DOI:
10.1039/C5RA14288A
(Paper)
RSC Adv., 2015,
5, 81087-81092
Self-fluorinated Bi3Ti2O8F formed by cross-linked nanosheets as a superior dye-sensitized photocatalyst†
Received
20th July 2015
, Accepted 14th September 2015
First published on 14th September 2015
Abstract
A new semiconductor photocatalyst Bi3Ti2O8F (BTOF) has been prepared by a hydrothermal method, whose surface is highly fluorinated by the intrinsic F ions. Each BTOF particle is formed by many cross-linked nanosheets (NS) and most of the NSs grow upright on each other. It is noted that the fluorinated surface exhibits strong electronegativity and superior adsorption capacity for cationic dyes selectively. In comparison with the TiO2 (Degussa P25), BTOF exhibits much higher photocatalytic activity for cationic dyes under both ultraviolet and visible light, which is demonstrated to be a process of dye sensitization photodegradation with good stability and recyclability. The strong dye sensitization effect of BTOF under visible light reveals the application potential in dye-polluted water treatment and dye-sensitized solar cells.
1. Introduction
Environmental pollution and solar energy conversion are becoming topical issues in the sustainable development of human society. Dye sensitization, as a promising solution to energy production and environmental pollutant control utilizing solar energy, has drawn much attention. Among various semiconductor photocatalysts, TiO2 has become the benchmark material for its outstanding performance in both dye-sensitized solar cells (DSSC)1–3 and dye-sensitized photodegradation (DSPD).4–7 In the process of dye sensitization, TiO2 acts as an electron acceptor and effectively promotes the separation of the photon-generated carriers in the excited dye molecules.8–10 For DSPD, the dissolved O2 in aqueous solution can easily capture electrons to form reactive species like O2˙−, ˙OOH and ˙OH, which can subsequently degrade dye molecules.11,12
It is known that the rate of charges transfer between photocatalyst and dye plays a crucial role in determining the photocatalytic activity of catalyst.13,14 In consideration of this, ensuring good interface contact between them and fabricating appropriate charge trapping centers in catalyst can be potential solutions to accelerate this process. To achieve this goal, many efforts, such as increasing the surface area, composites and surface modification have been made. Among these, surface fluorination of TiO2 is a promising approach to obtain active photocatalysts through increasing its adsorption capacity for dyes, and fabricating electron trapping centers, Ti–F groups.15,16 Yu17 and Pan18 pointed out that the formed Ti–F groups on the surface of TiO2 can attract the photogenerated electrons due to the strong electronegativity of the fluorine. Park19 pointed out that the surface fluorination of TiO2 enhances the OH radical mediated degradation but inhibits the hole transport. He et al.,20 emphasized the hydrophilia of the fluorinated surface of TiO2 where oxygen and water molecules are easily adsorbed. However, the enhancement of photocatalytic activity of fluorinated TiO2 is limited because that O ions in the TiO2 lattice are hard to be substituted by the foreign F ions, hence only a small amount of Ti–F groups can be formed in the surface of TiO2. On the other hand, the formed Ti–F groups can be washed away by the OH− ions through a simple ligand exchange reaction,20,21 which further decreases the amount of active sites and results in the gradual deactivation of fluorinated TiO2. Thus how to obtain sufficient and durable fluorinated catalyst remains a challenge.
In this work, we reported the synthesis of a fluoride photocatalyst Bi3Ti2O8F (BTOF) via a facile hydrothermal method. Surface analysis indicated that the intrinsic F ions can be firmly bonded to the surface of BTOF and heavily fluorinate the surface of BTOF. Each BTOF particle is formed by many cross-linked nanosheets (NS) and most of the NSs grow uprightly on each other. The specific three-dimensional cross structure of BTOF can accelerate the photodegradation rate by providing more active sites and facilitating the mass transfer process. In comparison with the TiO2 (Degussa P25), BTOF exhibits 100-fold improvement in the adsorption capacity, 1.25-fold and 4-fold improvement in photodegradation rate of cationic dyes under UV and visible light, respectively. To the best of our knowledge, BTOF has never been systematically prepared and studied as a photocatalyst before.
2. Experimental
2.1 Material preparation
The BTOF powders used in this study were prepared by a hydrothermal method. TiCl4, Bi(NO3)3·5H2O and NaF were analytical grade (purchased from Shanghai chemical industrial company) and were used without further purification. TiO2 (Degussa P25; BET surface area, 50 m2 g−1) powders were purchased from Degussa and provided a comparison. Deionized water was used in all experiments. In a typical procedure, 5 mmol of TiCl4 was dissolved in 35 ml of water, and then 7.5 mmol Bi(NO3)3·5H2O and 28.5 mmol NaF were added into the above solution in sequence under magnetic stirring, after that, the pH value of the turbid liquid was adjusted to about 9.5 with 2 M NaOH, then it was transferred into a 50 ml Teflon-lined autoclave and maintained at 180 °C for 6 h. After the reaction, the autoclave was cooled to room temperature and the white precipitate was collected and washed three times with water and ethanol, and then dried in an oven at 60 °C for 12 h.
2.2 Material characterization
Wide-angle angle X-ray diffraction (XRD) patterns of the BTOF powders were obtained on a D/Max-RBX-ray diffractomer (Rigaku, Japan) using Cu Kα radiation. The morphologies of the prepared samples were examined by scanning electron microscope (SEM; Hitachi, S-4800) and high resolution transmission electron microscope (TEM; JEOL 4000EX) images. X-ray photoelectron spectra (XPS) were measured on an Escalab 250Xi system (Thermo Scientific) using Mg Kα radiation of 1253.6 eV, the binding energy was calibrated with the reference to the C 1s peak at 284.8 eV. The diffuse reflectance spectrum of the sample was recorded in the range 200–700 nm on a Hitachi U-3010 spectroscope. Zeta potential measurement was performed using the Zetasizer 3000 HSA (Malvern Instruments), the solution concentration is 0.1 g L−1. The BET specific surface area of the samples was analyzed by nitrogen (N2) adsorption–desorption in a Quantacrome Autosorb-1-C instrument. Ultraviolet photoelectron spectroscopy (UPS) measurements were performed on BTOF film using a photoelectron spectrometer (Kratos Analytical, AXIS-ultra-DLD) with He (I) excitation of 21.22 eV and pass energy of 5 eV.
2.3 Adsorption and photodegradation testing
To evaluate dye adsorption and photodegradation performances of the samples, 20 mg powders (BTOF, or P25) were dispersed in 100 ml of a 20 μM dye aqueous solution. The time-varying concentrations of dyes were determined by measuring the absorbance of RhB at 550 nm, malachite green oxalate (MGO) at 618 nm and methyl orange (MO) at 462 nm with a spectrometer (Shimadzu, UV-1800) after removing the powders from each sample by centrifugation. The adsorption experiments were performed in dark at 25 °C with the suspension was magnetically stirred. In photodegradation experiments, a 300 W xenon lamp, equipped with a cut off filter of λ > 420 nm were used as the ultraviolet (UV) and visible (Vis) light source. Before illumination, the solution was kept in dark for 30 min to ensure establishment of an adsorption–desorption equilibrium. Photoluminescence (PL) spectra were recorded on an Edinburgh Instruments FLS920 fluorescence spectrometer equipped with xenon lamp.
3. Results and discussion
3.1 Phase and morphology
XRD patterns of the as-prepared sample are shown in Fig. 1a. The diffraction peaks match well with the standard patterns of BTOF phase (JCPDS no. 33-0220), which has a body-centered tetragonal structure with the lattice parameters of a = b = 3.318 Å, c = 24.97 Å. No other peaks were detected, indicating the high purity of the powders. The sharp intensity weakening of (004) peak at about 14.2° and intensity enhancement of (110) peak at about 33.1° imply the anisotropic growth of the BTOF, which is further confirmed by the SEM and TEM images. The overall view in Fig. 1b reveals that each BTOF particle is formed by cross-linked nanosheet (NS) and most of the NSs grow uprightly on each other. The three-dimensional cross structure of BTOF produces abundant open space and specific surface area, which can accelerate the photodegradation rate by providing more active sites and facilitating the mass transfer process. In addition, the particle size of the crystals was estimated to be about 500 nm, and no agglomeration was observed, hence improves the dispersion of catalyst in dye solution.
 |
| | Fig. 1 (a) XRD patterns of the BTOF powders; (b and d) TEM images of the BTOF sheets; (c) SEM images of the prepared BTOF nanoparticles. | |
Facet of the NSs is determined by TEM. Fig. 1d shows the top view of a horizontal and a vertical NS. In the horizontal sheet, two sets of planes perpendicular to each other are shown, both of which have a lattice spacing of 0.27 nm and correspond to the (110) and (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) planes, respectively. In the vertical NS, the planes have a lattice spacing of 0.623 nm, corresponding to the (004) planes. Combined with the XRD analysis, we concluded that the NSs have dominated (004) facets that are perpendicular to the c axis. The mechanism of facet mediation is speculated to be similar to the anisotropic growth of TiO2 in solution containing F ions,21 which is associated with the hindrance of fluoride on the extension of Ti–O–Ti bridging, thus the growth of BTOF crystal along the c axis is inhibited.
0) planes, respectively. In the vertical NS, the planes have a lattice spacing of 0.623 nm, corresponding to the (004) planes. Combined with the XRD analysis, we concluded that the NSs have dominated (004) facets that are perpendicular to the c axis. The mechanism of facet mediation is speculated to be similar to the anisotropic growth of TiO2 in solution containing F ions,21 which is associated with the hindrance of fluoride on the extension of Ti–O–Ti bridging, thus the growth of BTOF crystal along the c axis is inhibited.
3.2 Surface composition and chemical state
Fig. 2a shows the high-resolution XPS spectra of the F 1s and Ti 2p region. It can be seen that the F 1s spectrum was well fitted by two peaks: a stronger one at 680.5 eV and a weaker one at 684.0 eV, indicating two different chemical states of the F ions. The latter one corresponds to the Ti–F species in the surface, which is in agreement with the value measured in F–TiO2,22 while the value of 680.5 eV was rarely reported in former studies. In consideration of the existence of latticed F atoms inside the crystal, we can assign the signal at 680.5 eV to them. To distinguish the signal from external F atom from others, we dipped the BTOF powders in NH4F solution for introducing an increased number of adsorbed F ions in the surface. After separation and dry, the corresponding XPS spectra are provided in Fig. 2a. We can see that the peak at 684.0 eV become much stronger, indicating an increased number of surficial F atoms, while the intensity of the other one remains unchanged, corresponding to the unaffected internal F atoms, which reconfirms the above speculation. Besides, it is reported that F ions are weakly adsorbed on the surface of TiO2 and can hardly substitute the oxygen in TiO2 lattice,18 they can be easily washed away by the basic solution or long-term usage through a simple ion exchange, which will result in the gradual deactivation of photocatalyst. Interestingly, when BTOF was treated with the same method, the intensity of peak at 684.0 eV does not show much decrease compared with that of peak at 680.5 eV, that is, no F ions loss occurs for BTOF. It is noted that the fluorinated BTOF was prepared in alkaline solution in this work, thus we can ascribe the signal of surficial F to the bonded F ions instead of the adsorbed ones.
 |
| | Fig. 2 (a) F 1s XPS spectra of as prepared BTOF, NaOH-washed BTOF, NH4F-washed BTOF; (b) Ti 2p XPS spectra of BTOF and P25. | |
XPS spectra of Ti 2p for P25, BTOF are illustrated in Fig. 2b. The Ti 2p spectra for pure P25 have symmetrical peaks around 458.0 eV and 464.2 eV, which correspond to Ti 2p3/2 and Ti 2p1/2 respectively, suggesting that samples still maintain Ti4+ ions. While Ti 2p spectra of BTOF show an additional peak at 466.9 eV, which was also observed in TiF4. In consideration of that no TiF4 phase was detected, it maybe due to Ti–F in BTOF, consistent with the implication of the peak of F 1s at 680.5 eV. Moreover, the molar ratio of Ti–F species to the sum of titanium is calculated to be about 0.4, indicating a high fluorination degree of BTOF.
3.3 Adsorption
Fig. 3 presents the dynamic adsorption curves of BTOF (BET surface area, 45 m2 g−1) for dyes. For comparison, the results of P25 acquired in the same condition are shown as well. The time profiles of Ct/Co in Fig. 3a reveals an electrostatic adsorption mechanism of BTOF. To be specific, the BTOF powders show large adsorption capacity for cationic dyes like RhB and MGO. The calculated adsorption capacity of BTOF for RhB is 84.0 μmol g−1, about one hundred times higher than that of P25 (0.816 μmol g−1). Moreover, the adsorption process can finish in a few minutes, indicating that the BTOF powders can serve as efficient absorbents. When more BTOF powders are added, 100 mg for example, RhB can be entirely removed and clear water is obtained (inset of Fig. 3a). In contrast, the adsorption capacity of the BTOF for MO, an anionic organic dye, is negligible. The adsorption process of the BTOF exhibits electrostatic adsorption characteristics, which may be caused by some negative charges on the facets of BTOF NSs. To confirm this assumption, zeta potential measurement of the samples in aqueous solution with different pH values was carried out. As shown in Fig. 3b, zeta potential for the BTOF powders ranges from −9.0 mV at pH = 2.4 to −24.2 mV at pH = 7.7, while for P25, it changed from +38 to −2.1 mV over the same pH range. The negative zeta potential values within a broad pH range can well support the above assumption.
 |
| | Fig. 3 (a) Plots of dyes adsorption onto BTOF/P25. Inset, photos of discoloration of RhB solution caused by adsorption of BTOF. Co: the initial concentration of dye (RhB, or MO) before BTOF was added; C: the concentration of dye at different time; (b) zeta potential of BTOF and P25 solution over the same pH range. | |
But where do the charges originate from? It is known that BTOF belongs to Aurivillius phases having unique layer-stack structure.23 These compounds are generally constructed by regular intergrowth of (Bi2O2)2+ cationic layer and perovskite (An−1BnO3n+1)2− anionic layer, where A is K+, Pb2+, Bi3+, etc.; B is Ti4+, Nb5+, W6+, etc.; and n is an integer. It is well accepted that the alternative stacking of two oppositely charged layers can form intra-electric fields in the layered bismuth compounds.24–26 Fig. 4 illustrates the crystal structure of BTOF determined by Kodama,23 we can see that the BOF cationic layer and the perovskite anionic layer are both perpendicular to the c axis, and parallel to the exposed (004) facets of the NSs (inset of Fig. 4). We determined that the exposure of the anionic layers in (004) facets will endow NSs with negatively charged surfaces, which can well explain their selective adsorption for dyes. Moreover, those charged BTOF NSs can repel them from each other, avoiding the particle agglomeration, as mentioned in the above section.
 |
| | Fig. 4 Schematic drawing of the crystal structure of BTOF and the anisotropic growth of the BTOF NSs. | |
3.4 Photocatalytic activity
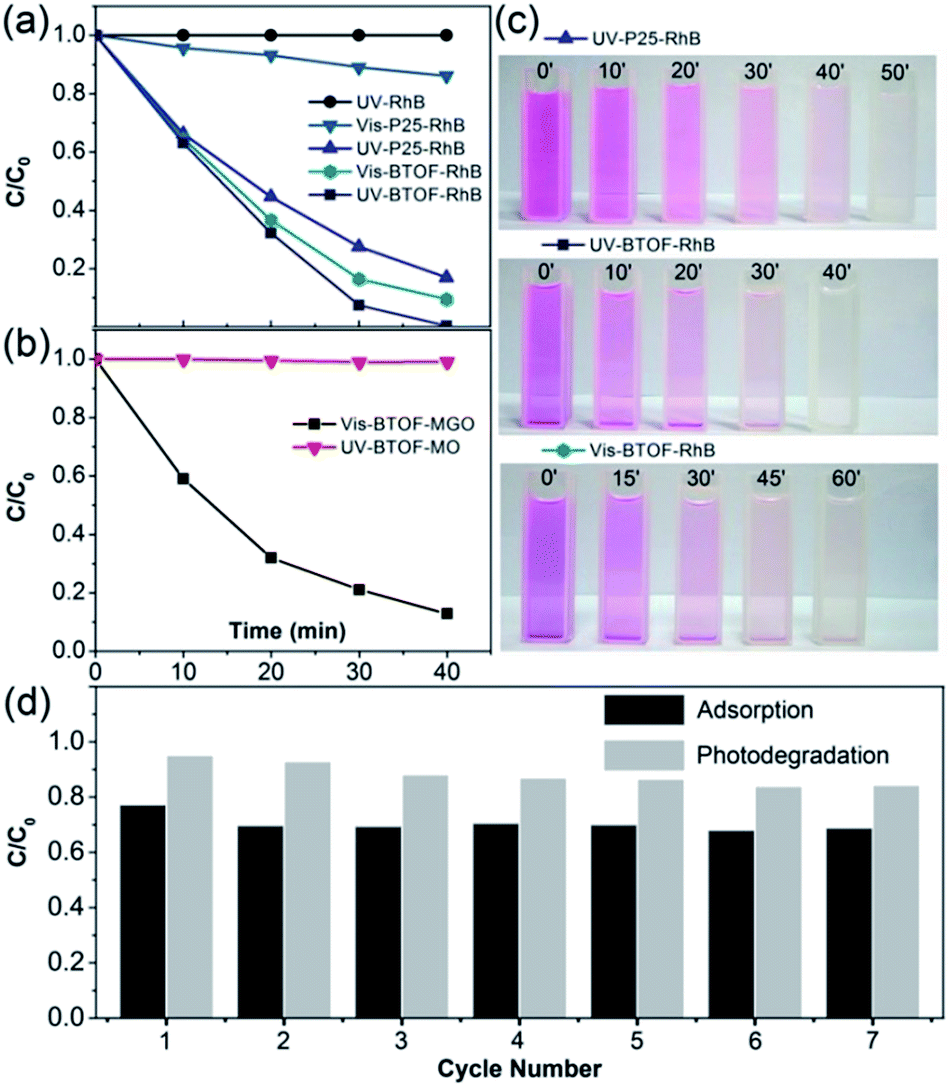
The photodegradation activities of the samples were evaluated by the degradation of RhB under UV light and visible light and compared with the well-known P25. The latter has become the benchmark photocatalyst as its prominent photocatalytic activity in UV light, as well as the fast dye-sensitized photodegradation for many kinds of dyes. In avoid of the complete adsorption and too fast degradation process, a low dosage ratio of photocatalyst was adopted, 20 mg BTOF for 100 ml of 20 μM dye solution, only one fifth of the mostly adopted dosage of photocatalyst. Under ultraviolet irradiation, BTOF exhibits higher degradation rate than P25, as shown in Fig. 5a. Nearly 100% RhB was degraded by BTOF after UV irradiation for 40 min, while around 80% by P25 under the same condition. For comparison, result of blank photolysis experiment for RhB is also provided (Fig. 5a) and degradation of RhB was negligible. Under visible irradiation (λ > 420 nm) for 40 min, activity of P25 decrease notably and only less than 20% RhB was degraded, while BTOF degrade about 80% of total RhB, which is even comparable with the activity of P25 under UV light. For visual comparison, photos of the RhB-photocatalyst suspension taken every few minutes are shown in Fig. 5c. BTOF also show considerable activity in degradation of other cationic dye like MGO, but no apparent activity for anionic dye like MO under both UV light and visible light (Fig. 5b). Besides, the photodegradation activity of BTOF was compared with other surface-fluorinated TiO2. Yu et al. researched the photocatalytic activity of surface-fluorinated TiO2 hollow microspheres, which exhibit the higher photocatalytic activity than pure TiO2 or P25.27 BTOF, with less dosage than surface-fluorinated TiO2, cost a shorter time to degrade more RhB instead.
 |
| | Fig. 5 Photodegradation over BTOF. (a and b) Variation of dyes concentrations as a function of illumination time under UV/visible light; (c) photos of the dye-photocatalyst suspension sampled every 10 min for UV-P25-RhB and UV-BTOF-RhB, 15 min for Vis-BTOF-RhB; (d) cycling adsorption capacity and photodegradation efficiency of BTOF for RhB. Co for adsorption: the initial concentration of dye (RhB, MGO, or MO) before BTOF were added; Co for photodegradation and C for adsorption: the concentration of dye when adsorption–desorption equilibrium was established in dark. C for photodegradation: the concentration of dye after 10 min visible irradiation. | |
By comparing the photodegradation and adsorption of BTOF for dyes, we find that the degradation rate of dyes relies heavily on the amount of adsorbed dyes, which indicates a dye-sensitized photooxidation mechanism. More details of the dye degradation process will be discussed in the next section.
The performances recyclability of BTOF in adsorption and photodegradation of cationic dyes were also monitored using RhB for seven cycles (each cycle lasts 40 min, 30 min for adsorption and 10 min for photodegradation under visible light. After each cycle, the residual RhB was removed thoroughly by UV irradiation, and the same amount of RhB was added for the next cycle.). Fig. 5d shows the cyclic performance of 50 mg BTOF powders for 100 ml of a 20 μM RhB aqueous solution. The adsorption amount and photodegradation rate decreased slightly in the second cycle and was stable thereafter, indicating that the prepared BTOF is recyclable both as adsorbent and photocatalyst.
3.5 Mechanism for photodegradation
The absorption spectrum in Fig. 6a illustrates that BTOF can only absorb photons with wavelength shorter than 374 nm. Further analysis of the adsorption spectrum reveals a band gap of 3.32 eV (inset of Fig. 6a), indicating that BTOF cannot harvest visible light (λ > 420 nm) and produce reactive species directly. It's known that dyes can harvest visible light, and be degraded through dye-sensitized photooxidization (DSPO). Hence, DSPO mechanism can well explain the degradation of cationic dyes under visible light.
 |
| | Fig. 6 (a) Ultraviolet-visible diffusive reflectance spectrum of the BTOF sample. Inset, band gap of BTOF; (b) UPS spectra of the BTOF. Insets, enlarged views of the indicated areas. | |
To accomplish interfacial electron transfer, the conduction band (CB) edge of the semiconductor photocatalyst should be lower than the lowest unoccupied molecular orbital (LUMO) level of excited dye.28 The energy value of the CB edge of BTOF with reference to the vacuum level is calculated to be −3.2 eV and based on the UPS spectrum of BTOF with respect to the He I photon energy (21.22 eV) shown in Fig. 6b. The LUMO level of RhB is calculated to be −3.08 eV according to the equation E (eV) = −4.5 − ENHE (V).28 And its redox potential versus a normal hydrogen electrode (ENHE, −1.42 V).29 Apparently, the lower CB edge of BTOF elicits a driving force for the photo-generated electrons to transfer from the dye to the BTOF.
More details of the photodegradation process are shown in the schematic diagram (Fig. 7). Firstly, the cationic dye is largely adsorbed on the surface of the BTOF powders through the electrostatic attraction. Once exposed to the light, the electrons in dyes can be excited from the highest occupied molecular orbital (HOMO) to the LUMO,30 then transferred into the lower conduction band of BTOF (eqn (1) and (2)). In this process, the holes are also formed from photoexcited RhB. It is noteworthy that the adsorption is the requisite of dye degradation because it offers the channel of electron transfer. Secondly, the excited electrons are scavenged by O2 at the active sites (Ti–F) in the surface of the BTOF NSs, which then produces superoxide radical O2˙− (eqn (3)). Similarly, holes react with H2O or hydroxyl ions to produce free hydroxyl radicals ˙OH (eqn (4)). At last, superoxide radical O2˙− and free hydroxyl radicals ˙OH, which are strong oxidants, can attack and decompose the dye molecules. Eventually, dye pollutants are degraded into water, carbon dioxide and mineral acids (eqn (5)).31
 |
| | Fig. 7 Schematic drawings of the detail of dye-sensitized photooxidation process. | |
What is the reason that the photocatalytic activity of BTOF is better than naked TiO2/P25 or surface-fluorinated TiO2/P25? One possible reason is that F− ions on the surface of BTOF could trap CB electrons and result in the reduction of the recombination rate of electron and hole, which plays a great important role in the degradation process.22,27 In all, the superior adsorption capacity and abundant Ti–F sites can reduce the recombination rate of electron and hole and accelerate the electron transfer rate between dye and catalyst, catalyst and O2, respectively, which eventually promotes the degradation of cationic dye under irradiation.
| | |
RhB + hν → hvb+ + ecb− electron and hole generation
| (1) |
| | |
ecb− + BTOF → evb− + BTOF + heat
| (2) |
| | |
H2O + hvb+ → ˙OHfree + H+
| (4) |
| | |
RhB + ˙OH + O2˙−→ CO2 + H2O + mineral acids
| (5) |
4. Conclusions
In this work, we prepared a new photocatalyst BTOF via a facile hydrothermal method and developed a self-fluorination treatment as a new method of surface modification of photocatalyst. BTOF NSs exhibit strong electro negativity and hydrophilic as the existence of abundant Ti–F groups in the surface, which contributes to its superior performances of adsorption and photodegradation for cationic dyes. In comparison with the well-known P25 used in dye photodegradation, it exhibits 100-fold improvement in adsorption capacity, 1.25-fold and 4-fold improvement in photodegradation rate of cationic dyes under UV and visible light, respectively. Moreover, the BTOF shows good recyclability in adsorption and degradation of cationic dyes as the stable fluorination by the domestic F atoms. As a low-cost, effective adsorbent and photocatalyst, BTOF shows great promise in dye-polluted water treatment.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (91333203), the Natural Science Foundation of Zhejiang province (LY14E020006) and the Doctorate Fund of the Ministry of Education (2011010110013).
Notes and references
- K. Tennakone, G. R. R. A. Kumara, A. R. Kumarasinghe, K. G. U. Wijayantha and P. M. Sirimanne, Semicond. Sci. Technol., 1995, 10, 1689 CrossRef.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- B. O'Regan and M. Grfitzeli, Nature, 1991, 353, 737–740 CrossRef PubMed.
- Z. Zhang, Y. Yuan, G. Shi, Y. Fang, L. Liang, H. Ding and L. Jin, Environ. Sci. Technol., 2007, 41, 6259–6263 CrossRef CAS.
- M. Saquib and M. Muneer, Desalination, 2003, 155, 255–263 CrossRef CAS.
- K. Sayama, K. Hara, N. Mori, M. Satsuki, S. Suga, S. Tsukagoshi, Y. Abe, H. Sugihara and H. Arakawa, Chem. Commun., 2000, 1173–1174 RSC.
- K. Sayama, S. Tsukagoshi, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga and H. Arakawa, J. Phys. Chem. B, 2002, 106, 1363–1371 CrossRef CAS.
- A. C. Khazraji, S. Hotchandani, S. Das and P. V. Kamat, J. Phys. Chem. B, 1999, 103, 4693–4700 CrossRef CAS.
- M. Stylidi, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2004, 47, 189–201 CrossRef CAS PubMed.
- L. Lucarelli, V. Nadtochenko and J. Kiwi, Langmuir, 1999, 16, 1102–1108 CrossRef.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS PubMed.
- G. Benkö, J. Kallioinen, J. E. I. Korppi-Tommola, A. P. Yartsev and V. Sundström, J. Am. Chem. Soc., 2001, 124, 489–493 CrossRef PubMed.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS PubMed.
- A. Hattori, M. Yamamoto, H. Tada and S. Ito, Chem. Lett., 1998, 8, 707–708 CrossRef.
- A. Hattori, M. Yamamoto, H. Tada and S. Ito, Langmuir, 1999, 15, 5422–5425 CrossRef CAS.
- J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Lett., 2002, 14, 3808–3816 CAS.
- J. H. Pan, X. Zhang, A. J. Du, D. D. Sun and J. O. Leckie, J. Am. Chem. Soc., 2008, 130, 11256–11257 CrossRef CAS PubMed.
- H. Park and W. Choi, J. Phys. Chem. B, 2004, 108, 4086–4093 CrossRef CAS.
- Z. Q. He, Q. L. Cai, F. Y. Hong, Z. Jiang, J. M. Chen and S. Song, Ind. Eng. Chem. Res., 2012, 51, 5662–5668 CrossRef CAS.
- K. Lv, B. Cheng, J. Yu and G. Liu, Phys. Chem. Chem. Phys., 2012, 14, 5349–5362 RSC.
- J. Yu, W. Wang, B. Cheng and B. L. Su, J. Phys. Chem. C, 2009, 113, 6743–6750 CAS.
- H. Kodama, F. Izumi and A. Watanabe, J. Solid State Chem., 1981, 36, 349–355 CrossRef CAS.
- W. F. Yao, H. Wang, X. H. Xu, S. X. Shang, Y. Hou, Y. Zhang and M. Wang, Mater. Lett., 2003, 57, 1899–1902 CrossRef CAS.
- H. Cheng, B. Huang, K. Yang, Z. Wang, X. Qin, X. Zhang and Y. Dai, ChemPhysChem, 2010, 11, 2167–2173 CrossRef CAS PubMed.
- H. Cheng, B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC.
- J. Yu, Q. Xiang, J. Ran and S. Mann, CrystEngComm, 2010, 12, 872–879 RSC.
- B. Li and H. Cao, J. Mater. Chem., 2011, 21, 3346–3349 RSC.
- T. Shen, Z. G. Zhao, Q. Yu and H. J. Xu, J. Photochem. Photobiol., A, 1989, 47, 203–212 CrossRef CAS.
- R. Vinu, S. Polisetti and G. Madras, Chem. Eng. J., 2010, 165, 784–797 CrossRef CAS PubMed.
- Y. Xie and C. Yuan, Appl. Catal., B, 2003, 46, 251–259 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14288a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.