
Regioselective synthesis of a vitamin K3 based dihydrobenzophenazine derivative: its novel crystal structure and DFT studies†
Dattatray Chadara,
Soniya S. Raoa,
Shridhar P. Gejjia,
Bharat Ugaleb,
C. M. Nagarajab,
Milind Nikaljea and
Sunita Salunke-Gawali*a
aDepartment of Chemistry, Savitribai Phule Pune University, Pune 411007, India. E-mail: sunitas@chem.unipune.ac.in; Fax: +91 2025693981; Tel: +91 2025601397 ext. 531
bDepartment of Chemistry, Indian Institute of Technology, Ropar, Rupnagar-140001, Punjab, India
First published on 1st September 2015
Abstract
A novel acid catalyzed regioselective Michael addition of o-phenylenediamine to vitamin K3 has been carried out to synthesize a dihydrobenzophenazine derivative viz. 6a-methyl-6a,7-dihydrobenzo[α]phenazin-5(6H)-one (1). The compound has been characterized using the single crystal X-ray diffraction and density functional theory.
The dihydrobenzophenazine (DHBP) derivatives, nitrogen containing heterocyclic compounds, have attracted significant attention in recent years owing to their potential applications as antibacterial,1 antimalarial,2 and antituberculosis3 properties in pharmaceutical chemistry. DHBP derivatives are comprised of a planar aromatic polycyclic structure rendered with topoisomerase I and II enzyme inhibition4,5 activities which facilitates their use as a potential anticancer target. The vital role of DHBP derivative in the field of medicine has provided impetus to undertake this study.6 We herewith report the synthesis and characterization of 6a-methyl-6a,7-dihydrobenzo[α]phenazin-5(6H)-one; 1 using precursor vitamin K3 displayed in Scheme 1.
 | ||
| Scheme 1 Molecular structures of vitamin K3(2-methyl-1,4-naphthoquinone) and 1 (6a-methyl-6a,7-dihydrobenzo[α] phenazin-5(6H)-one). | ||
It has been pointed out in the literature that the synthesis of DHBP derivatives were carried out by the reaction of naphthol,1 1,2-naphthoquinone,1 lawsone,7 laphocol8,9 with o-phenylenediamine, electrochemical10 and green chemistry approach.11 In the present endeavor, sulfuric acid was used to catalyze the Michael addition followed by condensation of o-phenylenediamine with vitamin K3, (2-methyl-1,4-naphthoquinone). The DHBP derivative 1 thus obtained was thoroughly characterized by single crystal X-ray diffraction, FT-IR, UV-visible, 1H, 13C-NMR, 2D-NMR, LC-MS experiments. To envisage the conformational characteristics we performed the calculations based on density functional theory.
Experiments have shown that the formation of product 6a-methyl-6a,7-dihydrobenzo[α]phenazin-5(6H)-one (1) is preferred (Path A) over 2 shown in the Path B of Scheme 2.
 | ||
| Scheme 2 Plausible mechanism for formation of 6a-methyl-6a,7-dihydrobenzo[α]phenazin-5(6H)-one (1). | ||
As shown in the Scheme 2 the protonation of the carbonyl group at C(4) position of α, β-unsaturated system in vitamin K3 favours the formation of nearly stable carbocation bound to methyl group, which influences the Michael addition of nucleophile to amino group at C(2) position of o-phenylenediamine thereby generating the intermediate species ‘A’ (Scheme 2). More often nucleophile addition prefer sterically less demanding approach to attack on Michael acceptor and hence it may be expected to give product 2 preferentially. On the contrary we observed the protonation favours the formation of product 1. Furthermore, protonation of the resulting enol in intermediate species A occurs subsequent to loss of hydrogen from the positively charged nitrogen species yielding the neutral species ‘B’. The other amino functionality in o-phenylenediamine orient itself closer enough to attack on the C(1) carbon of the carbonyl group in vitamin K3. Thus resulting condensation generates the C–N bond as indicated by intermediate ‘C’. The proton transfer facilitates the formation unstable aminol functionality (species D) subsequently loses water molecule yielding 1. The mass spectroscopic analysis of DHBP 1 showed the presence of relatively intense M + 1 molecular ion peak at m/z 263(Fig. S1 in ESI†). Its FT-IR spectroscopic analysis has indicative of a band at 3300 cm−1 (Fig. S3 in ESI†) assigned to νN–H stretching of the free –N–H moiety in DHBP derivative 1. The other intense FT-IR bands appeared at 1601 cm−1 and ∼1298 cm−1 were assigned to carbonyl (C(5)) and imino (C(12A)) stretching of the paranaphthoiminoquinone moiety, respectively.
The sharp singlet at δ = 1.06 in the measured 1H NMR spectrum (Fig. S6 in ESI†) in DMSO-d6 of 1 attributed to the methyl group attached to C(13). Moreover, the spectrum showed the characteristic geminal coupling pattern corresponding to non-equivalent methylene protons H(6A)and H(6B) which concomitantly emerge as a doublet of doublet at ∼δ = 2.8 and 3.4 respectively. The differences in chemical shifts between these two protons could be explained as per the conformation observed in the X-ray single crystal of DHBP derivative 1. The H(6A) proton has been appearing at slightly upfield than H(6B) proton; since it has been found to present in the shielding zone of the aromatic nucleus, whereas H(6B) proton has been seen away from shielding zone of the aromatic nucleus and in addition to that H(6B) proton also coming under the influence of deshielding zone of the adjacent carbonyl group at C(5) position. Thus overall effect on H(6B) proton indicative of the distinct downfield chemical shift at δ = 3.4. Normally, CH3–O appears at this chemical shift in the 1H NMR spectrum; the formation of product 1 is thus evident. The product 2 (Path B, Scheme 2) is expected to emerge with distinct pattern in its 1H NMR spectrum.
The Heteronuclear Single Quantum Coherence (gHSQC) experiments further revealed peaks (Fig. 1) confirm the presence of nonequivalent C(6) methylene protons and aliphatic carbon and its attached proton are correlated. As a result gHSQC study also supports to get the complete information related to structure of the DHBP derivative 1. The DEPT NMR experiment gives an idea about presence of CH3 (C(13)), CH (aromatic), CH2 (C(6), α-carbon) groups and quaternary carbon (C(6A)) (Fig. 2). Accordingly, we notice the normal 13C peak for CH3 and CH carbons; the peak for the quaternary carbon (C(6A)) disappears. The presence of inverted peak assigned to methylene at C(6) are supports the inference of regioselective addition of the o-phenylenediamine.
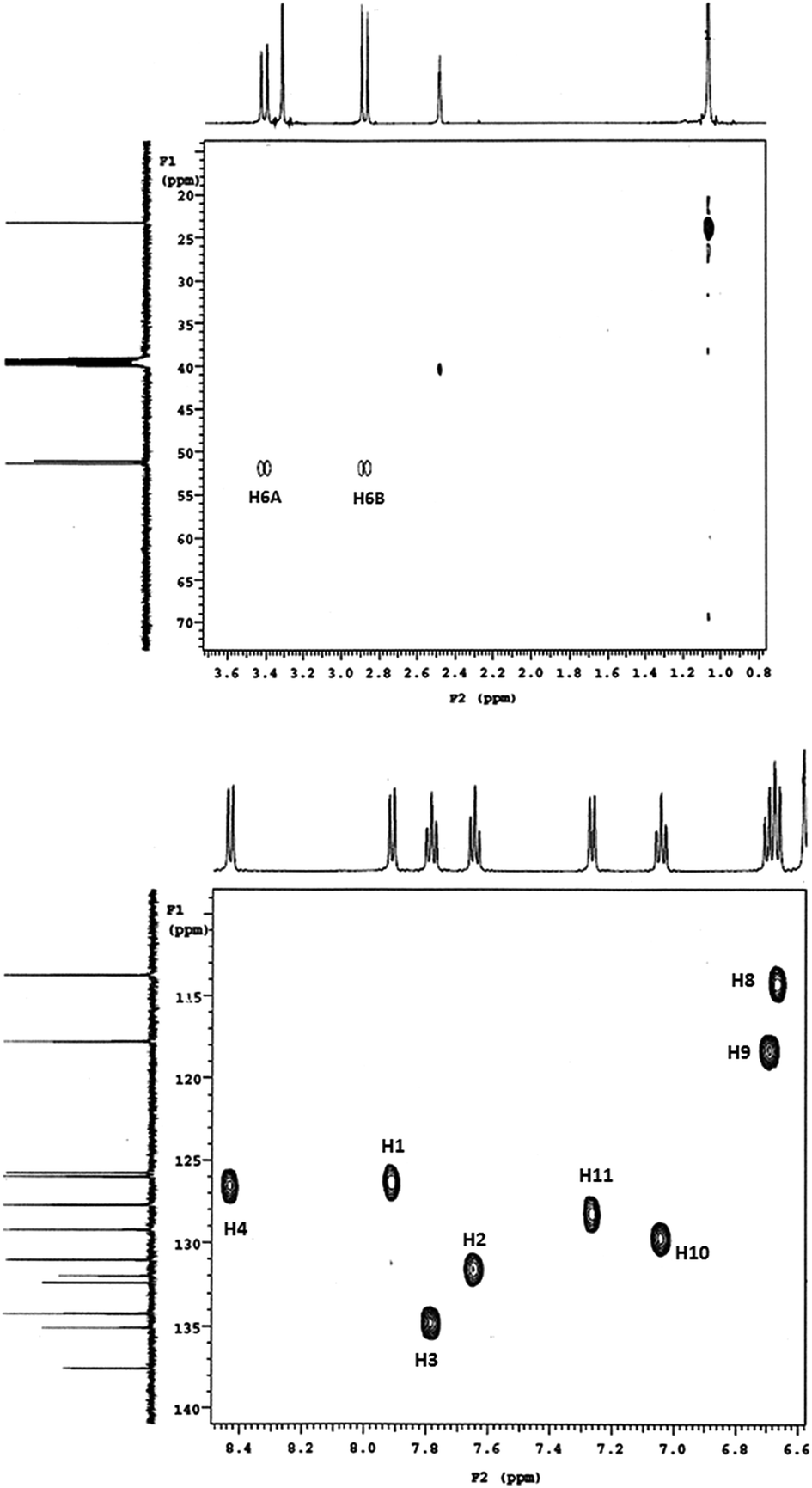 | ||
| Fig. 1 gHSQC NMR spectra of 1. | ||
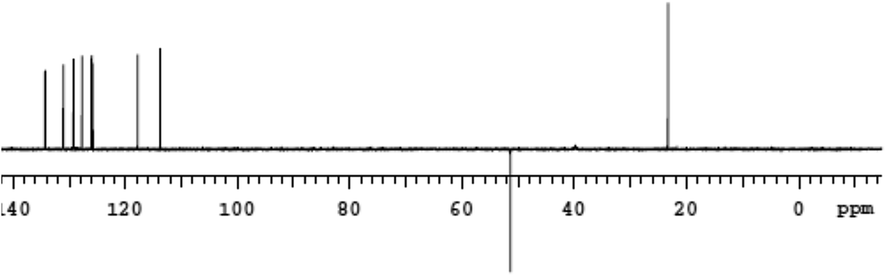 | ||
| Fig. 2 DEPT NMR spectra of 1. | ||
The UV-visible spectrum (Fig. S9 in ESI†) of the DHBP derivative 1 showed a broad band at ∼264–273 nm, indicative of the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N at C(12A) as well as aromatic rings. The appearance of UV band near 311 nm results from π–π* transition of the CO functionality.12
N at C(12A) as well as aromatic rings. The appearance of UV band near 311 nm results from π–π* transition of the CO functionality.12
The single crystal X-ray diffraction of DHBP derivative 1 shows the crystallization in orthorhombic space group Pbca which contains eight molecules in unit cell. Fig. 3 showed an ORTEP plot. Single crystal X-ray structure underlines the regioselective addition of o-phenelyenediamine at C(2) and the condensation with carbonyl group present at C(1) of the vitamin K3. The carbonyl bond distance (C(5)–O(1)) was 1.21 Å which is similar to that observed in oxidized form of naphthoquinone carbonyls.13 The sp3 hybridization of carbon atoms C(6) and C(6A) was thus inferred from the bond distance parameters viz. C(6)–C(6A); 1.519 Å, C(6A)–C(12A); 1.522 Å and C(6)–C(5); 1.494 Å. Thus, N(7) and N(12) atom centers are suggested to be sp3 and sp2 hybridized, respectively.14
 | ||
| Fig. 3 ORTEP plot of 1. Formula: C17H14N2O, formula weight: 262.30, orthorhombic crystal system, space group: Pbca, a/Å = 7.518(5), b/Å = 14.238(5), c/Å = 25.049(5), V (Å3) = 2681(2), Z = 8, R1 = 0.0363. Bond distances (Å): C(5)–O(1) = 1.217(2), C(5)–C(6) = 1.492(2), C(6)–C(6A) = 1.519(2), C(6A)–C(12A) = 1.521(2), C(6A)–C(7) = 1.453(2), N(7)–C(7A) = 1.375(2), N(12)–C(11A) = 1.285(2), N(12)–C(12A) = 1.405(2). ∠N(7)–C(6A)–C(6) = 112.3(1)°, ∠C(5)–C(6)–C(6A) = 114.0(1)°. | ||
The wire frame diagram of DHBP derivative 1 showed the presence of C–H⋯π interactions between the two aromatic rings A and D (Scheme 1) of the neighboring molecules. It help to facilitate the crystal network (Fig. 4), wherein the molecules from neighboring polymeric chain combine in head to head fashion revealing N–H⋯O interactions. The hydrogen bonding structural parameters are reported in Table 1.
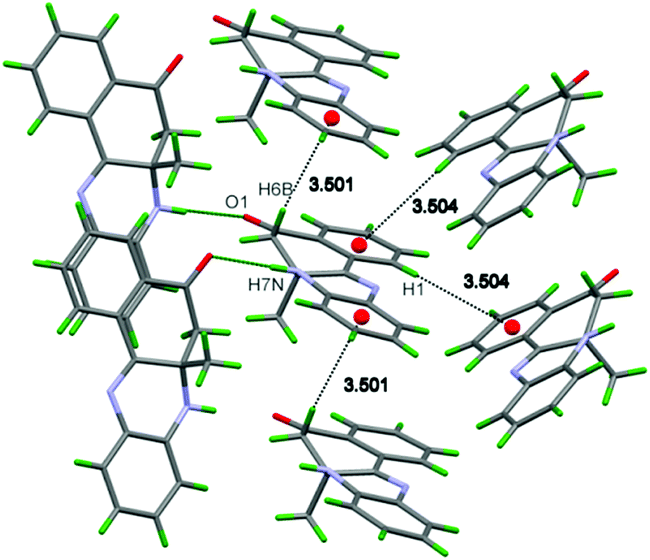 | ||
| Fig. 4 C–H⋯π and N–H⋯O interactions of (1). | ||
| D–H⋯A | D–H(Å) | H⋯A(Å) | D⋯A(Å) | ∠D–H⋯(Å) |
|---|---|---|---|---|
| a (i) −1/2 + x, 1/2 − y, 1 − z (ii) −1 + x, y, z (iii) −1/2 + x, y, 1.5 − z. | ||||
| N(7)–H(7A)⋯O(1)(i) | 0.90(2) | 2.12 (2) | 3.027(2) | 176(1) |
| C(6)–H(6B)⋯C(9)(ii) | 0.969(1) | 2.830(2) | 3.720(2) | 153.0(1) |
| C(1)–H(1)⋯C(2)(iii) | 0.930(1) | 2.817(2) | 3.621(2) | 145.4(1) |
The molecular structure of DHBP derivative 1 was optimized within the framework of density functional theory incorporating the B3LY P(Becke, Lee-Yang-Parr 3 parameter exchange correlation functional)15,16 (cf. Fig. 5) employing the Gaussian-09 program.17 The internally stored basis with diffuse functions being added on all the heavy atoms, designated with the 6-31+G(d,p) basis, was used. Stationary point structure thus obtained was confirmed to be local minimum on the potential energy surface through vibrational frequency calculations. Net atomic charges have been derived from population analysis based on the Hirshfeld partitioning scheme. The electron-rich regions of DHBP derivative 1 have further been characterized in terms of molecular electrostatic potential (MESP) which are portrayed in Fig. 6. Normal vibrations were assigned by visualizing displacements of atoms about their equilibrium (mean) positions combined with the potential energy distribution. The vibrational frequencies from the present DFT calculations are scaled by a factor of 0.9654; obtained from a comparison with the carbonyl stretching frequency in the measured spectra (1674 cm−1). The highest wave number vibration was assigned to –NH stretching appears at 3458 cm−1 which corresponds to (observed: 3300 cm−1) band in the experiment. Likewise the aromatic –CH stretching appear at the 3115 cm−1, 3106 cm−1 and 3060 cm−1, respectively (observed: 3051 cm−1), whereas the methylene stretching corresponds to the 2988 cm−1. The methyl vibrations appear ∼3015–2929 cm−1 (the lowest vibration arising from the locally symmetric –CH3 functionality). The experimental spectra revealed aromatic –CH and the methylene stretching in the region (observed: 2959, 2918 cm−1). Overall the calculated IR spectra are in fairly good agreement with those observed in the experiment.
 | ||
| Fig. 5 B3LYP/6-31+G (d,p) optimized structure of (1). Selected bond distance parameters (in Å) and Hirshfeld atomic charges (given in parentheses) are shown. | ||
 | ||
| Fig. 6 Molecular electrostatic potential surface of 1 (isosurface V = −52.5 kJ mol−1). | ||
The DHBP derivative 1 with both chair and boat conformers were optimized within the framework of density functional theory. The chair and boat forms of the isolated DHBP derivative 1 finally converged to the one structure portrayed in Fig. 5. Selected bond distance parameters (in Å) and Hirshfeld atomic charges (in parentheses) are depicted in Fig. 5. Structural parameters from the present theory are in consonant with those from the single crystal X-ray data reported in Table S2 of the ESI.† MESP analyses have shown that the electron-rich regions by and large, are localized near carbonyl oxygen and pyridinium nitrogen. It may, therefore, be inferred that crystal network extends via N–H⋯O interactions. MESP isosurface (V = −52.5 kJ mol−1) has been depicted in Fig. 6. The highest occupied molecular orbital (isosurface of −17.5 kJ mol−1) in the DHBP derivative 1 has been shown in Fig. S10 in the ESI.†
To summarize, a regioselective synthesis of 6a-methyl-6a,7-dihydrobenzo[α]phenazin-5(6H)-one i.e. dihydrobenzophenazine derivative 1 based on vitamin K3 has been reported in the present work. The regioselective yield of the product has been confirmed from the single crystal X-ray diffraction data and further supported by its electronic structure derived from the B3LYP/6-31+G(d,p) theory. The studies on reactions of 1,4-naphthoquinone derivatives with o-phenylenediamine are in progress in our laboratory.
SSG is grateful the Department of Biotechnology, India for the financial support. DC thanks the University Grants Commission, New Delhi, India for the Junior Research Fellowship. SSR acknowledges the financial support from the Savitribai Phule Pune University, Pune, India for the award of research fellowship through the potential excellence scheme.
Analytical data of 1, color: dark orange, yield: 0.459 g (30.13%), mp 152.27 °C. FT-IR (KBr; cm−1): 3300, 3051, 2959, 2918, 1674, 1600, 1477, 1450, 1427, 1365, 1338, 1298, 1263, 1209, 1167, 1108, 1076, 1057, 1026, 987, 943, 893, 873, 815, 795, 731, 680, 644, 586, 567, 594, 488, 432. 1H NMR (500 MHz, DMSO-d6), δ(ppm): 1.06 (3H, s), 2.84 (1H, d, J = 15.50 Hz), 3.40 (1H, d, J = 15.50 Hz), 6.67 (1H, d, J = 8.00 Hz), 6.69 (1H, d, J = 8.25 Hz), 7.04 (1H, d, J = 8.25 Hz), 7.25 (1H, d, J = 8.00 Hz), 7.64 (1H, d, J = 7.50 Hz), 7.78 (1H, d, J = 8.25 Hz), 7.90 (1H, d, J = 8.00 Hz), 8.43 (1H, d, J = 8.00 Hz), 13C NMR (125 MHz DMSO-d6), δ(ppm): C(1) = 125.79, C(2) = 131.05, C(3) = 134.28, C(4) = 126.01, C(4a) = 132.42, C(5) = 194.60, C(6) = 51.37, C(7) = 51.08, C(7A) = 132.12, C(8) = 113.78, C(9) = 117.79, C(10) = 129.26, C(11) = 127.74, C(11A) = 137.57, C(12A) = 155.80, C(12B) = 131.99. UV-vis; DMSO (λmax, nm): 437, 317, 271, 262. Anal. data. calc. for C17H14N2O: C, 77.84; H, 5.38, N, 10.68, found: C, 77.34; H, 5.40, 11.16.
Notes and references
- H. Hussain, S. Specht, S. R. Sarite, M. Saeftel, A. Hoerauf, B. Schulz and K. Krohn, J. Med. Chem., 2011, 54, 4913–4917 CrossRef CAS PubMed.
- M. E. Makgatho, R. Anderson, J. F. O'Sullivan, T. J. Egan, J. A. Freese, N. Cornelius and C. E. J. van Rensburg, Drug Dev. Res., 2000, 50, 195–202 CrossRef CAS.
- R. S. F. Silva, M. C. F. R. Pinto, M. O. F. Goulart, J. D. S. Filho, I. Neves Jr, M. C. S. Lourenco and A. V. Pinto, Eur. J. Med. Chem., 2009, 44, 2334–2337 CrossRef CAS PubMed.
- S.-T. Zhuo, C.-Y. Li, M.-H. Hu, S.-B. Chen, P.-F. Yao, S.-L. Huang, T.-M. Ou, J.-H. Tan, L.-K. An, D. Li, L.-Q. Gu and Z.-S. Huang, Org. Biomol. Chem., 2013, 11, 3989–4005 CAS.
- N. Vicker, L. Burgess, I. S. Chuckowree, R. Dodd, A. J. Folkes, D. J. Hardick, T. C. Hancox, W. Miller, J. Milton, S. Sohal, S. Wang, S. P. Wren, P. A. Charlton, W. Dangerfield, C. Liddle, P. Mistry, A. J. Stewart and W. A. Denny, J. Med. Chem., 2002, 45, 721–739 CrossRef CAS PubMed.
- D. Chadar, S. S. Rao, A. Khan, S. P. Gejji, K. S. Bhat, T. Weyhermüller and S. Salunke-Gawali, RSC Adv., 2015, 5, 57917–57929 RSC.
- B.-L. Yao, Y.-W. Mai, S.-B. Chen, H.-T. Xie, P.-F. Yao, T.-M. Ou, J.-H. Tan, H.-G. Wang, D. Li, S.-L. Huang, L.-Q. Gu and Z.-S. Huang, Eur. J. Med. Chem., 2015, 92, 540–553 CrossRef CAS PubMed.
- P. F. Carneiro, M. C. F. R. Pinto, T. S. Coelho, B. C. Cavalcanti, C. Pessoa, C. A. Simone, I. K. C. Nunes, N. M. de Oliveira, R. G. de Almeida, A. V. Pinto, K. C. G. de Moura, P. A. da Silva and E. N. da Silva Júnior, Eur. J. Med. Chem., 2011, 46, 4521–4529S CrossRef CAS PubMed.
- M. J. da Silva, M. C. F. R. Pinto, C. A. de Simone, J. G. Soares, J. R. M. Reys, J. D. S. de Filho, W. T. A. Harrison, C. E. M. Carvalho, M. O. F. Goulart, E. N. da Silva Júnior and A. V. Pinto, Tetrahedron Lett., 2011, 52, 2415–2418 CrossRef CAS PubMed.
- S. S. H. Davarani, A. R. Fakhari, A. Shaabani, H. Ahmar, A. Maleki and N. S. Fumani, Tetrahedron Lett., 2008, 49, 5622–5624 CrossRef PubMed.
- A. C. Sousa, M. C. Oliveira, L. O. Martins and M. P. Robalo, Green Chem., 2014, 16, 4127–4136 RSC.
- D. Chadar, M. Camilles, R. Patil, A. Khan, T. Weyhermüller and S. Salunke-Gawali, J. Mol. Struct., 2015, 1086, 179–189 CrossRef CAS PubMed.
- (a) R. Patil, D. Chadar, D. Chaudhari, J. Peter, M. Nikalje, T. Weyhermüller and S. Salunke-Gawali, J. Mol. Struct., 2014, 1075, 345–351 CrossRef CAS PubMed; (b) S. Pal, M. Jadhav, T. Weyhermüller, Y. Patil, M. Nethaji, U. Kasabe, L. Kathawate, V. B. Konkimalla and S. Salunke-Gawali, J. Mol. Struct., 2013, 1049, 355–361 CrossRef CAS PubMed; (c) O. Pawar, A. Patekar, A. Khan, L. Kathawate, S. Haram, G. Markad, V. Puranik and S. Salunke-Gawali, J. Mol. Struct., 2014, 105, 68–74 CrossRef PubMed; (d) S. Salunke-Gawali, O. Pawar, M. Nikalje, R. Patil, T. Weyhermüller, V. G. Puranik and V. B. Konkimalla, J. Mol. Struct., 2014, 1056–1057, 97–103 CrossRef CAS PubMed.
- L. Kathwate, P. V. Joshi, T. Dash, S. Pal, M. Nikalje, T. Weyhermüller, V. G. Puranik, V. B. Konkimalla and S. Salunke-Gawali, J. Mol. Struct., 2014, 1075, 397–405 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Jr Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. akajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian03, revision, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- D. J. Becke, Chem. Phys., 1993, 98, 5648–5652 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 LC-MS spectrum, Fig. S2–4 FT-IR figures, Fig. S5 DSC spectrum, Fig. S6–8 NMR, Fig. S9 UV-visible spectra, Fig. S10 HOMO–LUMO diagram, Fig. S11 optimized IR spectra, Table 1 and 2 crystallography data. CCDC 1037164. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra13169c |
| This journal is © The Royal Society of Chemistry 2015 |