
Plasmon induced Au particle and surface oxidation co-decorated BiOIO3 heteronanostructures with highly promoted photocatalysis and photoelectrochemical properties
Abstract
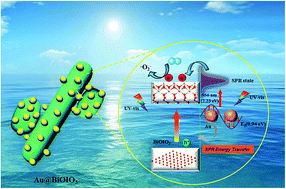
Plasmon induced Au nanoparticle and surface oxidation induced co-decorated BiOIO3 heterostructured nanocomposites have been developed via a facile in situ photosynthesis route. The structural and optical properties of the as-prepared photocatalysts were systematically characterized by XRD, XPS, TEM, SEM, UV-Vis DRS and PL. Fascinatingly, the introduction of Au nanoparticles induced not only an enhanced photoabsorption in the visible region, but also the microstructural variation of BiOIO3. The oxidative effect of HAuCl4 resulted in the formation of Bi4+/Bi5+, which led to the increased specific surface area of the products. The photocatalysis and photoelectrochemical properties of the samples were investigated by monitoring the photodecomposition of Rhodamine B (RhB) and photocurrent generation under UV-visible light illumination. The results revealed that Au@BiOIO3 presents drastically enhanced photoreactivity compared with the pristine BiOIO3. The highly improved photochemical properties are ascribed to the synergic contribution of the highly promoted generation and separation of charge carriers induced by the surface plasmon resonance (SPR) effect of Au particles, surface chemical state change, as well as the significantly high surface area that provides more reactive sites. These results are corroborated by the electrochemical impedance spectra (EIS), bode-phase spectra, PL spectra, active trapping and DMPO-assisted ESR measurements. This study not only provides evidence for the feasibility of metallic Au as a SPR co-catalyst of bismuth-based materials, but also furnishes new insights into the multiple effects for enhancing the photochemical properties.
 Please wait while we load your content...
Please wait while we load your content...

