α-bromoisobutyranilides. | ||
 | ||
| Fig. 9 Substitution rate for nitro substituted α-bromoisobutyranilides. | ||
The rate varied much less between the three chloro substituted compounds than it did for the nitro and methoxy compounds. The difference could be steric, or perhaps due to nitro and methoxy's capability for hydrogen bonding; both can bend with free rotation and contain free electron pairs.
However, one exception to the first principle of increased rate with more electron-withdrawing R groups is that bromo in the ortho position gives significantly faster reaction than chloro in the ortho position. As bromo and chloro are quite similar except for size, it appears that in this case we are observing steric facilitation of the nearby substitution by the larger bromo group managing to outweigh the normally stronger effect of rate increasing with electronegativity.
This contrasts with reported observations that ortho substituted phenacyl bromides are less reactive in nucleophilic substitution by pyridine or t-butylamine.41,42,65
Br–NO2 substitution at low nitrite concentration
Our reaction conditions used saturated sodium nitrite in DMF. To investigate the dependence of the rate on nitrite concentration, we sought to increase the solubility of nitrite ions in DMF by using a co-solvent. Kornblum reported that the addition of 8% urea to the DMF dissolved far more NaNO2 which further increased the rate of substitution.11 When we tried the reaction this way we observed a slight reduction in rate, and saw no increase in solubility. We are puzzled by Kornblum's use of urea. He may have intended it to react with free nitrous acid81 (which he discussed as responsible for the degradation of the desired nitro products), but there may have been reaction with some nitrite.Therefore we instead used a low concentration experiment to compare the substitution rate of 1 → 2 in a saturated solution of NaNO2 in DMF with rates observed in solutions that contained only 75% and 50% the concentration of a saturated NaNO2 solution. The reaction rate was lowered under these conditions (Fig. 10).
 | ||
| Fig. 10 Effect of nitrite concentration on 1 → 2 reaction rate. | ||
This change in rate is evidence against an SN1 mechanism as the rate-limiting step of an SN1 reaction would be the formation of a cationic intermediate, independent of the presence of nitrite ions. This clearly cannot be an exclusive SN1 process and we can declare that the nitrite must be taking part in the rate-limiting step.
Hammett plots
First-order plots of ln(%substrate) against time were prepared for the reactions of the compound library. The slope of these is −k′, where k′ is the pseudo-first-order rate constant. As has been mentioned, a dilution experiment showed that the reaction rate depends also on nitrite concentration, so these reactions are only pseudo first order due to the nitrite concentration's remaining effectively constant throughout the reaction. The nitrite concentration was the same in each experiment: ∼5 mmol of the α-bromoisobutyranilide reactant (1–2 g) with NaNO2 (4.00 g, 44.9 mmol) in 40 mL of DMF.Data and plots for individual compounds are shown in the ESI,† with linear regression analysis. The linearity of these first order plots is reasonably good, except for the fastest reactions, especially 1 → 2. In many cases, however, there was some noticeable deviation from linearity or accuracy at the longer time scales and using the earlier portion of the data (never fewer than nine data points) gave more accurate linearity and improved the R2 value considerably (these are shown in the ESI†). As well as the usual decline of analytical accuracy at lower concentrations, a small amount of the formed nitro product may degrade by further reaction with nitrite ions via a nitroso intermediate to produce the alkyl nitrite by-product (a process described by Kornblum82).
The k′ values obtained from these graphs for the meta and para substituted anilides were used to construct Hammett plots. The meta examples, when plotted (as log10k′/k′H) against ordinary σmeta values83 (which are based on Ka values for benzoic acids), gave a reasonable fit (R2 = 0.97) and showed a positive ρ value of 0.70, indicating that the transition state develops a negative charge relative to the starting species in the rate-determining step.
The para substituted compounds are more complex to consider because ‘through conjugation’ is possible, where a canonical form can be drawn that puts the charge right at the para position and potentially on the substituent itself. A Hammett plot against ordinary σ values (from benzoic acid Ka values) gave a fit that was not terribly good (R2 = 0.92). A plot using σ+ values84 (based on benzylic SN1 solvolysis, with a positive charge next to the ring), which have strong through-conjugation effects with electron-donating substituents gave a much worse fit (R2 = 0.76). We then tried σ− values,§ originally based on phenol Ka values, so a negative charge next to the ring and strong through-conjugation effects with electron-withdrawing substituents: this gave the best fit of all (R2 = 0.99) and a ρ of +0.67: in the phenol acidity standard ρ is 2.01.
This result implies that not only does this reaction have a negative charge on the transition state, but that charge can readily conjugate onto the ring.
A combined Hammett plot of both meta and para substituted compounds, using σmeta− and σpara− values,¶ gave R2 = 0.992 and a ρ value of 0.68 (Fig. 11).
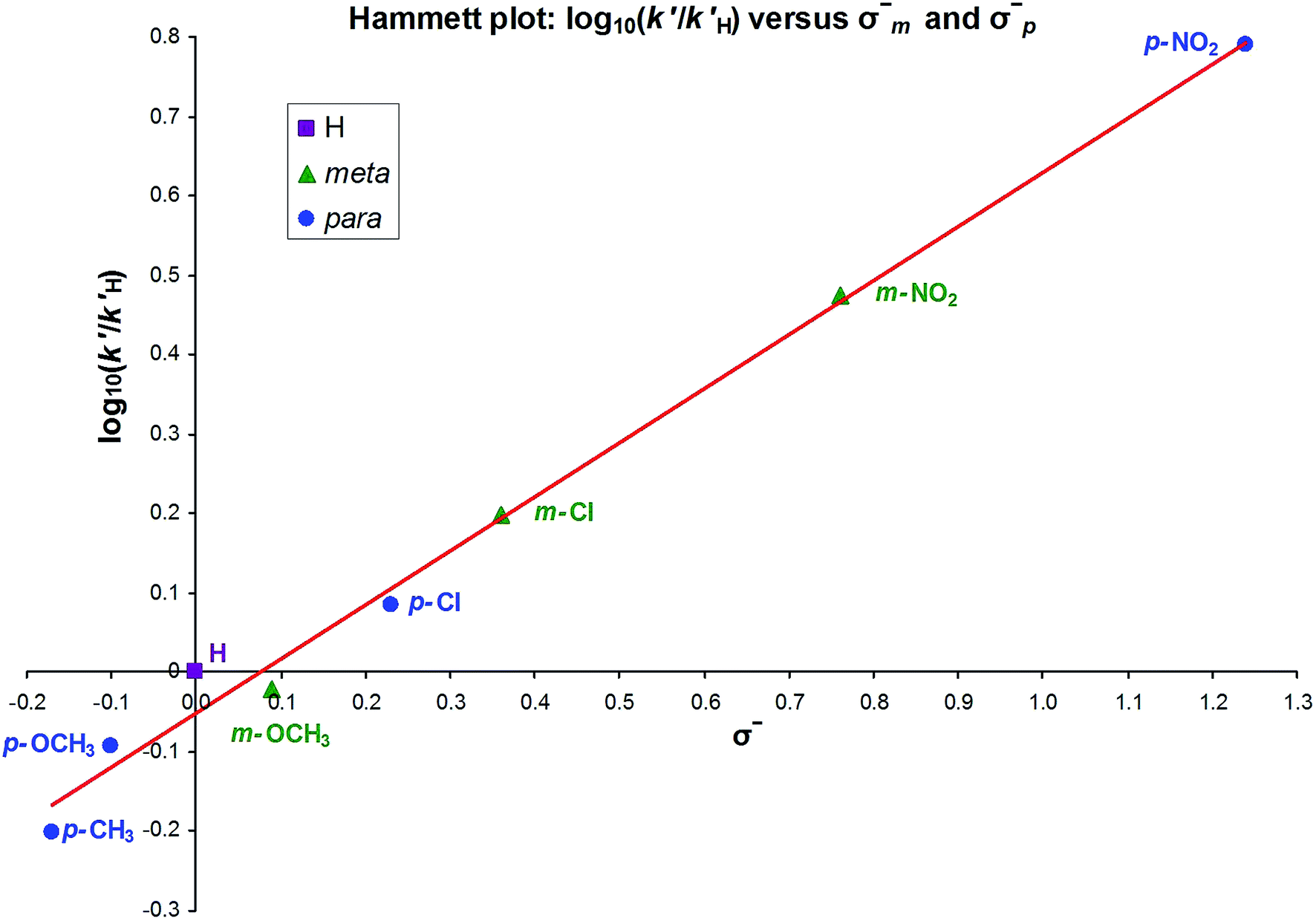 | ||
| Fig. 11 Hammett plot. | ||
The positive ρ implies a mechanism in which the nucleophile attacks first, before the leaving group leaves. One might think that the large ρ and the correlation with σ− implies the negative charge that forms must be on the nitrogen, but this isn't necessarily so. In anilides there is strong π character in the nitrogen–carbonyl bond and therefore the whole group has a π system that is planar with and conjugated with the aromatic ring's π system (Fig. 12).
 | ||
| Fig. 12 Planar and conjugated anilide π system. | ||
Therefore the negative charge that forms could be on the amide carbonyl, or on the position α to the carbonyl, as even there it will be conjugated with the ring (cf. hydrolysis of cinnamic esters, which has ρ = 1.27.).
If the mechanism started with deprotonation of the amide NH, where would it go next? One can only imagine forming an α-lactam, which would surely break open at the carbonyl. In any case, that mechanism wouldn't be available when the starting compound was an α-bromoester, and we know they also react.1,12 Hence it appears that it must start with at least partial addition at the carbonyl, or formation of an enolate. This provides several possibilities, each of which has several sub-possibilities:
(1) In the rate-determining step nitrite adds to the carbonyl as nitro, forming a negative oxygen. The carbonyl re-forms pushing the nitro to do a 1,2-shift onto the adjacent atom (like a semipinacol rearrangement), displacing the halogen (which may leave first to give either a carbocation or an epoxide). [cf. (ref. 41–47)] (Fig. 13).
 | ||
| Fig. 13 Possible mechanism number one. | ||
(2) In the rate-determining step nitrite adds to the carbonyl as nitrite, forming a negative oxygen. The carbonyl re-forms, pushing the nitrite nitrogen onto the adjacent atom in a four-centre reaction and displacing the halogen (which may leave first to give either a carbocation or an epoxide) (Fig. 14).
 | ||
| Fig. 14 Possible mechanism number two. | ||
(3) The negative oxygen formed in possibilities 1 or 2 could form an epoxide by displacing the adjacent bromide (which may leave first), then more nitrite could add at the other side of the epoxide. The carbonyl re-forms, pushing off the first nitrite. [cf. (ref. 40 and 48–55)] (Fig. 15).
 | ||
| Fig. 15 Possible mechanism number three. | ||
(4) The nitrite nucleophile bridges the carbonyl and the alpha carbon. Some electron density temporarily resides in the carbonyl π* antibonding orbital. The nucleophile finally displaces bromide in an SN2-like attack. [cf. (ref. 70–73)] (Fig. 16).
 | ||
| Fig. 16 Possible mechanism number four. | ||
(5) The rate-determining step is nucleophilic attack by nitrite (through nitrogen) at the bromine, with enolate as leaving group and forming nitryl bromide. The enolate formed could then react with the nitryl bromide to form the nitro product.
None of these options can be ruled out completely. The plausibility of mechanism 4 is supported by the molecular computations on reactions of (non-tertiary) α-halo carbonyls with nucleophiles,71–73 and by an observation of stereospecific substitution with second-order kinetics at a tertiary centre alpha to a carbonyl.94 The report from Edwards and Grieco for a long time remained an isolated witness, but is now joined by recent evidence of definite stereoinversion in tertiary α-chloroesters reacted with azide, thiols and fluoride.95,96 The bridging lowers the energy of the transition state, and, by delivering the nucleophile to the right position, may overcome the steric problems of tertiary SN2. The ρ value is similar to the values (ca. 1.05) obtained from reaction of phenacyl chlorides with carboxylate, which were interpreted as supporting a bridging mechanism,70 though the same paper references other substitutions of phenacyl halides in which the ρ value was much less.
However mechanism 5 is the only mechanism of these in which a full negative charge is directly part of the π system, supporting maximum through-resonance. Although nucleophilic attack on halogen as a means to substitution was considered by some early researchers32,41,67 it rarely appears in modern papers, though it is very similar to what happens in the specific reduction of α-halo carbonyls by soft nucleophiles.97 For example, it is commonly accepted that the α-halogenation of carbonyl compounds proceeds by the reaction of enol or enolate with molecular bromine, displacing bromide anion [cf. (ref. 98 and 99)]. This reaction is known to be reversible, so the principle of microscopic reversibility requires that the reduction of α-bromo carbonyl compounds by reaction with bromide occur by attack by bromide at the bromine, as has been pointed out by Altschul and Bartlett100 and Newman.101 Nitryl bromide is known to form and last for up to 30 min under some conditions.102
Br–NO2 substitution in the absence of O2
It has been suggested to us that as the reactions were carried out under air this substitution may be proceeding by means of a radical mechanism involving elemental oxygen (O2). A later experiment ruled out this proposal as the reaction proceeded at the same rate in the absence of oxygen.Conclusions
The Kornblum substitution has been robust from its inception and the few rules that govern it are widely known; mainly that it proceeds with primary or secondary but not tertiary carbons. We have re-addressed these rules for the examples shown here where the substitution is seen to proceed on tertiary halo-carbons that are α to a carbonyl (Fig. 17), albeit probably by a different mechanism to the standard Kornblum substitution (Fig. 18). | ||
| Fig. 17 Possible mechanism number five. | ||
 | ||
| Fig. 18 Kornblum substitution on a tertiary α-carbon. | ||
Many earlier researchers discussed substitution mechanisms in terms of a comparison between SN1 and SN2, a dichotomy that implied that these are the only two possibilities,103,104 and Kornblum himself wrote this way when discussing the reaction presented above.1,22,105 However, this type of Kornblum substitution does not behave like the usual SN1 or SN2 and it adds to the growing repertoire of substitutions that do not fit into the simple SN1/SN2 model that earlier researchers had leaned upon.106
Further, the reaction represents a good way to prepare alpha-nitro ketones, esters and amides which are versatile building blocks in organic synthesis as the nitro group may be reduced to an amino group. The preparation of our library of α-nitroisobutyranilides using the Kornblum substitution represents a far more convenient route to these compounds than what has been reported previously.13 Ono quoted the Kornblum substitution as high yielding for primary and secondary alkyl halides (50–70%), but low yielding (0–5%) for tertiary alkyl halides.4,5 We may now add to this that the Kornblum substitution is very high yielding (70–99%) for tertiary alkyl halides alpha to an anilide carbonyl.
Experimental section
General
IR spectra were measured from 4000–650 cm−1 using a Varian 1000 FTIR spectrometer with a diamond Attenuated Total Reflectance (ATR) attachment. Reaction rate was monitored using a Varian CP-3800 gas chromatograph equipped with an SGE Analytical Science BPX5 column (column width 0.25 mm, film width 0.25 μm) which was adjoined to a Varian Saturn 2200 GC/MS/MS. Accurate mass spectra were measured using a Waters GCT Premier HR-TOFMS equipped with an Agilent 7890 GC column. NMR spectra were obtained using a Bruker Avance 300 MHz spectrometer. Chemical shifts in 1H NMR spectra are relative to chloroform at 7.24 ppm; in 13C NMR spectra are relative to the central peak of deuterochloroform at 77.5 ppm.Rather than using IUPAC names, we have named the compounds as α-bromoisobutyranilides and α-nitroisobutyranilides in order to match the names given to them by Sayo et al.13 who have earlier reported the synthesis of some of the compounds in this category. Seven of the α-nitroisobutyranilides prepared by Sayo et al. have been prepared by our new method. Table 2 compares our measured melting points to those of Sayo et al.
| Compound | R |
M.p. (°C) | Sayo et al.13 | |
|---|---|---|---|---|
 |
6 | H | 104–107 | 104–105 |
| 8 | p-Me | 115–118 | 115–116 | |
| 14 | m-NO2 | 135–136 | 135–136 | |
| 16 | p-NO2 | 138–140 | 137.5–139 | |
| 22 | m-Cl | 125–128 | 134.5–136 | |
| 24 | p-Cl | 124–127 | 121–122.5 | |
| 30 | p-OMe | 69–71 | 73–74 |
Acylation reactions
The library of reactant compounds was prepared by reacting anilines (or in two cases, benzylamine and n-butylamine) with α-bromoisobutyryl bromide. All reactions were carried out at room temperature using ∼21.5 mmol (1.5–4 g) of the reactant amine, dissolved in 1,2-dichloroethane (35 mL). Oven-dried K2CO3 (3.00 g, 21.7 mmol) was added, then a 5% molar excess of α-bromoisobutyryl bromide was added last, dropwise. The flask was sealed and the reaction allowed to stir overnight at 700 rpm. The following morning the solvent was removed by rotary evaporation, and the residue was partitioned between ethyl acetate and water. The ethyl acetate fraction was dried (MgSO4), evaporated, and the residue, if solid, recrystallized from methanol.Characterization data for 1 are provided in our 2014 publication10 where it is given the correct IUPAC name of “2-bromo-N-[4-cyano-3-(trifluoromethyl)phenyl]-2-methylpropanamide”.
:1 hexanes/EtOAc, 0.74 in 65:35 hexanes/EtOAc and 0.95 in 1:1 hexanes/EtOAc; IR(cm−1): 3275, 3042, 2993, 2924, 1660 (CO), 1594, 1551, 1513, 1461, 1401, 1372, 1355, 1318, 1295, 1234, 1187, 1141, 962, 900, 862, 815, 767, 738, 677; 1H NMR (300 MHz, 26 mg: 0.4 mL CDCl3): δ 2.06 (6H, s, CH3), δ 7.15 (1H, tt, ArH4, J 2, J 8), δ 7.36 (2H, tt, ArH3, J 2, J 8), δ 7.54 (2H, dt, ArH2, J 2, J 8), δ 8.46 (1H, br, s, NH); 13C NMR (75 MHz, 137 mg: 0.4 mL CDCl3): δ 32.8 (s, C-3A/B), δ 63.2 (s, C-2), δ 120.3 (s, C-2′), δ 125.1 (s, C-4′), δ 129.3 (s, C-3′), δ 137.6 (s, C-1′), δ 170.2 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H12NOBr: 241.0102, observed: 241.0095.:1 hexanes/EtOAc, 0.78 in 65:35 hexanes/EtOAc and 0.93 in 1:1 hexanes/EtOAc; IR(cm−1): 3297, 3195, 3032, 3006, 2984, 2919, 1652 (CO), 1601, 1533, 1512, 1470, 1404, 1319, 1297, 1236, 1193, 1164, 1100, 1022, 1009, 946, 938, 893, 813, 767, 755, 696; 1H NMR (300 MHz, 35 mg: 0.4 mL CDCl3): δ 2.05 (6H, s, CH3), δ 2.33 (3H, s, ArCH3), δ 7.15 (2H, d, ArH3, J 8), δ 7.42 (2H, d, ArH2, J 8), δ 8.40 (1H, br, s, NH); 13C NMR (75 MHz, 135 mg: 0.4 mL CDCl3): δ 21.2 (s, ArCH3), δ 32.8 (s, C-3A/B), δ 63.4 (s, C-2), δ 120.3 (s, C-2′), δ 129.7 (s, C-3′), δ 134.8 (s, C-1′), δ 135.1 (s, C-4′), δ 170.1 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14NOBr: 255.0259, observed: 255.0254.:1 hexanes/EtOAc, 0.82 in 65:35 hexanes/EtOAc and 0.95 in 1:1 hexanes/EtOAc; IR(cm−1): 3189, 3117, 3076, 2974, 2937, 1696 (CO), 1680 (CO), 1605, 1592, 1467, 1449, 1365, 1298, 1271, 1239, 1199, 1170, 1144, 1105, 1086, 1050, 1016, 969, 947, 856, 763, 730, 700; 1H NMR (300 MHz, 30 mg: 0.4 mL CDCl3): δ 1.42 (3H, t, ethyl CH3), δ 2.07 (6H, s, CH3), δ 4.42 (2H, q, ethyl CH2), δ 7.12 (1H, td, ArH4, J 2, J 8), δ 7.56 (1H, td, ArH5, J 2, J 8), δ 8.08 (1H, dd, ArH6, J 2, J 8), δ 8.70 (1H, dd, ArH3, J 2, J 8), δ 11.90 (1H, br, s, NH); 13C NMR (75 MHz, 145 mg: 0.4 mL CDCl3): δ 14.4 (s, ethyl CH3), δ 32.1 (s, C-3A/B), δ 60.5 (s, C-2), δ 61.8 (s, ethyl CH2), δ 116.2 (s, C-2′), δ 120.5 (s, C-6′), δ 123.2 (s, C-4′), δ 131.2 (s, C-3′), δ 134.7 (s, C-5′), δ 141.4 (s, C-1′), δ 168.3 (s, ester CO), δ 171.0 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C13H15NO3Br: 312.0235, observed: 312.0222.:1 hexanes/EtOAc, 0.80 in 65:35 hexanes/EtOAc and 0.83 in 1:1 hexanes/EtOAc; IR(cm−1): 3320, 3118, 2985, 1701 (CO), 1606, 1584, 1544, 1496, 1458, 1427, 1391, 1374, 1335, 1268, 1221, 1145, 1112, 1077, 1044, 1009, 945, 891, 862, 787, 742, 681; 1H NMR (300 MHz, 32 mg: 0.4 mL CDCl3): δ 2.07 (6H, s, CH3), δ 7.23 (1H, td, ArH4, J 2, J 8), δ 7.68 (1H, td, ArH5, J 2, J 8), δ 8.25 (1H, dd, ArH3, J 2, J 8), δ 8.73 (1H, dd, ArH6, J 2, J 8), δ 11.34 (1H, br, s, NH); 13C NMR (75 MHz, 138 mg: 0.4 mL CDCl3): δ 32.2 (s, C-3A/B), δ 60.7 (s, C-2), δ 122.1 (s, C-3′), δ 124.0 (s, C-4′), δ 126.1 (s, C-6′), δ 134.7 (s, C-1′), δ 136.1 (s, C-5′), δ 137.0 (s, C-2′), δ 171.3 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N2O3Br: 285.9953, observed: 285.9949.:1 hexanes/EtOAc, 0.69 in 65:35 hexanes/EtOAc and 0.92 in 1:1 hexanes/EtOAc; IR(cm−1): 3370, 3090, 2980, 2931, 1694 (CO), 1590, 1525, 1484, 1418, 1392, 1374, 1349, 1315, 1298, 1243, 1152, 1108, 1079, 1007, 958, 893, 874, 813, 735, 673, 692, 673; 1H NMR (300 MHz, 45 mg: 0.4 mL CDCl3): δ 2.06 (6H, s, CH3), δ 7.51 (1H, t, ArH5, J 8), δ 7.90 (1H, dd, ArH6, J 2, J 8), δ 7.99 (1H, dd, ArH4, J 2, J 8), δ 8.46 (1H, t, ArH2, J 2), δ 8.66 (1H, br, s, NH); 13C NMR (75 MHz, 138 mg: 0.4 mL CDCl3): δ 32.4 (s, C-3A/B), δ 61.9 (s, C-2), δ 115.2 (s, C-2′), δ 119.6 (s, C-4′), δ 126.1 (s, C-6′), δ 130.0 (s, C-5′), δ 138.8 (s, C-1′), δ 148.7 (s, C-3′), δ 170.8 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N2O3Br: 285.9953, observed: 285.9963.:1 hexanes/EtOAc, 0.67 in 65:35 hexanes/EtOAc and 0.93 in 1:1 hexanes/EtOAc; IR(cm−1): 3406, 3115, 2929, 2931, 1698 (CO), 1612, 1596, 1534, 1496, 1404, 1334, 1300, 1243, 1194, 1177, 1142, 1101, 945, 882, 854, 831, 750, 691, 674; 1H NMR (300 MHz, 30 mg: 0.4 mL CDCl3): δ 2.05 (6H, s, CH3), δ 7.74 (2H, dt, ArH2, J 2, J 8), δ 8.23 (2H, dt, ArH3, J 2, J 8), δ 8.72 (1H, br, s, NH); 13C NMR (75 MHz, 122 mg: 0.4 mL CDCl3): δ 32.4 (s, C-3A/B), δ 62.1 (s, C-2), δ 119.7 (s, C-2′), δ 125.2 (s, C-3′), δ 143.5 (s, C-1′), δ 144.1 (s, C-4′), δ 170.7 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N2O3Br: 285.9953, observed: 285.9929.:1 hexanes/EtOAc, 0.85 in 65:35 hexanes/EtOAc and 0.96 in 1:1 hexanes/EtOAc; IR(cm−1): 3352, 2984, 2934, 1685 (CO), 1588, 1520, 1434, 1300, 1155, 1110, 1025, 939, 745, 683; 1H NMR (300 MHz, 144 mg: 0.4 mL CDCl3): δ 2.06 (6H, s, CH3), δ 7.01 (1H, td, ArH4, J 2, J 8), δ 7.33 (1H, td, ArH5, J 2, J 8), δ 7.56 (1H, dd, ArH3, J 2, J 8), δ 8.32 (1H, dd, ArH6, J 2, J 8), δ 9.04 (1H, br, s, NH); 13C NMR (75 MHz, 144 mg: 0.4 mL CDCl3): δ 32.7 (s, C-3A/B), δ 62.7 (s, C-2), δ 114.4 (s, C-2′), δ 121.8 (s, C-6′), δ 125.9 (s, C-5′), δ 128.6 (s, C-3′), δ 132.6 (s, C-4′), δ 135.8 (s, C-1′), δ 170.3 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11NOBr2: 318.9207, observed: 318.9194.:1 hexanes/EtOAc, 0.86 in 65:35 hexanes/EtOAc and 0.96 in 1:1 hexanes/EtOAc; IR(cm−1): 3365, 2985, 2934, 1686 (CO), 1593, 1514, 1439, 1304, 1154, 1111, 1054, 1034, 940, 746, 698; 1H NMR (300 MHz, 139 mg: 0.4 mL CDCl3): δ 2.04 (6H, s, CH3), δ 7.04 (1H, td, ArH4, J 2, J 8), δ 7.26 (1H, td, ArH5, J 2, J 8), δ 7.36 (1H, dd, ArH3, J 2, J 8), δ 8.30 (1H, dd, ArH6, J 2, J 8), δ 9.04 (1H, br, s, NH); 13C NMR (75 MHz, 139 mg: 0.4 mL CDCl3): δ 32.8 (s, C-3A/B), δ 62.9 (s, C-2), δ 121.5 (s, C-6′), δ 124.0 (s, C-2′), δ 125.4 (s, C-5′), δ 128.0 (s, C-3′), δ 129.4 (s, C-4′), δ 134.7 (s, C-1′), δ 170.4 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11NOClBr: 274.9713, observed: 274.9710.:1 hexanes/EtOAc, 0.78 in 65:35 hexanes/EtOAc and 0.94 in 1:1 hexanes/EtOAc; IR(cm−1): 3291, 2998, 2977, 2931, 1663 (CO), 1593, 1521, 1424, 1285, 1244, 1162, 1109, 919, 875, 860, 782, 758, 697, 682; 1H NMR (300 MHz, 31 mg: 0.4 mL CDCl3): δ 2.06 (6H, s, CH3), δ 7.14 (1H, dt, ArH4, J 2, J 8), δ 7.28 (1H, t, ArH5, J 8), δ 7.39 (1H, dq, ArH6, J 2, J 8), δ 7.70 (1H, t, ArH2, J 2), δ 8.47 (1H, br, s, NH); 13C NMR (75 MHz, 136 mg: 0.4 mL CDCl3): δ 32.8 (s, C-3A/B), δ 62.9 (s, C-2), δ 118.5 (s, C-6′), δ 120.5 (s, C-2′), δ 125.3 (s, C-4′), δ 130.3 (s, C-5′), δ 135.1 (s, C-3′), δ 139.0 (s, C-1′), δ 170.6 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11NOClBr: 274.9713, observed: 274.9686.:1 hexanes/EtOAc, 0.78 in 65:35 hexanes/EtOAc and 0.91 in 1:1 hexanes/EtOAc; IR(cm−1): 3285, 3188, 3122, 3071, 2988, 2941, 2895, 1656 (CO), 1591, 1552, 1529, 1478, 1459, 1418, 1398, 1372, 1351, 1305, 1287, 1254, 1240, 1187, 1142, 1092, 1074, 999, 962, 914, 904, 888, 864, 854, 792, 704, 683; 1H NMR (300 MHz, 43 mg: 0.4 mL CDCl3): δ 2.04 (6H, s, CH3), δ 7.30 (2H, dt, ArH3, J 2, J 8), δ 7.49 (2H, dt, ArH2, J 2, J 8), δ 8.45 (1H, br, s, NH); 13C NMR (75 MHz, 157 mg: 0.4 mL CDCl3): δ 32.6 (s, C-3A/B), δ 62.8 (s, C-2), δ 121.7 (s, C-2′), δ 129.3 (s, C-3′), δ 130.1 (s, C-4′), δ 136.2 (s, C-1′), δ 170.3 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11NOClBr: 274.9713, observed: 274.9706.:1 hexanes/EtOAc, 0.81 in 65:35 hexanes/EtOAc and 0.93 in 1:1 hexanes/EtOAc; IR(cm−1): 3380, 2983, 2936, 2839, 1677 (CO), 1600, 1522, 1486, 1459, 1433, 1336, 1290, 1250, 1218, 1176, 1157, 1110, 1047, 1026, 940, 773, 744; 1H NMR (300 MHz, 28 mg: 0.4 mL CDCl3): δ 2.06 (6H, s, CH3), δ 3.92 (3H, s, O–CH3), δ 6.90 (1H, dd, ArH3, J 2, J 8), δ 6.98 (1H, td, ArH5, J 2, J 8), δ 7.08 (1H, td, ArH4, J 2, J 8), δ 8.33 (1H, dd, ArH6, J 2, J 8), δ 9.13 (1H, br, s, NH); 13C NMR (75 MHz, 133 mg: 0.4 mL CDCl3): δ 32.5 (s, C-3A/B), δ 56.0 (s, O–CH3), δ 62.8 (s, C-2), δ 110.2 (s, C-3′), δ 119.5 (s, C-6′), δ 121.0 (s, C-5′), δ 124.4 (s, C-4′), δ 127.4 (s, C-1′), δ 148.5 (s, C-2′), δ 169.9 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14NO2Br: 271.0208, observed: 271.0195.:1 hexanes/EtOAc, 0.75 in 65:35 hexanes/EtOAc and 0.93 in 1:1 hexanes/EtOAc; IR(cm−1): 3454, 3340, 3003, 2961, 2942, 2897, 1660 (CO), 1597, 1546, 1528, 1510, 1462, 1442, 1414, 1375, 1355, 1299, 1232, 1183, 1172, 1141, 1111, 1032, 962, 902, 865, 823, 764; 1H NMR (300 MHz, 31 mg: 0.4 mL CDCl3): δ 2.10 (6H, s, CH3), δ 3.86 (3H, s, O–CH3), δ 6.75 (1H, dd, ArH4, J 2, J 8), δ 7.05 (1H, dd, ArH6, J 2, J 8), δ 7.30 (1H, dd, ArH5, J 2, J 8), δ 7.37 (1H, t, ArH2, J 2), δ 8.49 (1H, br, s, NH); 13C NMR (75 MHz, 31 mg: 0.4 mL CDCl3): δ 32.9 (s, C-3A/B), δ 55.7 (s, O–CH3), δ 63.5 (s, C-2), δ 105.8 (s, C-6′), δ 111.3 (s, C-2′), δ 112.4 (s, C-4′), δ 130.1 (s, C-5′), δ 139.0 (s, C-1′), δ 160.6 (s, C-3′), δ 170.3 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14NO2Br: 271.0208, observed: 271.0216.:1 hexanes/EtOAc, 0.72 in 65:35 hexanes/EtOAc and 0.91 in 1:1 hexanes/EtOAc; IR(cm−1): 3319, 3007, 2982, 2962, 2841, 1654 (CO), 1601, 1539, 1508, 1468, 1444, 1412, 1372, 1316, 1300, 1273, 1232, 1223, 1197, 1184, 1164, 1106, 1031, 952, 933, 890, 831, 809, 763, 751, 675; 1H NMR (300 MHz, 25 mg: 0.4 mL CDCl3): δ 2.07 (6H, s, CH3), δ 3.82 (3H, s, O–CH3), δ 6.90 (2H, dt, ArH3, J 2, J 8), δ 7.45 (2H, dt, ArH2, J 2, J 8), δ 8.40 (1H, br, s, NH); 13C NMR (75 MHz, 25 mg: 0.4 mL CDCl3): δ 32.4 (s, C-3A/B), δ 55.5 (s, O–CH3), δ 63.3 (s, C-2), δ 114.6 (s, C-3′), δ 121.8 (s, C-2′), δ 130.5 (s, C-1′), δ 156.8 (s, C-4′), δ 169.9 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14NO2Br: 271.0208, observed: 271.0197.:1 hexanes/EtOAc, 0.64 in 65:35 hexanes/EtOAc and 0.90 in 1:1 hexanes/EtOAc; IR(cm−1): 3300, 3065, 3030, 2973, 2939, 2920, 1642 (CO), 1533, 1495, 1471, 1453, 1418, 1355, 1292, 1195, 1102, 1081, 1014, 922, 826, 752, 729, 699, 693; 1H NMR (300 MHz, 23 mg: 0.4 mL CDCl3): δ 1.99 (6H, s, CH3), δ 4.47 (2H, d, CH2 J 8), δ 7.02 (1H, br, s, NH), δ 7.30 (2H, m, ArH2), δ 7.34 (2H, m, ArH3), δ 7.36 (1H, m, ArH4); 13C NMR (75 MHz, 125 mg: 0.4 mL CDCl3): δ 32.7 (s, C-3A/B), δ 44.5 (s, CH2), δ 62.9 (s, C-2), δ 127.7 (s, C-2′), δ 127.8 (s, C-4′), δ 129.0 (s, C-3′), δ 138.0 (s, C-1′), δ 172.2 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14NOBr: 255.0259, observed: 255.0265.O), 1528, 1465, 1437, 1370, 1301, 1282, 1225, 1190, 1112, 738; 1H NMR (300 MHz, 26 mg: 0.4 mL CDCl3): δ 0.96 (3H, t, alkyl4 J 8), δ 1.38 (2H, sextet, alkyl3 J 8), δ 1.54 (2H, sextet, alkyl2 J 8), δ 1.97 (6H, s, CH3), δ 3.28 (2H, sextet, alkyl1 J 8), δ 6.73 (1H, br, s, NH); 13C NMR (75 MHz, 147 mg: 0.4 mL CDCl3): δ 13.9 (s, C-4′), δ 20.2 (s, C-3′), δ 31.5 (s, C-2′), δ 32.8 (s, C-3A/B), δ 40.3 (s, C-1′), δ 63.3 (s, C-2), δ 172.0 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C8H16NOBr: 221.0415, observed: 221.0417.Substitution reactions
Unless otherwise stated, reactions were carried out at room temperature using ∼5 mmol (1–2 g) of the reactant bromo compound (1–2 g) with NaNO2 (4.00 g, 44.9 mmol) in DMF (40 mL) and a magnetic stirrer at 700 rpm.The rates were monitored by periodically removing 1 mL of the reacting mixture, placing it in dichloromethane (2 mL) and washing with water (4 × 3 mL) in a 5 mL screw cap vial. The dichloromethane layer was then dried (MgSO4) and analysed by GC-MS.
The time between each aliquot was determined for each reaction by trial in an initial rough experiment.
For preparative reactions, apart from the preparation of 2, which could be obtained by addition of water to the DMF reaction mixture, the substitutions were worked up on completion by the removal of DMF on a rotary evaporator with water bath at 70 °C and vacuum rigorously kept at 25 Torr, with Dow Corning high vacuum grease freshly applied to the joins and Keck clips used to hold the flask onto a non-reversible splash-guard. The residue was partitioned between water and ethyl acetate and the ethyl acetate fraction was evaporated. After TLC of the residue to determine a suitable eluent, the product was purified by column chromatography (40 mm diameter) on 43–60 μ silica (∼150 g) with pre-adsorption on ∼10 g of silica. This method typically produced 800–1300 mg of highly pure nitro substitution product as observed by NMR.
:1 ratio with DMF (86% yield when corrected for the DMF), m.p. 129–131 °C. A DMF free version of 2 could be prepared by repeated liquid/liquid extraction using water/ethyl acetate which provides a white amorphous powder of the same m.p. Characterization data for 2 are provided in our 2014 publication10 where it is given the correct IUPAC name of “N-[4-cyano-3-(trifluoromethyl)phenyl]-2-methyl-2-nitropropanamide”.:1 hexanes/EtOAc, 0.56 in 65:35 hexanes/EtOAc and 0.87 in 1:1 hexanes/EtOAc; IR(cm−1): 3256, 3199, 3136, 3076, 1655 (CO), 1598, 1549, 1538, 1492, 1459, 1440, 1399, 1373, 1352, 1321, 1266, 1233, 1189, 1143, 963, 894, 859, 752, 695, 666; 1H NMR (300 MHz, 26 mg: 0.4 mL CDCl3): δ 1.94 (6H, s, CH3), δ 7.17 (1H, tt, ArH4, J 2, J 8), δ 7.34 (2H, tt, ArH3, J 2, J 8), δ 7.48 (2H, dt, ArH2, J 2, J 8), δ 7.98 (1H, br, s, NH); 13C NMR (75 MHz, 26 mg: 0.4 mL CDCl3): δ 24.9 (s, C-3A/B), δ 91.6 (s, C-2), δ 120.8 (s, C-2′), δ 125.8 (s, C-4′), δ 129.5 (s, C-3′), δ 136.8 (s, C-1′), δ 164.7 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H12N2O3: 208.0848, observed: 208.0846.:1 hexanes/EtOAc, 0.61 in 65:35 hexanes/EtOAc and 0.87 in 1:1 hexanes/EtOAc; IR(cm−1): 3274, 3120, 3042, 2993, 2924, 2893, 2860, 1660 (CO), 1594, 1550, 1522, 1513, 1460, 1436, 1401, 1372, 1355, 1318, 1295, 1259, 1234, 1187, 1179, 1141, 961, 900, 862, 815, 770, 738, 678; 1H NMR (300 MHz, 21 mg: 0.4 mL CDCl3): δ 1.93 (6H, s, CH3), δ 2.32 (3H, s, ArCH3), δ 7.13 (2H, d, ArH3, J 8), δ 7.35 (2H, d, ArH2, J 8), δ 7.91 (1H, br, s, NH); 13C NMR (75 MHz, 42 mg: 0.4 mL CDCl3): δ 21.3 (s, ArCH3), δ 24.9 (s, C-3A/B), δ 91.6 (s, C-2), δ 121.1 (s, C-2′), δ 129.9 (s, C-3′), δ 134.3 (s, C-1′), δ 135.5 (s, C-4′), δ 164.9 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14N2O3: 222.1004, observed: 222.1006.:1 hexanes/EtOAc, 0.69 in 65:35 hexanes/EtOAc and 0.93 in 1:1 hexanes/EtOAc; IR(cm−1): 3177, 3120, 3082, 2991, 1699 (CO), 1685 (CO), 1608, 1594, 1551, 1529, 1466, 1455, 1366, 1351, 1303, 1277, 1251, 1238, 1182, 1139, 1090, 1016, 857, 763, 700; 1H NMR (300 MHz, 25 mg: 0.4 mL CDCl3): δ 1.42 (3H, t, ethyl CH3), δ 1.98 (6H, s, CH3), δ 4.41 (2H, q, ethyl CH2), δ 7.16 (1H, td, ArH4, J 2, J 8), δ 7.57 (1H, td, ArH5, J 2, J 8), δ 8.08 (1H, dd, ArH6, J 2, J 8), δ 8.66 (1H, dd, ArH3, J 2, J 8), δ 11.81 (1H, br, s, NH); 13C NMR (75 MHz, 68 mg: 0.4 mL CDCl3/0.1 mL d6-DMSO): δ 14.9 (s, ethyl CH3), δ 24.3 (s, C-3A/B), δ 62.5 (s, ethyl CH2), δ 92.3 (s, C-2), δ 118.9 (s, C-2′), δ 122.0 (s, C-6′), δ 125.2 (s, C-4′), δ 131.6 (s, C-3′), δ 135.1 (s, C-5′), δ 139.8 (s, C-1′), δ 166.5 (s, C-1), δ 168.2 (s, ester CO); GC-(EI) TOF-HRMS: calcd m/z for C13H15N2O5: 279.0981, observed: 279.0972.:1 hexanes/EtOAc, 0.70 in 65:35 hexanes/EtOAc and 0.90 in 1:1 hexanes/EtOAc; IR(cm−1): 3392, 2924, 2854, 1706 (CO), 1607, 1588, 1548, 1497, 1454, 1431, 1396, 1374, 1335, 1270, 1224, 1161, 1140, 1075, 898, 861, 854, 789, 742, 688; 1H NMR (300 MHz, 18 mg: 0.4 mL CDCl3): δ 2.00 (6H, s, CH3), δ 7.28 (1H, td, ArH4, J 2, J 8), δ 7.70 (1H, tt, ArH5, J 2, J 8), δ 8.27 (1H, dd, ArH3, J 2, J 8), δ 8.71 (1H, dd, ArH6, J 2, J 8), δ 11.09 (1H, br, s, NH); 13C NMR (75 MHz, 18 mg: 0.4 mL CDCl3): δ 24.6 (s, C-3A/B), δ 91.6 (s, C-2), δ 122.7 (s, C-3′), δ 124.9 (s, C-4′), δ 126.3 (s, C-6′), δ 134.1 (s, C-1′), δ 136.5 (s, C-5′), δ 137.2 (s, C-2′), δ 165.9 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N3O5: 253.0699, observed: 253.0705.:1 hexanes/EtOAc, 0.57 in 65:35 hexanes/EtOAc and 0.84 in 1:1 hexanes/EtOAc; IR(cm−1): 3347, 3093, 2923, 2854, 1659 (CO), 1617, 1553, 1532, 1458, 1434, 1401, 1373, 1350, 1317, 1287, 1262, 1234, 1192, 1145, 1089, 1079, 970, 909, 882, 856, 824, 809, 734, 693, 671; 1H NMR (300 MHz, 50 mg: 0.4 mL d6-DMSO): δ 1.93 (6H, s, CH3), δ 7.67 (1H, t, ArH5, J 8), δ 8.01 (1H, dd, ArH6, J 2, J 8), δ 8.07 (1H, dd, ArH4, J 2, J 8), δ 8.61 (1H, t, ArH6, J 2), δ 10.40 (1H, br, s, NH); 13C NMR (75 MHz, 50 mg: 0.4 mL d6-DMSO): δ 24.6 (s, C-3A/B), δ 92.5 (s, C-2), δ 115.7 (s, C-2′), δ 119.8 (s, C-4′), δ 127.4 (s, C-6′), δ 131.3 (s, C-5′), δ 140.2 (s, C-1′), δ 148.8 (s, C-3′), δ 167.5 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N3O5: 253.0699, observed: 253.0694.:1 hexanes/EtOAc, 0.45 in 65:35 hexanes/EtOAc and 0.87 in 1:1 hexanes/EtOAc; IR(cm−1): 3352, 1709 (CO), 1615, 1597, 1547, 1506, 1464, 1409, 1400, 1374, 1346, 1307, 1249, 1221, 1181, 1160, 1142, 1115, 898, 848, 829, 816, 752, 691; 1H NMR (300 MHz, 27 mg: 0.4 mL d6-DMSO): δ 1.94 (6H, s, CH3), δ 7.94 (2H, dt, ArH2, J 2, J 8), δ 8.29 (2H, dt, ArH3, J 2, J 8), δ 10.50 (1H, br, s, NH); 13C NMR (75 MHz, 27 mg: 0.4 mL d6-DMSO): δ 24.5 (s, C-3A/B), δ 92.5 (s, C-2), δ 121.2 (s, C-2′), δ 125.7 (s, C-3′), δ 144.0 (s, C-4′), δ 145.1 (s, C-1′), δ 167.4 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N3O5: 253.0699, observed: 253.0702.:1 hexanes/EtOAc, 0.69 in 65:35 hexanes/EtOAc and 0.91 in 1:1 hexanes/EtOAc; IR(cm−1): 3398, 3339, 2997, 2925, 1699 (CO), 1590, 1548, 1519, 1464, 1436, 1398, 1373, 1346, 1299, 1237, 1207, 1167, 1143, 1121, 1047, 1026, 896, 855, 750; 1H NMR (300 MHz, 26 mg: 0.4 mL CDCl3): δ 1.98 (6H, s, CH3), δ 7.04 (1H, td, ArH4, J 2, J 8), δ 7.34 (1H, td, ArH5, J 2, J 8), δ 7.56 (1H, dd, ArH3, J 2, J 8), δ 8.24 (1H, dd, ArH6, J 2, J 8), δ 8.52 (1H, br, s, NH); 13C NMR (75 MHz, 66 mg: 0.4 mL CDCl3): δ 24.9 (s, C-3A/B), δ 91.4 (s, C-2), δ 114.8 (s, C-2′), δ 122.6 (s, C-6′), δ 126.8 (s, C-5′), δ 128.9 (s, C-3′), δ 132.8 (s, C-4′), δ 134.9 (s, C-1′), δ 164.9 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N2O3Br: 285.9953, observed: 285.9961.:1 hexanes/EtOAc, 0.70 in 65:35 hexanes/EtOAc and 0.90 in 1:1 hexanes/EtOAc; IR(cm−1): 3352, 2998, 2922, 2852, 1697 (CO), 1594, 1549, 1518, 1467, 1441, 1398, 1373, 1347, 1302, 1238, 1168, 1144, 1128, 1055, 1035, 897, 856, 751, 690; 1H NMR (300 MHz, 21 mg: 0.4 mL CDCl3): δ 1.97 (6H, s, CH3), δ 7.11 (1H, td, ArH4, J 2, J 8), δ 7.30 (1H, td, ArH5, J 2, J 8), δ 7.40 (1H, dd, ArH3, J 2, J 8), δ 8.26 (1H, dd, ArH6, J 2, J 8), δ 8.58 (1H, br, s, NH); 13C NMR (75 MHz, 39 mg: 0.4 mL CDCl3): δ 24.9 (s, C-3A/B), δ 91.5 (s, C-2), δ 122.2 (s, C-6′), δ 124.2 (s, C-2′), δ 126.2 (s, C-5′), δ 128.2 (s, C-3′), δ 129.5 (s, C-4′), δ 133.8 (s, C-1′), δ 164.7 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N2O3Cl: 242.0458, observed: 242.0462.:1 hexanes/EtOAc, 0.72 in 65:35 hexanes/EtOAc and 0.89 in 1:1 hexanes/EtOAc; IR(cm−1): 3385, 3188, 3122, 3071, 2988, 2941, 2895, 1656 (CO), 1590, 1552, 1528, 1478, 1459, 1418, 1398, 1372, 1351, 1304, 1287, 1254, 1240, 1231, 1187, 1142, 1092, 1074, 999, 914, 904, 888, 854, 791, 704, 682; 1H NMR (300 MHz, 18 mg: 0.4 mL CDCl3): δ 1.82 (6H, s, CH3), δ 6.99 (1H, d, ArH4, J 8), δ 7.14 (1H, td, ArH5, J 2, J 8), δ 7.43 (1H, d, ArH6, J 8), δ 7.64 (1H, m, ArH2), δ 9.38 (1H, br, s, NH); 13C NMR (75 MHz, 18 mg: 0.4 mL CDCl3/2 drops d6-DMSO): δ 24.7 (s, C-3A/B), δ 91.2 (s, C-2), δ 118.9 (s, C-6′), δ 121.0 (s, C-2′), δ 124.7 (s, C-4′), δ 129.8 (s, C-5′), δ 134.2 (s, C-3′), δ 139.3 (s, C-1′), δ 165.8 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N2O3Cl: 242.0458, observed: 242.0475.:1 hexanes/EtOAc, 0.67 in 65:35 hexanes/EtOAc and 0.90 in 1:1 hexanes/EtOAc; IR(cm−1): 3303, 3195, 3126, 3057, 3002, 2924, 2854, 1664 (CO), 1599, 1547, 1533, 1492, 1460, 1400, 1379, 1353, 1308, 1287, 1241, 1188, 1145, 1087, 1014, 960, 904, 864, 820, 747, 708, 695, 667; 1H NMR (300 MHz, 21 mg: 0.4 mL CDCl3): δ 1.94 (6H, s, CH3), δ 7.30 (2H, d, ArH3, J 8), δ 7.44 (2H, d, ArH2, J 8), δ 8.01 (1H, br, s, NH); 13C NMR (75 MHz, 21 mg: 0.4 mL CDCl3): δ 25.0 (s, C-3A/B), δ 91.7 (s, C-2), δ 122.3 (s, C-2′), δ 129.5 (s, C-3′), δ 131.0 (s, C-4′), δ 135.5 (s, C-1′), δ 164.8 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C10H11N2O3Cl: 242.0458, observed: 242.0477.:1 hexanes/EtOAc, 0.65 in 65:35 hexanes/EtOAc and 0.89 in 1:1 hexanes/EtOAc; IR(cm−1): 3331, 3043, 3005, 2964, 2936, 2901, 2838, 1675 (CO), 1594, 1553, 1521, 1493, 1460, 1432, 1403, 1375, 1357, 1322, 1287, 1262, 1220, 1177, 1142, 1112, 1042, 1025, 963, 899, 862, 849, 780, 748, 739, 724, 666; 1H NMR (300 MHz, 19 mg: 0.4 mL CDCl3): δ 1.95 (6H, s, CH3), δ 3.91 (3H, s, O–CH3), δ 6.90 (1H, dd, ArH3, J 2, J 8), δ 6.97 (1H, td, ArH5, J 2, J 8), δ 7.10 (1H, td, ArH4, J 2, J 8), δ 8.28 (1H, dd, ArH6, J 2, J 8), δ 8.62 (1H, br, s, NH); 13C NMR (75 MHz, 57 mg: 0.4 mL CDCl3): δ 24.9 (s, C-3A/B), δ 56.2 (s, O–CH3), δ 91.6 (s, C-2), δ 110.4 (s, C-3′), δ 120.3 (s, C-6′), δ 121.4 (s, C-5′), δ 125.3 (s, C-4′), δ 126.9 (s, C-1′), δ 148.7 (s, C-2′), δ 164.4 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14N2O4: 238.0954, observed: 238.0952.:1 hexanes/EtOAc, 0.55 in 65:35 hexanes/EtOAc and 0.85 in 1:1 hexanes/EtOAc; IR(cm−1): 3276, 3223, 3154, 3007, 2943, 2838, 1665 (CO), 1614, 1597, 1539, 1489, 1451, 1427, 1397, 1373, 1344, 1320, 1301, 1277, 1267, 1208, 1182, 1149, 1031, 953, 844, 788, 764, 749, 727, 686; 1H NMR (300 MHz, 28 mg: 0.4 mL CDCl3): δ 1.93 (6H, s, CH3), δ 3.80 (3H, s, O–CH3), δ 6.72 (1H, dd, ArH4, J 2, J 8), δ 6.96 (1H, dd, ArH6, J 2, J 8), δ 7.22 (1H, t, ArH5, J 8), δ 7.26 (1H, d, ArH2, J 2), δ 7.98 (1H, br, s, NH); 13C NMR (75 MHz, 120 mg: 0.4 mL CDCl3): δ 24.6 (s, C-3A/B), δ 55.5 (s, O–CH3), δ 91.5 (s, C-2), δ 106.8 (s, C-6′), δ 111.6 (s, C-2′), δ 113.2 (s, C-4′), δ 130.0 (s, C-5′), δ 138.0 (s, C-1′), δ 160.4 (s, C-3′), δ 165.2 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14N2O4: 238.0954, observed: 238.0954.:1 hexanes/EtOAc, 0.48 in 65:35 hexanes/EtOAc and 0.80 in 1:1 hexanes/EtOAc; IR(cm−1): 3346, 3003, 2961, 2939, 2898, 2840, 1660 (CO), 1597, 1545, 1530, 1510, 1462, 1440, 1414, 1403, 1375, 1356, 1311, 1299, 1268, 1232, 1184, 1173, 1141, 1112, 1031, 962, 901, 863, 850, 824, 764; 1H NMR (300 MHz, 28 mg: 0.4 mL CDCl3): δ 1.92 (6H, s, CH3), δ 3.79 (3H, s, O–CH3), δ 6.86 (2H, d, ArH3, J 8), δ 7.37 (2H, d, ArH2, J 8), δ 7.87 (1H, br, s, NH); 13C NMR (75 MHz, 120 mg: 0.4 mL CDCl3): δ 24.9 (s, C-3A/B), δ 55.8 (s, O–CH3), δ 91.5 (s, C-2), δ 114.5 (s, C-3′), δ 122.9 (s, C-2′), δ 129.8 (s, C-1′), δ 157.5 (s, C-4′), δ 164.9 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14N2O4: 238.0954, observed: 238.0971.:1 hexanes/EtOAc, 0.49 in 65:35 hexanes/EtOAc and 0.76 in 1:1 hexanes/EtOAc; IR(cm−1): 3295, 3088, 3028, 3003, 2930, 1652 (CO), 1547, 1496, 1453, 1427, 1405, 1374, 1356, 1312, 1288, 1236, 1209, 1162, 1077, 1055, 1029, 1000, 865, 747, 732, 698, 671; 1H NMR (300 MHz, 19 mg: 0.4 mL CDCl3): δ 1.85 (6H, s, CH3), δ 4.45 (2H, d, CH2 J 8), δ 6.46 (1H, br, s, NH), δ 7.22–7.37 (5H, m, ArH2−6); 13C NMR (75 MHz, 55 mg: 0.4 mL CDCl3): δ 24.8 (s, C-3A/B), δ 44.4 (s, CH2), δ 91.0 (s, C-2), δ 127.8 (s, C-2′), δ 128.1 (s, C-4′), δ 129.1 (s, C-3′), δ 137.5 (s, C-1′), δ 167.2 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C11H14N2O3: 222.1004, observed: 222.1001.O), 1620, 1542, 1465, 1440, 1403, 1374, 1355, 1301, 1287, 1205, 1157, 867; 1H NMR (300 MHz, 21 mg: 0.4 mL CDCl3): δ 0.92 (3H, t, alkyl4 J 8), δ 1.32 (2H, sextet, alkyl3 J 8), δ 1.49 (2H, sextet, alkyl2 J 8), δ 1.83 (6H, s, CH3), δ 3.27 (2H, sextet, alkyl1 J 8), δ 6.15 (1H, br, s, NH); 13C NMR (75 MHz, 21 mg: 0.4 mL CDCl3): δ 14.0 (s, C-4′), δ 20.2 (s, C-3′), δ 24.9 (s, C-3A/B), δ 31.5 (s, C-2′), δ 40.3 (s, C-1′), δ 91.1 (s, C-2), δ 167.0 (s, C-1); GC-(EI) TOF-HRMS: calcd m/z for C8H16N2O3: 188.1161, observed: 188.1173.:1 or greater ratio was desired, 84 mg (0.025 mmol) of 1 was used in all three reactions. The reactions were monitored by GC-MS using the same extraction method and instrument as had been used to monitor the other substitution reactions. Aliquots were taken at ∼1 h intervals to obtain five data points for each reaction; all fifteen GC-MS sample vials were run on the same GC-MS on the same day.Acknowledgements
We wish to acknowledge and thank RMIT for the unwavering support of the synthetic organic laboratory. We wish to thank Frank Antolasic for assistance with mass spectrometry experiments. We wish to thank Steve Diver at the University of Buffalo for suggesting the oxygen-free experiment.References
- N. Kornblum, M. E. Chalmers and R. Daniels, The reaction of silver nitrite with α haloesters, J. Am. Chem. Soc., 1955, 77(24), 6654 CrossRef CAS.
- N. Kornblum, H. O. Larson, D. D. Mooberry, R. K. Blackwood, E. P. Oliveto and G. E. Graham. A new method for the synthesis of aliphatic nitro compounds. Chemistry and Industry, 1956, p. 443 Search PubMed.
- N. Kornblum, R. K. Blackwood and J. W. Powers, A new synthesis of α nitroesters, J. Am. Chem. Soc., 1957, 79(10), 2507 CrossRef CAS.
- N. Ono, The nitro group in organic synthesis, Wiley publishing, 1991 Search PubMed.
- N. Ono, The nitro group in organic synthesis, Wiley publishing, ISBNs: 0-471-31611-3 (Hardback); 0-471-22448-0 (Electronic), 2001 Search PubMed.
- V. A. Mamedov, A. A. Kalinin, A. T. Gubaidullin, I. A. Litvinov and Ya. A. Levin, The Kornblum reaction of α-sunstituted 3-benzyl-1,2-dihydro-2-oxoquinoxalines. Synthesis and structure of 3-benzoyl-2-oxo-1,2-dihydroquinoxaline, Chem. Heterocycl. Compd., 2002, 38(12), 1504–1510 CrossRef CAS.
- S. T. Staben, X. Linghu and F. D. Toste, Enantioselective Synthesis of Hydroxyenones by Chiral Base-Catalyzed Kornblum DeLaMare Rearrangement, J. Am. Chem. Soc., 2006, 128(39), 12658–12659 CrossRef CAS PubMed.
- P. Sykes, A guidebook to mechanism in organic chemistry, Longman publishing, London, 5th edn, 1981 Search PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Published by John Wiley & Sons, 6th edn (Textbook), p. 571, 2007, and references therein Search PubMed.
- M. J. Leonard, A. R. Lingham, J. O. Niere, N. R. C. Jackson, P. G. McKay and H. M. Hügel, Alternative synthesis of the anti-baldness compound RU58841, RSC Adv., 2014, 27(4), 14143–14148 RSC.
- N. Kornblum, H. O. Larson, R. K. Blackwood, D. D. Mooberry, E. P. Oliveto and G. E. Graham, A new method for the synthesis of aliphatic nitro compounds, J. Am. Chem. Soc., 1956, 78(7), 1497–1501 CrossRef CAS.
- N. Kornblum and J. W. Powers, Production of 2 nitroesters, US pat., number 2,816,909, 1957.
- H. Sayo, H. Ohomori, T. Umeda and M. Masui, Bull. Chem. Soc. Jpn., 1971, 45, 203–208 CrossRef.
- L. W. Kissinger and H. E. Ungnade, Derivatives of nitromethylamines. I. Nitromethyl isocyanates, J. Org. Chem., 1957, 22, 1662–1665 CrossRef.
- G. Gelbard and S. Colonna, Anionic activation in polymer-supported reactions; Nucleophilic substitution with anion-exchange resins; I. Synthesis of alkyl phenyl ethers, nitrocarboxylic esters, and α-alkyl-β-dicarbonyl compounds, Synthesis, 1977, 2, 113–116 CrossRef.
- S. R. Sandler and W. Karo, Sourcebook of advanced organic laboratory preparations (Textbook), Published by Academic Press Inc., San Diego, ISBN 0-12-618506-9, 1992, p. 160 Search PubMed.
- B. Mehta and M. Mehta, Organic chemistry (Textbook), published by Prentice-Hall of India, ISBN 81-203-2441-2, 2005, p. 757 Search PubMed.
- A. Bahl and B. S. Bahl. Advanced organic chemistry (Textbook), Published by S. Chand & Company Ltd, ISBN 81-219-3515-6, 2008, p. 837 Search PubMed.
- S. Winstein, S. Smith and D. Dawish, Alleged SN2 Finkelstein substitutions of t-butyl bromide, Tetrahedron Lett., 1959, 1(16), 24–31 CrossRef.
- M. B. Smith andJ. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, published by John Wiley & Sons, 6th edn (Textbook), 2007, p. 516, Footnote about Kornblum’s rule Search PubMed.
- I. Fleming, Frontier Orbitals and Organic Chemical Reactions (Textbook), published by Wiley, London, 1976, pp. 41–43 Search PubMed.
- V. A. Glushkov, I. V. Mashevskaya, V. I. Sokol, E. V. Feshina and O. A. Maiorova, Synthesis and structure of 5,5-dialkyl-3-arylamino-2-thiohydantoins, Chem. Heterocycl. Compd., 1997, 33(7), 783–788 CrossRef CAS.
- J. W. Thorpe and J. Warkentin, Stereochemical and Steric Effects in Nucleophilic Substitution of α-Halo Ketones, Can. J. Chem., 1973, 51, 927–935 CrossRef CAS.
- E. Buncel and H. Wilson, The Reactivity Selectivity Principle: Should It Ever Be Used?, J. Chem. Educ., 1987, 64, 475–480 CrossRef CAS.
- A. Slator and D. F. Twiss, The Chemical Dynamics of the Reactions between Sodium Thiosulphate and Organic Halogen Compounds. Part. III., J. Chem. Soc., Trans., 1909, 95, 93–103 RSC.
- H. T. Clarke, The Relation between Reactivity and Chemical Constitution of Certain Halogen Compounds, J. Chem. Soc., Trans., 1910, 97, 416–429 RSC.
- J. B. Conant and W. R. Kirner, The relation between the structure of organic halides and the speed of their reaction with inorganic iodides. I. The problem of alternating polarity in chain compounds, J. Am. Chem. Soc., 1924, 46, 232–252 CrossRef CAS.
- J. B. Conant, W. R. Kirner and R. E. Hussey, The relation between the structure of organic halides and the speeds of their reaction with inorganic iodides. III. The influence of unsaturated groups, J. Am. Chem. Soc., 1925, 47, 488–501 CrossRef CAS.
- F. G. Bordwell and W. T. Brannen Jr, The Effect of the Carbonyl and Related Groups on the Reactivity of Halides in SN2 Reactions, J. Am. Chem. Soc., 1964, 86, 4645–4650 CrossRef CAS.
- D. J. Pasto, K. Garves and M. P. Serve, The Mechanism of Solvolysis of Phenacyl Halides in Various Solvents, J. Org. Chem., 1967, 32, 774–778 CrossRef CAS.
- J. P. Guthrie and J. Cossar, The chlorination of acetone: a complete kinetic analysis, Can. J. Chem., 1986, 64, 1250–1266 CrossRef CAS.
- J. W. Baker, Anomalies in the Reactivities of Side-chain Halogens with Special Reference to Reaction Mechanism, J. Chem. Soc., 1933, 1128–1133 RSC.
- A. Streitweiser, Solvolytic Displacement Reactions At Saturated Carbon Atoms, Chem. Rev., 1956, 56(4), 571–752 CrossRef.
- A. J. Sisti and S. Lowell, Mechanism of the bimolecular nucleophilic displacement reaction on α-halocarbonyl compounds, Can. J. Chem., 1964, 42, 1896–1900 CrossRef CAS.
- A. Halvorsen and J. Songstad, The Reactivity of 2-Bromo-1-phenylethanone (Phenacyl Bromide) toward Nucleophilic Species, J. Chem. Soc., Chem. Commun., 1978, 327–328 RSC.
- W. Forster and R. M. Laird, J. Chem. Soc., Perkin Trans. 2, 1982, 135–138 RSC.
- S. S. Shaik, α- and β-Carbon Substituent Effect on SN2 Reactivity. A Valence-Bond Approach, J. Am. Chem. Soc., 1983, 105, 4359–4367 CrossRef CAS.
- I. Lee, H. J. Koh, Y. S. Park and H. W. Lee, Reactivity–selectivity relationship and kinetic solvent isotope effects in nucleophilic substitution reactions, J. Chem. Soc., Perkin Trans. 2, 1993, 1575–1582 RSC.
- A. W. Erian, S. M. Sherif and H. M. Gaber, The Chemistry of α-Haloketones and Their Utility in Heterocyclic Synthesis, Molecules, 2003, 8, 793–865 CrossRef CAS PubMed.
- M. Katayama, K. Sasagawa and H. Yamataka, Experimental study on the reaction pathway of α-haloacetophenones with nucleophiles: direct substitution or carbonyl addition?, J. Phys. Org. Chem., 2012, 25, 680–685 CrossRef CAS PubMed.
- J. W. Baker, Mechanism of Aromatic Side-chain Reactions with Special Reference to the Polar Effects of Substituents. The ortho-Effect in the Reaction of Phenacyl Bromides with Pyridine, J. Chem. Soc., 1938, 445–448 RSC.
- J. W. Baker, Mechanism and kinetics of aromatic sidechain substitution. Interpretation of reaction data by the method of relative energy levels, Trans. Faraday Soc., 1941, 37, 632–644 RSC.
- H. J. Koh, K. L. Han, H. W. Lee and I. Lee, Kinetics and Mechanism of the Pyridinolysis of Phenacyl Bromides in Acetonitrile, J. Org. Chem., 2000, 65, 4706–4711 CrossRef CAS PubMed.
- I. Lee, H. W. Lee and Y. K. Yu, Kinetics and Mechanism of the Aminolysis of Phenacyl Bromides in Acetonitrile. A Stepwise Mechanism with Bridged Transition State, Bull. Korean Chem. Soc., 2003, 24, 993–998 CrossRef CAS.
- K. S. Lee, K. K. Adhikary, H. W. Lee, B.-S. Lee and I. Lee, Nucleophilic substitution reactions of α-chloroacetanilides with benzylamines in dimethyl sulfoxide, Org. Biomol. Chem., 2003, 1, 1989–1994 CAS.
- S. Dey, K. K. Adhikary, C. K. Kim, B.-S. Lee and H. W. Lee, Nucleophilic Substitution Reactions of α-Chloroacetanilides with Pyridines in Dimethyl Sulfoxide, Bull. Korean Chem. Soc., 2005, 26, 776–780 CrossRef CAS.
- S. Dey, “Kinetics and Mechanism of the Pyridinolysis of α-Chloroacetanilides in Dimethyl Sulfoxide and the Aminolysis of Aryl Phenyl Chlorothiophosphates with Anilines in Acetonitrile”, Masters Thesis, Inha University, Republic of Korea, 2005.
- A. M. Ward, Investigations on the Bivalency of Carbon. Part II. The Displacement of Chlorine from Desyl Chloride. Benzoin Diethylacetal, J. Chem. Soc., 1929, 1541–1553 RSC.
- T. I. Temnikova and E. N. Kropacheva, Zh. Obshch. Khim., 1949, 19, 1917 CAS.
- B. F. Hofferth, Chem. Abstr., 44, 1929, 1950 Search PubMed.
- C. L. Stevens, W. Malik and R. Pratt, Isolation of an Epoxyether from the Reaction of an α-Haloketone with Base, J. Am. Chem. Soc., 1950, 72, 4758–4760 CrossRef CAS.
- C. L. Stevens, M. L. Weiner and R. C. Freeman, Epoxyethers. III. Reaction of Desyl Chloride with Base, J. Am. Chem. Soc., 1953, 75, 3977–3980 CrossRef CAS.
- M. O. Funk and E. T. Kaiser, Hydrolysis of 1-Benzyl-3-bromoacetylpyridinium Bromide. Evidence for Neighboring Group Participation, J. Am. Chem. Soc., 1977, 99, 5336–5340 CrossRef CAS.
- J. C. Hummeln and H. Wynberg, Alkaloid assisted asymmetric synthesis IV: additional routes to chiral epoxides, Tetrahedron Lett., 1978, 19, 1089–1092 CrossRef.
- R. Hoffman, Synthetic transformations using arenesulfonyloxy groups, first as electrophiles, then as leaving groups, Tetrahedron, 1991, 47, 1109–1135 CrossRef CAS.
- M. J. S. Dewar, The electronic theory of organic chemistry, Clarendon Press, Oxford, 1949, p. 73 CrossRef CAS; P. D. Bartlett and E. N. Trachtenburg, 5,7-Dinitro-3-coumaranone and the Mechanism of the Bimolecular Nucleophilic Displacement Reaction in Phenacyl Compounds, J. Am. Chem. Soc., 1958, 80, 5808–5812 CrossRef CAS.
- D. J. McLennan and A. Pross, The Mechanism for Nucleophilic Substitution of α-Carbony1 Derivatives. Application of the Valence-bond Configuration Mixing Model, J. Chem. Soc., Perkin Trans. 2, 1984, 981 RSC.
- D. Kost and K. Alviram, The SN2 Transition State. 6: Breakdown of the Reactivity–Selectivity Principle in the SN2 Reaction of α-Halocarbonyl Compounds. A Molecular Orbital Analysis [1], Isr. J. Chem., 1985, 26(4), 349–353 CrossRef CAS PubMed.
- T. I. Yousaf and E. S. Lewis, Enolate Structures Contributing to the Transition State for Nucleophilic Substitution on α-Substituted Carbonyl Compounds, J. Am. Chem. Soc., 1987, 109, 6137–6142 CrossRef CAS.
- I. Lee, C. S. Shim, S. Y. Chung and H. W. Lee, “Resonance Shunt” Phenomenon in Nucleophilic Substitution of α-Carbonyl Derivatives Demonstrated by the Cross Interaction Constants, Bull. Korean Chem. Soc., 1987, 8, 350–351 CAS.
- I. Lee, C. S. Shim, S. Y. Chung and H. W. Lee, Nucleophilic substitution reactions of phenacyl benzenesulphonates with anilines in methanol–acetonitrile mixtures, J. Chem. Soc., Perkin Trans. 2, 1988, 975–981 RSC.
- I. Lee and I. C. Kim, Cross interaction Constants As a Measure of the Transition State Structure (Part 2). Nucleophilic Substitution Reactions of Phenacyl Bromides with Aniline in Methanol-Acetonitrile Mixtures, Bull. Korean Chem. Soc., 1988, 9, 133–135 CAS.
- I. Lee, S. W. Hong and J. H. Park, Cross Interaction Constants as a Measure of the Transition State Structure. (Part 10), Mechanism of Reactions between Phenacyl Benzenesulfonates with N,N-Dimethylanilines, Bull. Korean Chem. Soc., 1989, 10, 459–462 CAS.
- I. Lee, C. S. Shim and H. W. Lee, Cross-interaction constants as a measure of the transition state structure (part V). The transition [sic] state structure for reactions of phenacyl benzenesulphonates with benzylamines in methanol, J. Phys. Org. Chem., 1989, 2, 484–490 CrossRef CAS PubMed.
- D. M. Kalendra and B. R. Sickles, Diminished Reactivity of Ortho-Substituted Phenacyl Bromides toward Nucleophilic Displacement, J. Org. Chem., 2003, 68, 1594–1596 CrossRef CAS PubMed.
- E. D. Hughes, Mechanism and kinetics of substitution at a saturated carbon atom, Trans. Faraday Soc., 1941, 37, 603–631 RSC.
- E. D. Hughes, Reactions of halides in solution, Q. Rev., Chem. Soc., 1951, 5, 245–269 RSC.
- J. W. Baker, Reactions of ω-Substituted Acetophenone Derivatives. Part II. The Mechanism of the Interaction of ω-Halogenoacetophenones with Primary and Tertiary Bases, J. Chem. Soc., 1932, 1148–1157 RSC.
- R. G. Pearson, S. H. Langer, F. V. Williams and W. J. McGuire, Mechanism of the Reaction of α-Haloketones with Weakly Basic Nucleophilic Reagents, J. Am. Chem. Soc., 1952, 74, 5130–5132 CrossRef.
- C. Srinivasan, A. Shunmugasundaram and N. Arumugam, Kinetics of the Reactions of Phenacyl Bromide and of para-substituted Phenacyl Bromides with Benzoate and Substituted trans-Cinnamate Ions, J. Chem. Soc., Perkin Trans. 2, 1985, 17–19 RSC.
- A. S. Morkovnik, L. N. Divaeva and V. A. Anisimova, Mechanism of the reaction of neutral and anionic N-nucleophiles with α-halocarbonyl compounds, Russ. Chem. Bull., 2007, 56(6), 1194–1209 CrossRef CAS.
- A. Fábián, F. Ruff and Ö. Farkas, Mechanism of nucleophilic substitutions at phenacyl bromides with pyridines. A computational study of intermediate and transition state, J. Phys. Org. Chem., 2008, 21, 988–996 CrossRef PubMed.
- S. Itoh, N. Yoshimura, M. Sato and H. Yamataka, Computational Study on the Reaction Pathway of α-Bromoacetophenones with Hydroxide Ion: Possible Path Bifurcation in the Addition/Substitution Mechanism, J. Org. Chem., 2011, 76, 8294–8299 CrossRef CAS PubMed.
- B. K. Carpenter, Intramolecular Dynamics for the Organic Chemist, Acc. Chem. Res., 1992, 25, 520–528 CrossRef CAS.
- B. K. Carpenter, Dynamic Behavior of Organic Reactive Intermediates, Angew. Chem., Int. Ed. Engl., 1998, 37, 3340–3350 CrossRef.
- B. K. Carpenter, Nonexponential decay of reactive intermediates: new challenges for spectroscopic observation, kinetic modeling and mechanistic interpretation, J. Phys. Org. Chem., 2003, 16, 858–868 CrossRef CAS PubMed.
- D. H. Ess, S. E. Wheeler, R. G. Iafe, L. Xu, N. Çelebi-Ölçüm and K. N. Houk, Bifurcations on Potential Energy Surfaces of Organic Reactions, Angew. Chem., Int. Ed. Engl., 2008, 47, 7592–7601 CrossRef CAS PubMed.
- A. J. Fry and Y. Migron, A convenient new synthesis of α-fluorocarbonyl compounds, Tetrahedron Lett., 1979, 20(36), 3357–3360 CrossRef.
- G. Richard, Bull. Soc. Chim. Fr., 1938, 5, 286 CAS.
- N. Kornblum, R. A. Smiley, R. K. Blackwood and D. C. Iffland, The mechanism of the reaction of silver nitrite with alkyl halides. The contrasting reactions of silver and alkali metal salts with alkyl halides. The alkylation of ambident anions, J. Am. Chem. Soc., 1955, 77(23), 6269 CrossRef CAS.
- A. Lasalle, C. Roizard, N. Midoux, P. Bourret and P. J. Dyens, Removal of nitrogen oxides (NOx) from flue gases using the urea acidic process: kinetics of the chemical reaction of nitrous acid with urea, Ind. Eng. Chem. Res., 1992, 31(3), 777–780 CrossRef CAS.
- N. Kornblum and J. H. Eicher, A new reaction of α nitroesters, J. Am. Chem. Soc., 1956, 78(7), 1494–1497 CrossRef CAS.
- J. Shorter, Compilation and critical evaluation of structure-reactivity parameters and equations – Part I: Values of σm, and σp based on the ionization of substituted benzoic acids in water at 25 °C (Technical Report), Pure Appl. Chem., 1994, 66, 2451–2510 CrossRef CAS.
- H. C. Brown and Y. Okamoto, Electrophilic Substituent Constants, J. Am. Chem. Soc., 1958, 80, 4979 CrossRef CAS.
- C. Hansch, A. Leo and R. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 97, 165–195 CrossRef.
- C. Hansch, A. Leo and D. H. Hoekman, Exploring QSAR: Hydrophobic, electronic and steric constants (ACS Professional Reference Book), volume 2 of ed. C. Hansch and A. Leo, Exploring QSAR, American Chemical Society, 1995 Search PubMed.
- F. G. Bordwell and P. J. Boutan, Conjugative Effects in Divalent Sulfur Groupings, J. Am. Chem. Soc., 1956, 78, 854–860 CrossRef CAS.
- M. M. Fickling, A. Fischer, B. R. Mann, J. Packer and J. Vaughan, Hammett Substituent Constants for Electron-withdrawing Substituents: Dissociation of Phenols, Anilinium Ions and Dimethylanilinium Ions, J. Am. Chem. Soc., 1959, 81, 4226–4230 CrossRef CAS.
- L. A. Cohen and W. M. Jones, A Study of Free Energy Relationships in Hindered Phenols. Linear Dependence for Solvation Effects in Ionization, J. Am. Chem. Soc., 1963, 85, 3397–3402 CrossRef CAS.
- L. A. Cohen and W. M. Jones, A Study of Free Energy Relationships in Hindered Phenols. Correlation of Spectral Properties with Substituent Constants, J. Am. Chem. Soc., 1963, 85, 3402–3406 CrossRef CAS.
- A. Fischer, G. J. Leary, R. D. Topsom and J. Vaughan, Ionic Dissociation of 4-Substituted Phenols and 2,6-Dichloro- and 2,6-Dimethyl-phenols in Organic Solvents, J. Chem. Soc. B, 1967, 846–851 RSC.
- G. Chuchani and A. Frohlich, The pKa Values of Mono-substituted Phenols and Benzenethiols and the Conjugation of Substituents having a Strong +K Effect, J. Chem. Soc. B, 1971, 1417–1420 RSC.
 (A study on the linear free energy relationships for amines: I. A relatively complete set of new σ− values for anilines) Acta Chim. Sin., 32, 107–121, 1966, 136, 137.
(A study on the linear free energy relationships for amines: I. A relatively complete set of new σ− values for anilines) Acta Chim. Sin., 32, 107–121, 1966, 136, 137.- O. E. Edwards and C. Grieco, SN2 Displacement at Tertiary Carbon, Can. J. Chem., 1974, 52, 3561–3562 CrossRef CAS.
- K. Shibatomi, Y. Soga, A. Narayama, I. Fujisawa and S. Iwasa, Highly Enantioselective Chlorination of β-Keto Esters and Subsequent SN2 Displacement of Tertiary Chlorides: A Flexible Method for the Construction of Quaternary Stereogenic Centers, J. Am. Chem. Soc., 2012, 134, 9836–9839 CrossRef CAS PubMed.
- R. Y. Liu, M. Wasa and E. N. Jacobsen, Enantioselective synthesis of tertiary α-chloro esters by non-covalent catalysis, Tetrahedron Lett., 2015, 56(23), 3428–3430 CrossRef CAS PubMed.
- M. Penso, S. Mottadelli and D. Albanese, Reductive dehalogenation of α-haloketones promoted by hydroiodic acid and without solvent, Synth. Commun., 1993, 23(10), 1385–1391 CrossRef CAS PubMed.
- J. W. Thorpe and J. Warkentin, Acetate-catalyzed Bromination and Exchange Reactions of 2-Butanone, Can. J. Chem., 1972, 50, 3229–3232 CrossRef CAS.
- R. A. Cox and J. Warkentin, Kinetics of Bromination of Acetone, Bromoacetone, and 1,1-Dibromoacetone, Can. J. Chem., 1972, 50, 3233–3238 CrossRef CAS.
- R. Altschul and P. D. Bartlett, A quantitative study of the so-called “positive halogen” in ketones and esters, J. Org. Chem., 1940, 5, 623–636 CrossRef CAS.
- M. S. Newman, Concerning the Reduction of α-Bromoketones by Hydrogen Bromide, J. Am. Chem. Soc., 1951, 73, 4993–4994 CrossRef CAS.
- D. Scheffler, H. Grothe and H. Willner, Properties of pure nitryl bromide. Thermal behavior, UV/Vis and FTIR spectra, and photoisomerization to trans–BrONO in an Argon matrix, Inorg. Chem., 1997, 36(3), 335–338 CrossRef CAS.
- J. Hine, Carbon dichloride as an intermediate in the basic hydrolysis of chloroform. A mechanism for substitution reactions at a saturated carbon atom, J. Am. Chem. Soc., 1950, 72(6), 2438–2445 CrossRef CAS.
- S. Winstein, E. Grunwald and H. W. Jones, The correlation of solvolysis rates and the classification of solvolysis reactions into mechanistic categories, J. Am. Chem. Soc., 1951, 73(6), 2700–2707 CrossRef CAS.
- N. Kornblum, W. J. Jones and D. E. Hardies, Stereochemistry and mechanism in reactions of silver salts with alkyl halides. The reaction of silver nitrite with alkyl halides, J. Am. Chem. Soc., 1966, 88(8), 1704–1707 CrossRef CAS.
- F. Michael Akeroyd, Why was a fuzzy model so successful in physical organic chemistry?, International Journal for Philosophy of Chemistry, 2000, 6, 161–173 Search PubMed.
- A. J. Gordon and R. A. Ford, The chemist’s companion. A handbook of practical data, techniques, and references (Textbook), Published by John Wiley & Sons, New York, ISBN 0-471-3190-7, 1972, p. 438 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14798k |
| ‡ The following content is taken, in part, from the PhD thesis of the primary author, Matthew Leonard. |
| § We have normally used the preferred σp− values of Hansch, Leo and colleagues.85,86 However there is disagreement concerning the best value of σp− for methoxy. Hansch et al. prefer −0.26, which is essentially σp (−0.27); but this and similar values are only obtained when the anilinium acidity is used as the basis of measurement. When using aqueous phenols, p-methoxyphenol's acidity requires a σp− in the range −0.10 to −0.135,87–92 and these values give the best fit to our data. |
| ¶ We used Chuchani and Frohlich's values of σm− and σp− for methoxy92 and Zeng's values of σm− for chloro and nitro.93 The remaining σp− values are from Hansch, Leo and colleagues.85,86 |
| This journal is © The Royal Society of Chemistry 2015 |