
Effect of preparation of Fe–Zr–K catalyst on the product distribution of CO2 hydrogenation
Abstract
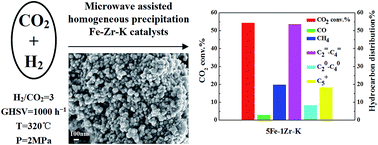
The precursors of Fe–Zr–K catalysts were prepared by microwave assisted homogeneous precipitation followed by incipient wetness impregnation. The obtained mesoporous particles were small and uniformly shaped. After reduction, the catalyst showed high activity for the selective production of light olefins from CO2 hydrogenation, superior to the catalyst obtained by co-precipitation. Characterization indicated that with Zr addition, the reducibility, surface basicity and surface atomic composition of the samples varied with the Zr content. The CO2 adsorption ability of the samples was increased and the samples became hard to reduce as a result of the interaction between iron species and zirconia. In the case of using 5Fe–1Zr–K under selected conditions of 1000 h−1, 320 °C and 2 MPa, the CO2 conversion and the selectivity of C2–C4 olefins reached 54.36% and 53.63%. The olefin/paraffin ratio in the C2–C4 hydrocarbon product fraction reached 6.44. The selectivity of CO and C5+ hydrocarbons were 3% and 19.78%, respectively. The catalytic activity remained stable up to 92 h in time-on-stream reaction.
 Please wait while we load your content...
Please wait while we load your content...

